

Abstract
The Escherichia coli single-stranded DNA binding protein (SSB) is a central player in DNA metabolism where it organizes genome maintenance complexes and stabilizes single-stranded DNA (ssDNA) intermediates generated during DNA processing. Due to the importance of SSB and to facilitate real-time studies, we developed a dual plasmid expression system to produce novel, chimeric SSB proteins. These chimeras, which contain mixtures of histidine-tagged and fluorescent protein(FP)-fusion subunits, are easily purified in milligram quantities and used without further modification, a significant enhancement over previous methods to produce fluorescent SSB. Chimeras retain the functionality of wild type in all assays, demonstrating that SSB function is unaffected by the FPs. We demonstrate the power and utility of these chimeras in single molecule studies providing a great level of insight into the biochemical mechanism of RecBCD. We also utilized the chimeras to show for the first time that RecG and SSB interact in vivo. Consequently, we anticipate that the chimeras described herein will facilitate in vivo, in vitro and single DNA molecule studies using proteins that do not require further modification prior to use.
Keywords: single stranded DNA binding protein (SSB), fluorescent reporter, DNA replication, DNA repair, DNA recombination
Introduction
The Escherichia coli (E. coli) single-stranded DNA binding protein (SSB) is essential to all aspects of DNA metabolism stabilizing ssDNA intermediates generated during DNA processing and organizing genome maintenance complexes.1–5 The protein, like the majority of eubacterial SSBs, exists as a stable homo-tetramer with a monomer Mw of 18,843 Da.6 Each monomer is divided into two domains defined by proteolytic cleavage: an N-terminal portion comprising the first 135 residues and a C-terminal domain that includes residues 136 to 177. The N-terminal domain contains elements critical to tetramer formation and the oligonucleotide/oligosaccharide binding-fold critical to the binding of ssDNA.7 DNA binding occurs via the wrapping of ssDNA around the SSB tetramer using an extensive network of electrostatic and base-stacking interactions with the phosphodiester backbone and nucleotide bases, respectively.7–9
The C-terminus, which is highly disordered even when the protein is bound to ssDNA10 has received considerable attention over the past few years as it contains a highly conserved region responsible for mediating interactions with several key proteins involved in DNA metabolism.11–20 Early studies indicated that the last 26 amino acids of SSB were critical to this interaction.16,21 Recent work has shown that it is only the C-terminal 11 residues that are critical for this interaction, which is central to the role of SSB as an organizer of dynamic, genome maintenance complexes.5
Because of its importance in DNA metabolism, real-time visualization of SSB in living cells and at the single DNA-molecule level is essential to further understanding its function and the dynamics of its interactions with partner proteins. As ssb is essential, mutagenesis to add fluorophores or to attach fluorescent proteins (FPs) must be carefully done. This is challenging as mutations in the monomer are present in the stable homo-tetramer, could perturb ssDNA binding or interactions with partner proteins and, mutations in the C-terminus can render cells inviable.22 Previous work has demonstrated that ssb can be mutated at internal sites to produce mono-cysteine variants which when functionalized, can be used as sensitive reporters of DNA unwinding and in DNA binding studies.23,24 These mutant and modified SSB proteins are limited to only in vitro studies however. In vivo studies have been done with fluorescent SSBs but these used uncharacterized fusions or carefully constructed fusions with FPs optimized for eukaryotic cells. Further, characterization of the protein resulting from the fusion was not provided, although the fusion strain exhibited wild-type cell-cycle parameters but demonstrated genetic instability.25
To circumvent the above-mentioned problems and to produce carefully designed, SSB proteins that have broad utility, we developed a dual plasmid expression system. This system produces novel SSB proteins that are active, chimeric, fluorescent, and can be used in many studies without additional modification. These chimeric proteins bind ssDNA as rapidly as wild type, maintain their interactions with other DNA binding proteins of DNA metabolism, function as reporters of nascent ssDNA in vitro, and enable real-time visualization of SSB in vivo and on single molecules of DNA.
Results and Discussion
Design of chimeric and fluorescent SSB proteins
The approach to produce active and fluorescent, chimeric SSB proteins consists of a dual plasmid system. The first plasmid expresses N-terminal his6-SSB and the second expresses a C-terminal fusion of SSB to an auto- FP (Fig. 1A). When these proteins are expressed in the same cell, a heterogeneous population of SSB tetramers is produced that can be separated into distinct species using nickel-column chromatography. The rationale for this approach is as follows. First, an N-terminal his6-ssb construct was required to facilitate protein purification. His6-SSB subunits have wild type C-termini and should therefore retain interactions with partner proteins. Next, we selected the ssb C-terminus as the site to add FPs. Even though this region is critical to SSB function, it is flexible and is positioned away from the body of the protein.7,10 We reasoned that addition of a FP at the C-terminus would position it away from the body of the SSB tetramer and therefore, have minimal effects on tetramer formation and binding to either ssDNA or partner proteins (Fig. 1B). To position the FP even further away from the core of SSB, we added an additional six glycine residues to the C-terminus and then fused ssb-gly6 in separate constructs in-frame, to the N-terminus of either the cyan (CFP), green (GFP), or yellow (YFP), FPs (FP). Novel, monomeric auto-FPs were selected that have been optimized for expression and folding in E. coli, and have improved brightness and FRET Förster radii relative to previous variants.26 Selection of E.coli-optimized FPs is important so that the accumulation of fluorescence parallels protein expression, resulting in close correlation of biological function with fluorescence. As a control, we also used commercial superglo-GFP (sgGFP) that had the A206K mutation introduced to minimize GFP-mediated dimer formation.27–29
Figure 1.
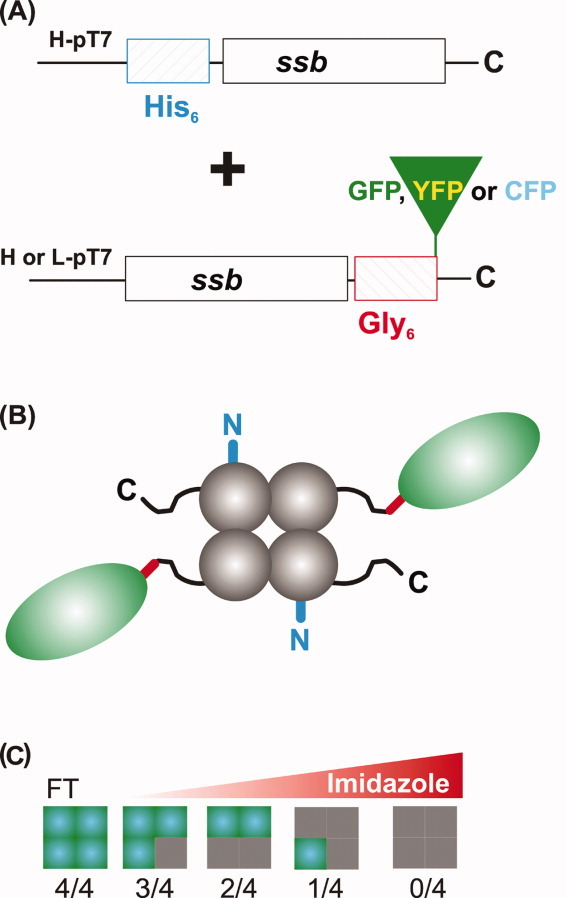
Scheme for production of FP-tagged SSB chimeras. A: The dual plasmid system showing the SSB-FP fusion and his6-SSB constructs. The designations L-pT7 and H-pT7 refer to low and high copy number plasmids, respectively. SSB expression in each plasmid is under the control of T7 promoters. B: Schematic of the 2/4 SSB chimera. N-terminal his6-tag (blue), the C-terminal extension (black), gly6-linker (red), and the FP (green oval) are shown. C: Application of cleared cell lysates to a nickel column permits separation of the heterogeneous pool of SSB proteins. Gray squares, his6-SSB subunits; green, SSB-FP fusions. FT, flow through. Values below each tetramer indicate the number of fusion subunits per tetramer. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To create a situation where subunit mixing would occur in vivo resulting in tetramers with different ratios of “wild type” to SSB-fusions, two expression strategies were adopted and used separately. The first, designed for protein purification for in vitro studies, used two high copy number vectors expressing the his6-SSB and SSB-FPs at equivalent levels (data not shown). This strategy, designated H/H (H = high copy number) is required to ensure that all possible ratios of SSB to SSB-FP would be obtained in the same cell (Fig. 1C). The second strategy, designated H/L (L = low copy number) is anticipated to facilitate in vivo studies where SSB fluorescence will be monitored. The unequal level of expression should lead to incorporation of only one or two fusions per tetramer to ensure that the resulting SSB proteins contained several subunits with wild type C-termini. Finally, as over-expression of auto-FPs can lead to misfolded protein lacking the desired fluorescence properties, an E. coli strain containing a mutation in lacY was used to provide IPTG concentration-dependent, homogeneous levels of induction (Table I).
Table I.
Strains and Plasmids
| Strains and plasmids | Relevant characteristics | Source |
|---|---|---|
| Strains | ||
| 27771 | Tuner (λDE3) harboring pET28a-ssb & pET21a-ssb-gly6-cfp; H/Ha | This study |
| 27772 | Tuner (λDE3) harboring pET28a-ssb & pET21a-ssb-gly6-yfp; H/H | This study |
| 27773 | Tuner (λDE3) harboring pET28a-ssb & pET21a-ssb-gly6-gfp; H/H | This study |
| 27774 | Tuner (λDE3) harboring pET28a-ssb & pET21a-ssb-gly6-msg-gfp; H/H | This study |
| 27764 | Tuner (λDE3) harboring pET28a-ssb & pWKS130-ssb-gly6-gfp; H/La | This study |
| 27632 | Tuner (λDE3) harboring pET15b-ssb & pWKS130-ssb-gly6-cfp; H/L | This study |
| 27634 | Tuner (λDE3) harboring pET15b-ssb & pWKS130-ssb-gly6-yfp; H/L | This study |
| 27636 | Tuner (λDE3) harboring pET15b-ssb & pWKS130-ssb-gly6-msg-gfp; H/L | This study |
| 27738 | Tuner (λDE3) harboring pET21a+-ssb & pWKS130-ssb-gly6-msg-gfp & pACYCDuet-mCherry-recG; H/L/L | This study |
| Plasmids | ||
| pET21a+-ssb | Expression of wtSSB; Ampr | Keck Lab |
| pET15b-ssb | Expression of N-terminal His6-SSB; Ampr | This study |
| pET28a-ssb | Expression of N-terminal His6-SSB; Kmr | This study |
| pSCFP3A-C1 | Monomeric CFP expression plasmid; Kmr | (26) |
| pSGFP3A-C1 | Monomeric GFP expression plasmid; Kmr | (26) |
| pSYFP3A-C1 | Monomeric YFP expression plasmid; Kmr | (26) |
| pQBI-T7-GFP | Superglo GFP (sgGFP) expression plasmid; Ampr | QBiogene |
| pET21a-ssb-gly6-cfp | High copy number plasmid expression of SSB-gly6-CFP; Ampr | This study |
| pET21a-ssb-gly6-gfp | High copy number plasmid expression of SSB-gly6-GFP; Ampr | This study |
| pET21a-ssb-gly6-yfp | High copy number plasmid expression of SSB-gly6-YFP; Ampr | This study |
| pWKS130-ssb-gly6-cfp | Low copy number plasmid expression of SSB-gly6-CFP; Kmr | This study |
| pWKS130-ssb-gly6-gfp | Low copy number plasmid expression of SSB-gly6-GFP; Kmr | This study |
| pWKS130-ssb-gly6-yfp | Low copy number plasmid expression of SSB-gly6-YFP; Kmr | This study |
| pWKS130-ssb-gly6-msg-gfp | Low copy number plasmid expression of SSB-gly6-msgGFPA206K; Kmr | This study |
| pACYCDuet- mCherry-recG | Low copy number plasmid expression of His6-mCherry-RecG; Cmr | Unpublished |
H: high copy number plasmid; L: Low copy number plasmid
Purification of chimeric and fluorescent SSB proteins
Following over-expression and cell lysis, cleared cell lysates were applied to a nickel column to isolate individual tetramers. As homo-tetrameric SSB-fusions do not contain a histidine tag they are expected to elute in the column flow through (Fig. 1C). The remaining tetramers bind to the resin with different affinities due to the presence of one to four histidine tags, with the homo-tetrameric, his6-SSB binding with the greatest affinity. By applying an imidazole gradient, tetramers of different composition are eluted with increasing concentrations of imidazole [Figs. 1(C) and 2(A, B)]. Fraction pooling was done carefully as the intervening regions contain mixtures of different ratio chimeras.
Figure 2.
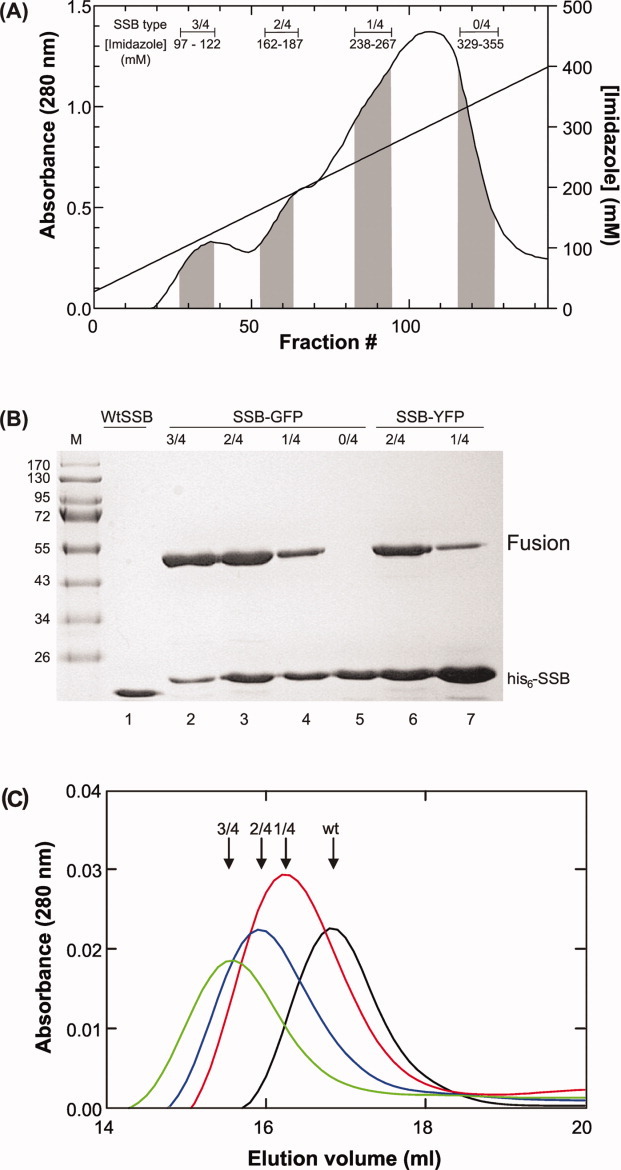
FP-tagged SSB chimeras can be easily purified. A: SSB chimeras elute at different concentrations of imidazole. A Ni2+-column elution profile from a 1L, H/H (his6-SSB/SSB-GFP) cell lysate is shown. The elution positions of each chimera as determined by SDS-PAGE are indicated (gray-shaded regions). Intervening regions contain mixtures of the two species flanking the region. B: Purified proteins are chimeric. SDS-PAGE gel of final protein pools. Lanes 2–5 are from the profile shown in panel (a); lanes 6 and 7 are from a separate preparation from cells expressing his6-SSB and SSB-YFP fusion protein from H/L plasmid constructs. C: Chimeras elute from the Superose 6 gel filtration column at distinct positions. The elution positions of Wt and chimeric SSBs are indicated. Wt eluted as a homotetramer with Mw of 76 kDa. The chimeras elute at positions corresponding to octamers with Mws of 198, 249, and 333 kDa for 1/4, 2/4, and 3/4 chimeras, respectively. Predicted Mws as determined from the primary amino acid sequence are 110, 135, and 161 kDa, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We expected the mass ratio of fluorescent to his6-SSB subunits to be ∼ 7:1 for 3/4 chimera (i.e., three fusion and one his-tag subunits); ∼ 2:1 for the 2/4, and 1:1.4 for the 1/4 protein. To determine these ratios in the purified chimeras, densitometric analysis of SDS-PAGE gels was used. The results show that mixing occurred in vivo, as expected and that the ratio of fusion to his6-SSB subunits is within experimental error, exactly as predicted (Fig. 2B). Similar results were obtained with all four FP fusions (data not shown). When the H/L plasmids were used for over-expression, only the 2/4 and 1/4 chimeric SSBs were obtained in significant quantities with the majority of SSB protein eluting as homo-tetrameric his6-SSB (Fig. 2B and data not shown). Therefore, mixing occurs in vivo with both plasmid systems, but different yields of each chimeric SSB are obtained. The properties of the chimeras are listed in Table II.
Table II.
Biochemical and Biophysical Properties of Chimeric SSB Fluorescent Proteins
| Chimeric SSB fluorescent proteins | Molecular mass (kDa)a | Calculated ɛ280 (M−1 cm−1)b | # of SSB FP fusion subunits/tetramer | Subunit mass ratio (His-SSB:SSB-FP)C | Excitation/Emission peak (nm)d |
|---|---|---|---|---|---|
| SSB-GFP 1/4 | 110.09 | 133,855 | 1 | 1.4 : 1 | 488/509 |
| SSB-GFP 2/4 | 135.62 | 155,870 | 2 | 1 : 2.2 | 488/509 |
| SSB-GFP 3/4 | 161.16 | 177,885 | 3 | 1 : 6.6 | 488/509 |
| SSB-YFP 1/4 | 110.07 | 135,345 | 1 | 1.4 : 1 | 515/528 |
| SSB-YFP 2/4 | 135.59 | 158,850 | 2 | 1 : 2.2 | 515/528 |
| SSB-YFP 3/4 | 161.11 | 182,355 | 3 | 1 : 6.6 | 515/528 |
| SSB-CFP 1/4 | 110.09 | 137,865 | 1 | 1.4 : 1 | 413/428 |
| SSB-CFP 2/4 | 135.62 | 163,890 | 2 | 1 : 2.2 | 413/428 |
| SSB-CFP 3/4 | 161.16 | 189,915 | 3 | 1 : 6.6 | 413/428 |
Tetramer molecular weights were calculated from the amino acid sequence of each subunit present in the tetramer.
The calculated extinction coefficient of the tetramer represents the sum total extinction coefficient of the individual fusion and his6-SSB subunits. An ɛ = 49,975 M−1 cm−1 was used for the GFP fusion monomers, ɛ = 51,465 M−1 cm−1 for YFP and ɛ = 53,985 M−1 cm−1 for CFP fusion monomers. The extinction coefficient for the his6-SSB is ɛ = 27, 960 M−1 cm−1. All extinction coefficients were calculated from the predicted amino acid sequence of subunits.28
The subunit mass ratios were determined by densitometric analysis of SDS-PAGE gels. The expected the mass ratio of fluorescent to his6-SSB subunits is 6.6:1 for 3/4 chimera (i.e., 3 fusion and one his-tag subunits); 2.2:1 for the 2/4, and 1:1.4 for the 1/4 protein.
The excitation and emission maxima are from the associated fluorescent proteins. These values did not change appreciably in the fusions.
It is conceivable that subunit mixing does not occur in vivo as expected but only takes place once cells are lysed. To test this, two separate cultures of his-SSB and SSB-GFP were grown to early log phase and induced with IPTG. Four hours later, the cultures were combined and cells harvested together by centrifugation. Next, lysis of the mixed culture was done, and the cleared cell lysate applied to the nickel column. Following extensive washing, the bound proteins were eluted in a single step.
The results show that when his-tagged wild type and SSB-GFP tetramers were mixed, the amount of SSB-GFP eluted from the column is extremely low, whereas there is an abundance of his-SSB (Supporting Information Figure 1B). In contrast, when the proteins are expressed in the same cell, there are almost equal amounts of his-SSB and SSB-GFP in the cleared cell lysate and further, there is a significant amount of each subunit in the pooled fraction eluted from the column (Supporting Information Figure 1C). Therefore, mixing occurs in vivo when subunits are coexpressed.
The analysis of the SDS-PAGE gels demonstrates that both fluorescent and nonfluorescent subunits are present in the final pool of protein. SDS-PAGE does not show whether both subunits are present in the same tetramer however. Therefore, it is necessary to ascertain whether a single chimeric species is present and to determine its molecular weight. To achieve these goals, aliquots of purified proteins were subjected to gel filtration chromatography.
The results show that the wild type SSB eluted from the column at ∼ 17 mL with the 1/4, 2/4 and 3/4 chimeras eluting increasingly earlier from the column, consistent with the incorporation of 1, 2, and 3 fluorescent subunits per tetramer, respectively (Fig. 2C). Analysis of Kav values revealed that the molecular weight of each chimera is consistent with an SSB octamer (data not shown). Dimer formation is attributed in part to the histidine tag blocking the SSB N-terminus, which functions to prevent octamer formation and to the presence of the FPs, which can induce dimer formation.30,31
Characterization of SSB chimeras
ssDNA binding – site size and salt stability
Following Mw characterization, the chimeric SSBs were evaluated for the capability to bind ssDNA by monitoring the quenching of the intrinsic fluorescence of tryptophan residues of SSB that occurs on binding to ssDNA.32 Control experiments using wild type and homo-tetrameric, his6-SSB proteins were done first producing the expected site size of 7 to 10 nt per SSB monomer (Table III). If the presence of the SSB-fusions negatively affects ssDNA binding, this could manifest as a change in the site size relative to wild type. This would suggest an inability of the fusion subunits to bind ssDNA or possibility, an inability of the ssDNA to fully wrap around the protein. Therefore, fluorescence quenching experiments using the C-, G- and YFP chimeras were done to evaluate ssDNA binding. The results show that the presence of as many as three FPs per tetramer does not significantly affect the site size (Table III). However, the stability of the SSB-DNA complex is affected by the N-terminal his-tag, consistent with previous work.30 This is reflected in a twofold reduction in the salt-titration midpoint (STMP) relative to wild type (Table III). Stability is restored when fusion proteins (with wild type N-termini) are introduced into the tetramer, with the STMP of the 3/4 chimera being the same as wild type. The effect of the his-tag on complex stability may not be that critical as the STMP is 430 ± 10 mM, well above that of partner proteins.19
Table III.
ssDNA Binding Properties of SSB Proteins
| Protein | Subunit composition (F:W)a | Site size (nt)b | STMP (mM)c |
|---|---|---|---|
| Wild type | 0:4 | 7 ± 2 | 710 ± 15 |
| His6-SSB | 0:4 | 10 ± 1 | 430 ± 10 |
| SSB-CFP/Wt-SSB | 1:4 | 10 ± 1 | - |
| SSB-CFP/Wt-SSB | 2:4 | 10 ± 2 | - |
| SSB-GFP/Wt-SSB | 1:4 | 6 ± 1 | 470 ± 14 |
| SSB-GFP/Wt-SSB | 2:4 | 9 ± 1 | 600 ± 30 |
| SSB-GFP/Wt-SSB | 3:4 | 9 ± 1 | 730 ± 7 |
| SSB-YFP/Wt-SSB | 1:4 | 13 ± 1 | - |
| SSB-YFP/Wt-SSB | 2:4 | 11 ± 1 | - |
F, fusion of SSB-auto-fluorescent protein; W, wild type or his6-SSB.
Data are from 2 to 4 independent titrations done on separate days.
Values are from 2 to 3 separate titrations done on the separate days.
ssDNA binding – association kinetics
Although the SSB chimeras bind ssDNA producing complexes similar to wild type, the presence of the auto- FPs could affect the rate of DNA binding, an effect that could not be readily discerned in equilibrium binding studies (Table III). Knowledge of association rate constants is important not only for in vitro studies but also in vivo where mixtures of chimeras are present. If the rate of binding of chimeras is inhibited by the FP, then the capability to monitor formation of ssDNA intermediates and or loading of partner proteins onto the DNA would be impaired and would instead be dominated by the more rapidly binding wild type homotetramers. This would then result in delays in appearance of fluorescence signal due to faster binding by the unmodified wild type.
Therefore, to measure the kinetics of association of SSB proteins to ssDNA, a series of stopped-flow experiments were done. Here, mixing of either wild type, or in separate experiments, chimeric SSB proteins (at 10 nM tetramer) with oligo d(T)70 at concentrations ranging from 75 to 125 nM was done and the reaction progress monitored by following the quenching of the intrinsic fluorescence of SSB that occurs on DNA binding.33
The results show that for wild type, each time course shows a single-exponential decrease in tryptophan fluorescence, similar to what has been shown previously (Fig. 3 and Ref. 33). Observed rates (kobs) obtained from single-exponential fits of the time courses as a function of ssDNA concentration yield a rate constant of (7.01 ± 1.69) × 108 M−1 s−1, similar to the previously published rate constant of (9.47 ± 0.30) × 108 M−1 s−1 for wild type SSB (Table IV and Ref. 33). Identical experiments were done with the 1/4 and 2/4 GFP, and YFP chimeras. As for wild type, the time course of binding of each chimeric protein to the oligo d(T)70 substrates shows a single-exponential decrease in fluorescence (Fig. 3). Analysis of time courses of binding yields second-order association rate constants for binding of chimeric SSB proteins to oligo d(T)70 that are virtually identical to that of the wild type protein (Table IV). Therefore, the association of chimeric SSB proteins with ssDNA is not affected by fusion of the protein to GFP or YFP.
Figure 3.
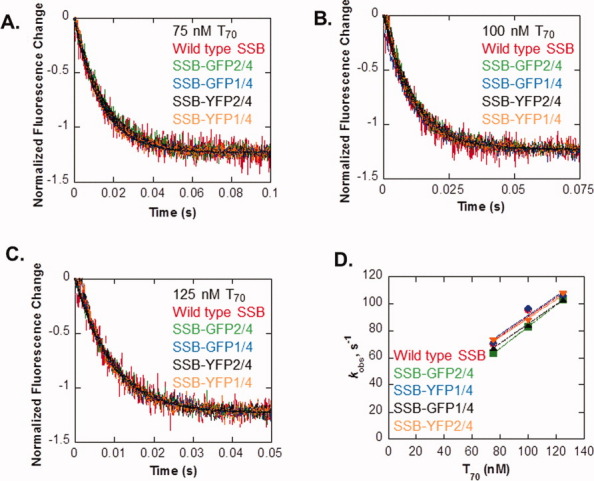
SSB chimeras associate with ssDNA as rapidly as wild type protein. The change in fluorescence over time is shown for 10-nM wild type SSB (red), SSB-GFP2/4 (green), SSB-GFP1/4 (blue), SSB-YFP2/4 (black), and SSB-YFP1/4 (orange) mixing with 75 nM T70 (A), 100 nM T70 (B), and 125 nM T70 (C). Rates obtained by single exponential fits of the data in A–C are plotted versus DNA concentration (D). Linear fits of the data provided the second-order association rate constants in Table III. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table IV.
Kinetic Constants for SSB Bindinga
| Wild type SSB | GFP1/4-SSB | GFP2/4-SSB | YFP1/4-SSB | YFP2/4-SSB | |
|---|---|---|---|---|---|
| kassoc, M−1 s−1 | (7.01 ± 1.69) × 108 | (7.03 ± 1.85) × 108 | (7.93 ± 0.09) × 108 | (6.90 ± 0.62) × 108 | (7.16 ± 0.24) × 108 |
Experiments were done as described in Materials and Methods using 10 nM SSB proteins and varying concentrations of d(T)70. The values for the second order rate constants were obtained from linear fits to the data in Figure 3D.
ssDNA binding – reporting formation of nascent ssDNA intermediates
The protection of ssDNA intermediates resulting from a DNA transaction is central to SSB function.5. Therefore to determine whether the SSB chimeras can bind to nascent ssDNA, we tested them in real-time DNA helicase assays using the E.coli RecBCD enzyme.34 This DNA helicase is the most rapidly moving and highly processive helicase characterized to date.35,36 Furthermore, RecBCD unwinding in the presence of wild type SSB has been well documented, in particular, by monitoring the quenching of the intrinsic fluorescence of SSB, which occurs on binding to ssDNA. This analysis is important as it may more accurately reflect nascent ssDNA binding by SSB in vivo, which is different from binding of phage ssDNA used to characterize the site size (Table III).
The rate of DNA unwinding by RecBCD in the presence wild type SSB was 199 ± 22 bp/s, consistent with previous work (Fig. 4 and Ref. 34). The rate of DNA unwinding in the presence of each 2/4 chimera was within experimental error the same as wild type yielding values of 157 ± 39, 195 ± 24, and 172 ± 14 bp/s for the C-, G- and YFP chimeras, respectively. Therefore, SSB chimeras accurately report formation of nascent ssDNA intermediates by a DNA helicase and furthermore, they do not inhibit DNA unwinding by RecBCD enzyme.
Figure 4.
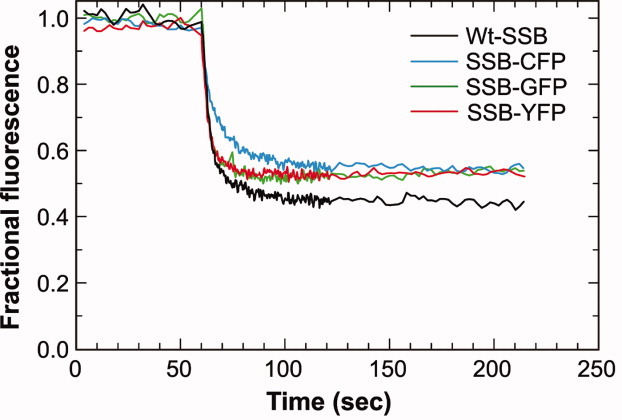
SSB chimeras function as reporters of nascent ssDNA in vitro. Typical unwinding traces from experiments in the presence of wild type or 2/4 SSB-chimeras are shown. The extent of unwinding for Wt (n = 5) is 53% and for the chimeras 52 ± 2% (n = 3 each). In all assays, RecBCD was added at t = 60 s to initiate DNA unwinding. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
SSB chimeras physically and functionally interact with a partner protein
Previous work has demonstrated physical and functional interactions between SSB and several enzymes of DNA metabolism.17,19,20 These interactions are mediated via the essential SSB C-terminus. To determine if chimeras retain this capability we examined the physical interaction between RecG and SSB in vitro. Protein coprecipitation assays are used to demonstrate interactions between the C-terminus of SSB and cognate proteins.19 This assay relies on the capability of SSB to precipitate in low concentrations of ammonium sulfate, whereas partner proteins do not. Thus, coprecipitation provides evidence of protein–protein interactions and elimination of the C-terminus of SSB abrogates these interactions.18,19
The control experiments show that as before, wild type SSB and RecG coprecipitate with >70% of the input RecG appearing in the pellet (Fig. 5A and Ref. 19). Similar results are obtained with His6-SSB indicating that the histidine tag does not affect the binding of the protein to RecG. More importantly, each of the SSB chimeras binds to RecG indicating that the C-termini of the his6-subunits within each chimera are accessible for binding to cognate proteins.
Figure 5.
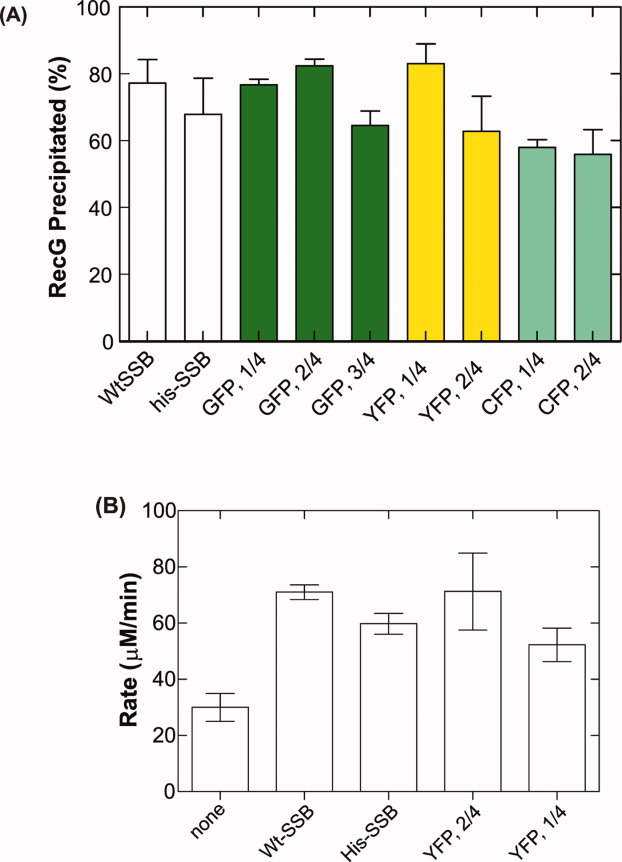
SSB chimeras maintain physical and functional interactions with RecG. A: Chimeras bind to RecG as efficiently as wild type. The amount of RecG precipitated is expressed as a fraction of the total amount present in the reaction. Data are from two to five experiments done on separate days. B: SSB chimeras stabilize the binding of RecG to ssDNA as effectively as Wt-SSB. RecG, ssDNA-dependent ATPase assays were done in the presence or absence of SSB as indicated.19 The data represent two to four independent NaCl titrations. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
It is conceivable, that the purified chimeras are unstable and that the coprecipitation results from rearrangement into homotetramers followed by binding to the wild type subunits. Therefore, the stability of the heterotetramers is an important issue to address. This was evaluated using crosslinking using dimethylsuberimidate followed by analysis using denaturing PAGE as described previously for wild type SSB (Supporting Information Fig. 2; Ref. 22). The results show that chemical crosslinking leads to the appearance of a covalently connected heterotetramer with a Mw = 138KDa (complex 1); a heterotrimeric complex consisting of 2 wt and 1 SSB-GFP subunit (complex 2) and a heterodimer (complex 3). These results are similar to what was observed for wild type SSB and support the notion that the purified heterotetramers are stable complexes.22
In addition to a physical interaction, functional interactions between SSB and repair helicases also play a key role in tetramer function.17,19,20 To determine if these interactions are maintained in the chimeras, we monitored the effects of the SSB chimeras on the STMP of the ATPase activity of RecG. Previous work showed that wild type SSB increases the titration midpoint of RecG from 35 to 70 mM NaCl.19,37 This increase is consistent with SSB stabilizing the binding of RecG to ssDNA. Therefore, if the presence of the fusion subunits negatively affects the functional interaction between chimera SSB and RecG, a failure to stabilize binding of RecG to ssDNA would be observed and manifest as an inability to increase the STMP of the enzyme.
Control experiments demonstrate that His6-SSB stimulates the STMP of RecG by a factor of two, similar to that observed for wild type and consistent with previous results (Fig. 5B and Ref. 19). Importantly, the results also show that SSB chimeras containing either one or two fusions subunits per tetramer are also able to increase the STMP of RecG 2-fold (Fig. 5B). Thus, the functional interaction between SSB and RecG is unaffected by the presence of auto-fluorescent fusion proteins in tetramers.
SSB chimeras report formation of nascent ssDNA in a single DNA molecule assay
We have demonstrated that the SSB chimeras contain fluorescent reporter subunits at expected ratios and that the chimeric tetramers bind to ssDNA as efficiently as wild type SSB. To determine whether the fluorophores enable visualization of SSB, we used an established single DNA-molecule, unwinding assay shown in schematic form in Figure 6A.38 In this assay, the 2/4 SSB-GFP chimera that was used to report formation of nascent ssDNA (Fig. 4), was also used to report DNA unwinding by the E.coli RecBCD enzyme on single molecules of DNA.
Figure 6.

SSB chimeras report DNA unwinding on single molecules of DNA. A: A schematic of the single DNA molecule assay for binding of SSB-FP to nascent ssDNA in real time. A microfluidic chamber the size of a microscope slide is viewed from the top. The device contains two fluid streams that flow parallel to one another with minimal mixing. Solutions are introduced into the chamber using a syringe pump. The position of an optically trapped DNA molecule is highlighted by the gray-shaded sphere in the center of the flow cell. The X indicates the position of the trap. The arrow above the flow cell indicates the direction of flow and the direction in which the attached DNA molecules are stretched by fluid flow. The arrow to the left of the inset indicates the direction the motorized stage is moved once a DNA-bead complex has been trapped. The result of this motion is to translate the trapped DNA (green strand, inset) from the sample (S) to the reaction stream (R) of the flow cell where it can be condensed (indicated as shortening of the dsDNA concomitant with formation of a toroid-like structure). Inset, binding to nascent ssDNA produced by the helicase activity of RecBCD. The 2/4 SSB-GFP chimera (or in separate assays, the 2/4 SSB-YFP chimera) was added to DNA beads followed by addition of magnesium acetate and ATP. RecBCD was added last and the reaction mix was immediately transferred to the syringe and introduced into the flow cell. DNA-bead complexes which contained partially unwound DNA molecules were translated into the adjacent stream to visualize SSB-FP bound to the nascent ssDNA. B: Selected, sequential frames from a video recording of the unwinding of a single dsDNA molecule by RecBCD. The direction of unwinding is from right to left. Numbers at the lower right of each frame indicate real time. Arrows indicate the position of a bright spot correlating with binding of SSB-YFP (2/4). The DNA molecule is faintly visible due to bleed through of the YOYO-1 fluorescence into the YFP channel. The movie is available as Supporting Information. C: Analysis of the movie in (B). Upper panel, a record of position; lower panel, instantaneous velocity. The time-dependent position of the bright spot is obtained by smoothing with a second-order Savitzky-Golay filter with a time constant of 1 s (red line). The instantaneous velocity for RecBCD is determined by using a differential average and velocities are averaged to obtain rates (n = 4).39 Arrows indicate pauses. D, E: Individual movies frames from single DNA-molecule unwinding reactions. DNA molecules in each panel are of different lengths as they were visualized at different stages of the unwinding reaction. In panel (b), SSB is visible at the free end of the DNA distal to the bead (arrow). F: Analysis of the fluorescence intensity of the DNA-bead complexes in panels (b) and (c). Intensity was measured using the line profile tool of Image Pro. The intensity due to the bead is indicated. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Bacteriophage λ DNA substrates were constructed as described and then attached at low density to 1-μm streptavidin-coated, polystyrene beads.39 The fluorescent dye YOYO-1 was bound to the DNA and these fluorescent-labeled, single molecules of DNA were reacted at 23°C with RecBCD and ATP in the presence of the chimeric SSB protein (Fig. 6A, inset). During the course of the reaction, the DNA-bead complex was optically trapped in the first stream of the Y-shaped, microfluidic, laminar flow cell (Fig. 6A and Refs. 38–40). Visualization buffer containing ATP without SSB was introduced into the second stream under conditions of laminar flow, creating a situation where the two streams flowed parallel to one another with minimal mixing. Once a DNA-bead complex was trapped, the stage was moved so that the partially unwound DNA is translated from the sample to the reaction stream of the flow cell, where continued unwinding could be visualized. It was necessary to trap and translate partially unwound DNA molecules due to the high fluorescence background from the SSB chimera that precludes clear visualization in the sample stream.
The results show the first demonstration of an SSB protein monitoring the production of ssDNA by a DNA helicase in real time as the SSB chimera binds to the nascent unwound strands of DNA produced by the DNA helicase activity of RecBCD (Fig. 6B). Binding is visualized as a region of enhanced fluorescence distal to the bead in the flow (Fig. 6B, arrows). Analysis of the movie in Figure 5B shows that DNA unwinding occurs at a rate of 951 ± 39bp/s, consistent with in vitro bulk-phase and single molecule studies (Fig. 6C and Refs. 38 and 41). Furthermore, and as we showed in Figure 4, the chimeric SSB does not inhibit translocation and DNA unwinding by RecBCD as the calculated rate is within experimental error, identical to that reported previously.38 The analysis also shows that during the course of the reaction, the rate of unwinding varies from >2,000 bp/s to less than 50 bp/s. Further, RecBCD also pauses transiently and backtracks briefly before continuing to unwind the DNA duplex (Fig. 6C; t = 6 s; arrows). Previous studies of RecBCD translocation and DNA unwinding showed that the rate of unwinding is constant.38,42 In addition, pauses were only detected at the recombination hotspot chi.43 The improved visualization afforded by the SSB chimera enables a higher level of resolution so that the rate changes and pauses at sites other than chi as well as enzyme backtracking can be clearly observed. A more extensive analysis of RecBCD unwinding at this higher level of resolution is currently underway.
The accumulation of fluorescence at the free end of the DNA distal to the bead in the flow is not due to the fluorescence signal from the DNA stained with YOYO-1. Instead it is due to the binding of the SSB chimera to the nascent ssDNA. To demonstrate this, the comparison of two DNA molecules unwound in the presence or absence of SSB-GFP is presented [Fig. 6(D,E)]. In these reactions, the DNA stained with YOYO-1 is clearly visible. A region of enhanced fluorescence on the DNA distal to the bead in the flow is visible only in the reaction containing fluorescent SSB [Fig. 6D, arrow]. The presence of the fluorescent chimera results in a fivefold greater fluorescence signal at the free end of the DNA where unwinding occurs, relative to the unwound part of the fluorescent DNA molecule. In contrast, and consistent with previous results,38 no fluorescence enhancement is observed at the unwinding site in the absence of the SSB chimera [Fig. 5(E,F)]. In these reactions, the fluorescence intensity across the DNA molecule is relatively uniform. In summary, the fluorescent and chimeric SSB proteins accurately report formation of ssDNA by a DNA helicase in real time on single molecules of DNA.
SSB chimeras enable visualization of SSB in vivo
The above-mentioned data demonstrate the utility of the fluorescent SSB chimeras for in vitro studies. To determine whether chimeras can be used as reporters of SSB in vivo, cells expressing the SSB-GFP chimera were transformed with a compatible, low copy number vector encoding a his6-mCherry-RecG fusion. In these experiments, the H/L SSB plasmid system was used with wild type SSB replacing his6-SSB to minimize octamer formation. The H/L combination was used to ensure that SSB tetramers would contain at least one fluorescent reporter subunit with the remaining subunits available for interaction with RecG. Cells were grown to early log phase, expression induced with 100-μM IPTG and analyzed using wide-field, epi-fluorescence microscopy.
The images show that SSB fluorescence is observed primarily in clusters that are often observed at opposing poles of the cell consistent with previous results (Fig. 7, arrows 1 and 2, and Ref. 44). In contrast, RecG fluorescence presents as a more uniform distribution within cells. Analysis of these images results in a Pearson's correlation coefficient of 0.88 indicating that RecG and SSB colocalize in vivo (Fig. 7D, arrows 3 and 4). These results suggest that RecG and SSB interact in vivo under conditions of exponential cell growth in the absence of exogenous DNA damage.
Figure 7.
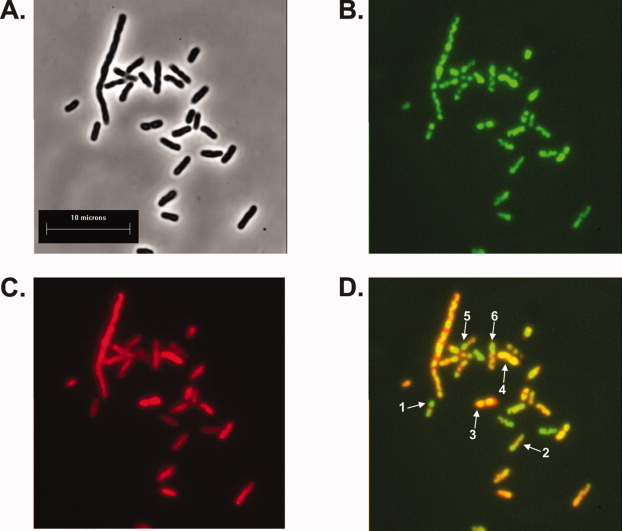
SSB chimeras can be visualized in vivo. Representative microscopy images showing colocalization of SSB and RecG. A: phase contrast image; B: the same field of cells viewed in epi-fluorescence using a GFP filter; C: the same cells viewed using a texas red filter, and D: merge of panels B and C. The Pearson's correlation coefficient for this image is 0.88. Arrows 1 and 2, SSB-GFP foci which do not colocalize with mcherry RecG; arrows 3 and 4, SSB foci which clearly colocalize with RecG and arrows 5 and 6, cells where expression of both proteins occurs but colocalization is not observed. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To further explore the interaction between SSB and RecG in vivo, 1L cultures of the SSB/SSB-YFP/his6-mcherry RecG cells were grown under identical conditions to those used for the microscopy. Clarified cell lysates from these cultures were applied to a nickel column and eluted with an imidazole gradient. The fractions which were eluted with the gradient contained both his6-mCherry-RecG and the SSB chimera, demonstrating that fluorescent tetramers retain functional interactions with partner proteins in vivo (unpublished observations). To test whether this interaction requires the C-terminus of SSB, experiments were repeated using SSBΔC8/SSB-YFP/his6-mcherry RecG cells. SSBΔC8 lacks the terminal eight residues critical for protein–protein interactions and does not bind RecG, RecQ, ExoI, or PriA in vitro.17–19,45 The results show that when SSB is replaced with SSBΔC8, mcherryRecG and only a very small fraction of YFP fluorescence eluted from the column. To determine whether these in vivo interactions are affected by the presence of the FPs, experiments were repeated with 1L cultures of SSB/his6-RecG and separately, SSBΔC8/his6-RecG cells. As for the FPs, RecG, and SSB coeluted from the nickel column and coelution required the C-terminus of SSB (unpublished observations). Finally, the colocalization data are also consistent with preliminary in vitro studies demonstrating fluorescence resonance energy transfer between the 2/4 SSB-GFP chimera and mCherry-RecG bound simultaneously to a model fork substrate (unpublished observations). Collectively, these data suggest that SSB and RecG interact in vivo and that the FPs do not inhibit functional interactions between these two proteins. Furthermore, these in vivo results are consistent with previous work showing that SSB and RecG interact in vitro.19 The above-mentioned unpublished results will be published separately.
Conclusions
We have developed a simple and rapid method to produce active, chimeric and fluorescent SSB proteins. These carefully designed chimeras retain the functionality of wild type both in vivo and in vitro, while simultaneously providing real-time visualization. These properties are provided by the distinct C-termini: wild type for the his-tagged subunit and fluorescent for the fusion subunit. These features, combined with the histidine tag, enable rapid purification of milligram quantities of FPs. We anticipate that the chimeras described herein will facilitate in vivo, in vitro, and single DNA molecule studies using proteins that do not require further modification. The use of proteins that do not require additional modifications to enable visualization is a significant advantage over previous methods to produce fluorescent SSB derivatives.23,24 The proteins produced in this study contain a defined number of fluorophores with the final preparation containing undetectable levels of unmodified, non- FP that can contaminate in vitro labeling reactions.23,24 Furthermore, and relevant to the in vivo situation where tetramer mixtures exist, the chimeric proteins bind to ssDNA as rapidly as wild type. This means that the chimeras compete on equal footing for ssDNA binding with wild type and therefore, should provide accurate monitoring of formation of ssDNA intermediates and/or binding to cognate proteins.
To demonstrate the utility of the heterotetramers, we have used purified SSB chimeras to provide a greater level of insight into the mechanism of translocation and DNA unwinding by RecBCD on single molecules of DNA. The results show that RecBCD pauses, backtracks, and changes translocation velocity on DNA substrates that do not contain the recombination hotspot chi. We have also used the chimeras to demonstrate for the first time that RecG and SSB interact in vivo. These findings, combined with the careful and detailed characterization of the chimeras, demonstrate the power and utility of these SSB proteins in DNA metabolic studies.
The design of the SSB fusion genes used in this study positions the auto- FP well away from the body of the tetramer resulting in minimal perturbation in protein function. Further, the fusion gene design permits facile in-frame insertion of other FPs in the same site at the C-terminus, permitting labeling of SSB in virtually any color. In principle, mixed-color chimeras could also be produced using three compatible plasmids. This would facilitate simultaneous visualization of the dynamic interaction of a single SSB tetramer with two different partner proteins using multicolor FRET. Finally, the dual plasmid system combined with nickel column chromatography can be readily adapted to any homo-multimeric protein where differential labeling or tagging of subunits or mixing of wild type and mutant subunits is required.
Materials and Methods
Cloning of his6-SSB and ssb-fusions
A pET21a+ plasmid over-expressing wild type SSB was a gift from Dr. James Keck (UW Madison). The ssb gene from this vector was subcloned into pET15b (Novagen) using NdeI and HindIII to create N-terminal his6-ssb. To create C-terminal auto- FP fusions, the stop codon of wild type ssb in pET21a+ was changed to glycine using the Lightning QuickChange mutagenesis kit (Stratagene). This places the ssb gene in frame with the C-terminal his6-tag of the vector. Following DNA sequencing, annealed oligonucleotides encoding a five glycine linker including an NgoMIV site were inserted downstream of the C-terminal glycine using HindIII and XhoI. The positive clones were verified by restriction enzyme mapping. Then, in separate reactions, genes encoding monomeric cyan, yellow, and green FPs were amplified using PCR and inserted into the XhoI site of ssb-gly6-his6 using Infusion cloning (Clontech). The design of oligonucleotides for PCR retained the stop codon of each of the FPs so that the resulting ssb-gly6-fusions are out of frame with the C-terminal his6-tag of the vector. The sources of the monomeric FPs are pSCFP3A-C1 for CFP, pSGFP2-C1 for GFP and pSYFP2-C1 for YFP.26 To ensure we had selected the correct FPs an additional source of GFP was used. pQBI-T7-GFP (QBiogene) contained the superglo GFP (sgGFP) gene, which was modified using the Lightning QuickChange mutagenesis kit to contain the A207K mutation. Cloning procedures using monomeric sgGFP A207K (msgGFP) were the same as that of the other FPs. SSB chimeras containing GFP2-C1 or msgGFP behaved in identical fashion and were used interchangeably in assays.
Dual-plasmid expression systems
To facilitate coexpression of his6-SSB and fusion constructs within the same cell, we used dual expression systems. In the first, both his6 and the SSB fusion were expressed from high copy number plasmids (H/H, H = high) using 500-μM IPTG, whereas in the second, the SSB fusion was expressed from a low copy number vector (H/L, L = low) using either 500 or 1000 μM IPTG. For the H/H system, to ensure compatible expression with SSB fusions on pET21a+ (AmpR), his6-ssb was subcloned from pET-15b (AmpR) into pET-28a (KanR) using NcoI and HindIII. The resulting clone was verified using restriction enzyme mapping. Then, the two high copy number plasmids were sequentially transformed into Tuner (λDE3) for expression. For the H/L system, the ssb-fusions were subcloned into the BamHI and EcoRI sites of the low copy number vector pWKS130 using Infusion cloning.46 Then pET15b-his6-ssb and pWKS130 carrying the ssb fusion, were sequentially transformed into Tuner (λDE3) pLysS for expression. Tuner (λDE3) and Tuner (λDE3) pLysS cells (Novagen) gave similar results in expression studies.
Purification of wild type SSB protein
Wild type SSB protein was purified from strain K12ΔH1Δtrp as described.47 The concentration of purified SSB protein monomers was determined at 280 nm using ɛ = 27, 960 M−1 cm−1.
Purification of histidine-tagged SSB proteins
His6-SSB and SSB chimeras were purified using the same protocol. Cells were grown to an OD600 = 0.4, IPTG added and growth continued for an additional 4 h. Cells were harvested by centrifugation and resuspended in 50-mM Tris-HCl (pH8) containing 20% sucrose and protease inhibitors (Novagen). Lysis at 4°C was initiated by the addition of lysozyme (1 mg/mL, final) and benzonase (35 U/mL, final) followed by stirring for 30 min, culminating with the addition of deoxycholate (0.05%, final) and stirring for an additional 30 min. Next, the concentration of NaCl was increased to 500 mM, imidazole to 30 mM and the solution stirred for an additional 30 min. Cell lysates were clarified by centrifugation at 35,000 × G for 90 min at 4°C. SSB proteins were found in the supernatant as determined by SDS-PAGE and visual inspection for fluorescent chimeras.
Cleared cell lysates were diluted and loaded directly onto a 20-mL Ni-sepharose high performance column (GE Healthcare) equilibrated in binding buffer containing 30-mM imidazole. The column was washed until the baseline reached zero and the proteins subsequently eluted using a linear imidazole gradient (30–400 or 500 mM). Proteins were identified by SDS-PAGE. To ensure that chimera pools were homogeneous, pooled fractions were diluted in binding buffer without imidazole and rechromatographed using the same linear gradient. The resulting preparations were greater than 95% homogeneous, did not contain detectable levels of either ss- or dsDNA nucleases and were used without additional purification. Typically a single preparation yielded 40–60 mg/L of all SSB types with the relative amounts of each chimera varying between the H/H and H/L strains. The variation is due to the different levels of expression of the FP fusions (data not shown).
Fractions containing His6-SSB or SSB chimeras with different ratios of SSB-fusion:His6-SSB were pooled, dialyzed overnight against Buffer A (20-mM Tris-HCl (pH8.0), 1-mM EDTA and 500-mM NaCl), followed by dialysis for 8hr against buffer A plus 50% glycerol and storage of aliquots at −80°C. The concentration of His6-SSB in monomers was determined at 280 nm using ɛ = 27, 960 M−1 cm−1. The concentration of chimeras in tetramers was determined at 280 nm using the sum total extinction coefficient of the fusion and his6-SSB subunits (Table II). All extinction coefficients were calculated from the amino acid sequence of subunits.48
Analysis of chimeras by gel filtration
Gel filtration chromatography was performed using a 24 mL Tricorn, Superose 6 column (GE Biosciences). Prior to gel filtration, samples (5 μM tetramer) were diluted to 500 μL with running buffer (20-mM Tris-OAc (pH7.5), 1-mM DTT, 150-mM NaCl and 10-mM Mg(OAc)2) and immediately injected onto the column. A calibration curve was determined by measuring the elution volumes of the following standards chromatographed in the same running buffer: carbonic anhydrase (29 KDa), wild type SSB (76 KDa), alcohol dehydrogenase (150 KDa), β-amylase (200 KDa) and apoferritin (443 KDa), and calculating their corresponding Kav values using blue dextran to determine the void volume.49 Then, the Kav value for each chimera was calculated and the molecular weights determined from the calibration curve.
Analysis of ssDNA binding
The site size of SSB proteins was determined by monitoring the quenching of the intrinsic fluorescence of SSB that occurs on binding to ssDNA.32 Fluorescence data were collected as a function of time using a Varian Cary Eclipse Fluorescence spectrophotometer equipped with a multicell holder which was connected to a PCB150 peltier controlled water circulator, which maintained temperature within ±0.5°C. The excitation and emission wavelengths were set at 290 and 340 nm, respectively; the excitation slit width was 2.5 and the emission slit was 20 nm. Assays (500 μL) were done at 20°C and contained 25-mM Tris-acetate (pH 7.5), 1-mM DTT and 1-μM SSB (either wild type or chimera). Reactions were initiated by the addition of M13 mp7 ssDNA (1μM nucleotides, final). Subsequent additions of ssDNA were done once the fluorescence signal had stabilized. The final concentration of ssDNA was corrected according to the final volume of the reaction mixture following each addition.
For determination of the STMP of ssDNA binding, the same reaction buffer was used as for site-size determination. First, stoichiometric complexes of 1 or 2 μM SSB and M13 mp7 ssDNA were formed. Then, NaCl was added in 200 or 400 mM increments with the final [NaCl] corrected according to the final volume of the reaction mixture following each addition. Each addition of NaCl was performed only when the fluorescence signal had stabilized.
Fluorescence stopped-flow kinetics
All concentrations indicated are after mixing. The rate of SSB (10 nM) association with varying concentrations of oligo d(T)70 in 10-mM Tris, pH 8.1, 0.1-mM EDTA, 0.2-M NaCl was measured by rapidly mixing in a SX.18MV stopped flow reaction analyzer (Applied Photophysics) maintained at 25°C. The change in tryptophan fluorescence over time was monitored with excitation at 280 nm through 2-mm slits. Emission was measured after a 320-nm cutoff filter. Data were fit to a single exponential using Kaleidagraph software.
DNA helicase assays
The unwinding of dsDNA was monitored using a fluorescence based assay as described previously.50 In this assay, the helicase activity of RecBCD was assayed by observing the quenching of the intrinsic fluorescence of SSB protein that occurs upon binding to single stranded DNA (ssDNA).34 Assays (500 μL) were done at 20°C and contained 25-mM Tris-acetate (pH 7.5), 2-mM magnesium acetate, 1-mM DTT, 1-mM ATP, 1-μM SSB (either wild type or chimera), 2.4-nM molecules of DNA and 2-nM RecBCD. Reactions were initiated by the addition of RecBCD and monitored for 5 min. The fluorescence data were collected as a function of time using a Varian Cary Eclipse Fluorescence spectrophotometer equipped with a multicell holder, which was connected to a PCB150 peltier controlled water circulator, which maintained temperature within ±0.5°C. The excitation and emission wavelengths were set at 290 and 340 nm, respectively; the excitation slit width was 2.5 and the emission slit was 20 nm. All rates are reported in nM bp/s/nM RecBCD.34
Coprecipitation assays
Coprecipitation of SSB and RecG was done as described previously.19 Reactions were done on ice in a volume of 20 μL and contained 20-mM Tris-HCl (pH 7.5), 150-mM NaCl, 5% glycerol, 1-mM DTT, 25-μM SSB monomers. Where present, RecG was added to a final concentration of 20 μM. Following mixing, tubes were incubated on ice for 15 min, mixed with an equal volume of ammonium sulfate precipitation buffer [reaction buffer plus 2.6 M (NH4)2SO4] and incubated for an additional 15 min. The mixtures were subjected to centrifugation at 15,000 × G at 4°C for 1 min. The supernatant was transferred to a fresh tube and the pellet was washed three times followed by an identical centrifugation run. The final pellet was resuspended in 40 μL of 2× SDS-PAGE loading buffer, and 10 μL of loading buffer was also added to the supernatant. Thereafter, both pellet and supernatant were boiled for 2 min and 20 μL of each sample subjected to electrophoresis. Gels were stained with Coomassie brilliant blue, destained, and photographed. Quantitation of gels was done using Image Quant Software (GE Healthcare).
ATPase assays
The hydrolysis of ATP and STMP assays were monitored using a coupled spectrophotometric assay.37,19 The standard reaction buffer contained 20-mM Tris-acetate (pH 7.5), 1-mM DTT, 0.3-mM NADH, 7.5-mM PEP, 20-U/mL pyruvate kinase, 20-U/mL lactate dehydrogenase, 100-nM RecG, 1-mM ATP, and 10-mM magnesium acetate. Assays were performed in a reaction volume of 150 μL. The concentration of M13 ssDNA was 36-μM nucleotides, SSB proteins (either wild type or chimeras) were present at 1.8 μM and RecG was at 100 nM. Reactions were initiated by the addition of RecG following a 5-min incubation of all other components. Once a steady-state rate of ATP hydrolysis was achieved, NaCl was added in 50 or 75-mM increments. This was repeated until all ATP hydrolysis of RecG ceased. The resulting hydrolysis rate in each steady-state region was calculated and expressed as a percent of the steady-state rate in the absence of NaCl. The total volume used to calculate final concentration of NaCl was adjusted after each addition to correct for the additions themselves. These rates were subsequently graphed to determine the concentration of NaCl resulting in a 50% reduction in the rate of ATP hydrolysis, which corresponds to the salt-titration mid-point.
Single DNA molecule optical trapping and video, fluorescence-microscopy
Reactions were performed in two-stream, micromachined flow cells with inlet ports connected to 1-mL Hamilton Gas Tight syringes controlled by a syringe pump (PHD 2000, Harvard Apparatus, MA). Flow cells were held in place on a Märzhäuser stage in a Leica DMIRE2 fluorescence microscope. The DMIRE2 was modified to mate with a Laser Manipulator LM-02 (Solar TII, Belarus) containing galvano-scanning mirrors (VM500, GSI Lumonics, MA). Optical traps were formed by passing an Nd:YVO4 infrared laser (wavelength = 1064 nm, 5 W, Spectra Physics) through the LM-02, which split the beam into S- and P-components to form fixed and mobile optical traps in the focal plane of the microscope. The position and motion of the mobile trap was controlled by the scanning mirrors. The resulting, independent laser beams were focused through an oil-immersion objective lens (Plan Apo 100×, 1.4 NA, Leica) to a position 18 μm above the lower surface of the flow cell. Fluorescent DNA-bead complexes were excited using an XCite120 lamp (XFO) using a GFP-3035B fluorescence cube (Semrock, Rochester, NY). To visualize the SSB-YFP chimera, a YFP-2427A fluorescence filter cube was used (Semrock). Fluorescence images were captured at video rate by an EB-CCD camera (Hamamatsu) and recorded on digital videotape (DV184) using a digital VCR (Sony DSR-11).
Bacteriophage λ DNA-bead complexes were prepared as described.39,51 Sample syringes contained 30 mM NaHCO3 (pH 8), 100 mM DTT, 20% sucrose, 9.9 × 107 DNA-bead complexes, 0.2 μM YOYO-1, 2 mM Mg(OAc)2, 2.4-mM ATP, 177-nM RecBCD, and 10 nM SSB-GFP or separately, SSB-YFP (2/4 chimera). Reaction syringes contained 30-mM NaHCO3 (pH 8), 30-mM DTT, 20% sucrose, 2-mM Mg(OAc)2 and 2.4-mM ATP. Reactions were done at 23°C at a linear flow velocity of 74 μm/s.
Single molecule data analysis
Downloading of movies to an IBM compatible computer and subsequent data analyses were done as follows. Images were captured on an IBM compatible computer interfaced with the DSR-11 via a PIXCI SV5 frame-grabber controlled by XCAP software (EPIX). Captured videos were converted into individual, sequential frames to make measurements using Image-Pro Plus v6.1 (Media Cybernetics). Calibration of the microscope optics was achieved using a Stage Micrometer (Leica), resulting in 8.54 pixels per micron. Under the assay conditions used, the average corrected length of individual, stretched, fluorescent λ DNA molecules was 18 μm (n = 100). DNA lengths were corrected for slight bead blooming (i.e., beads appeared 1.2× larger than anticipated). Thus for all calculations, as λ DNA is 48,504 bp in length, the average number of bp/μm is 2695.
The rate of DNA unwinding was calculated by measuring the position of the SSB chimera (the bright spot at the free end of the DNA) using the Object Tracking function of Image Pro. Data were exported from Image Pro and analyzed using GraphPad Prism (v 5.1). To ascertain the time-dependent position of the DNA-bound, SSB chimera, the data from individual trajectories were smoothed with a second-order Savitzky-Golay filter with a time constant of 0.2 s as described previously.39,51,52 Then, the instantaneous velocity for each bright spot was determined by using a differential average over the length of the movie from the point at which bright spots were observed until they ceased moving entirely. The instantaneous velocities of RecBCD molecules were then averaged to obtain the steady-state rate of DNA unwinding.
Fluorescence microscopy
Cells containing the high/low dual plasmids for the SSB-msgGFP chimera expression were transformed with a vector expressing his6-mcherry-RecG (details to be published elsewhere). These were grown to an OD600 = 0.4 and induced with 100-μM IPTG and grown for an additional 3 h. Cultures were subjected to centrifugation and resuspended in 10 mM MgSO4. Ten milliliter of cells were deposited on poly-L-lysine treated coverslips and allowed to bind for 10 min. Unbound cells were removed by aspiration, followed by two washes with 10 mM MgSO4. Dried coverslips were placed onto a 20-mL water drop on a microscope slide, sealed with nail polish and imaged using epi-fluorescence microscopy.
Cells were viewed on an inverted microscope (Nikon Diaphot 300) equipped with a dry condenser (0.85 NA) for phase contrast optics. Fluorescence images were obtained using a 100×, 1.25 NA plan achromat objective. Light from a mercury arc lamp was attenuated by a 10% transmittance neutral density filter and heat filter. One second exposure, 16 bit images were captured after 4× projection by a cooled, back-thinned CCD camera (Princeton Instruments, Princeton, NJ) controlled by IP lab software (Signal Analytics, Vienna, VA). Computer-controlled shutters were placed in the transmitted and epi light paths to limit illumination. Captured images were analyzed using Image-Pro Plus v6.1 (Media Cybernetics).
Acknowledgments
We would like to thank Joe Golebiowski and Mark Sutton for comments. Work on this project was supported by a Susan G. Komen Breast Cancer Foundation Grant # BCTR0601350, National Institutes of Health Grants R24 GM 080599, and R24 GM080599 (P.R.B., A,K.B., and K.D.R.).
Supplementary material
References
- 1.Chase JW, Williams KR. Single-stranded DNA binding proteins required for DNA replication. Annu Rev Biochem. 1986;55:103–136. doi: 10.1146/annurev.bi.55.070186.000535. [DOI] [PubMed] [Google Scholar]
- 2.Meyer RR, Laine PS. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev. 1990;54:342–380. doi: 10.1128/mr.54.4.342-380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohman T, Ferrari M. Escherichia coli single-stranded DNA-binding protemultiple DNA-binding modes and cooperativities. Annu Rev Biochem. 1994;63:527–570. doi: 10.1146/annurev.bi.63.070194.002523. PMID: 7979247 {Medline} [DOI] [PubMed] [Google Scholar]
- 5.Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol. 2008;43:289–318. doi: 10.1080/10409230802341296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sancar A, Williams KR, Chase JW, Rupp WD. Sequences of the ssb gene and protein. Proc Natl Acad Sci USA. 1981;78:4274–4278. doi: 10.1073/pnas.78.7.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghunathan S, Kozlov A, Lohman T, Waksman G. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat Struct Biol. 2000;7:648–652. doi: 10.1038/77943. [DOI] [PubMed] [Google Scholar]
- 8.Chrysogelos S, Griffith J. Escherichia coli single-strand binding protein organizes single-stranded DNA in nucleosome-like units. Proc Natl Acad Sci USA. 1982;79:5803–5807. doi: 10.1073/pnas.79.19.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuznetsov S, Kozlov A, Lohman T, Ansari A. Microsecond dynamics of protein-DNA interactions: direct observation of the wrapping/unwrapping kinetics of single-stranded DNA around the E. coli SSB tetramer. J Mol Biol. 2006;359:55–65. doi: 10.1016/j.jmb.2006.02.070. [DOI] [PubMed] [Google Scholar]
- 10.Savvides SN, Raghunathan S, Futterer K, Kozlov AG, Lohman TM, Waksman G. The C-terminal domain of full-length E. coli SSB is disordered even when bound to DNA. Prot Sci. 2004;13:1942–1947. doi: 10.1110/ps.04661904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umezu K, Kolodner RD. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem. 1994;269:30005–30013. [PubMed] [Google Scholar]
- 12.Glover B, McHenry C. The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J Biol Chem. 1998;273:23476–23484. doi: 10.1074/jbc.273.36.23476. PMID: 9722585 {Medline} [DOI] [PubMed] [Google Scholar]
- 13.Genschel J, Curth U, Urbanke C. Interaction of E. coli single-stranded DNA binding protein (SSB) with exonuclease I. The carboxy-terminus of SSB is the recognition site for the nuclease. Biological Chemistry. 2000;381:183–192. doi: 10.1515/BC.2000.025. [DOI] [PubMed] [Google Scholar]
- 14.Handa P, Acharya N, Varshney U. Chimeras between single-stranded DNA-binding proteins from Escherichia coli and Mycobacterium tuberculosis reveal that their C-terminal domains interact with uracil DNA glycosylases. J Biol Chem. 2001;276:16992–16997. doi: 10.1074/jbc.M100393200. [DOI] [PubMed] [Google Scholar]
- 15.Kantake N, Madiraju MV, Sugiyama T, Kowalczykowski SC. Escherichia coli RecO protein anneals ssDNA complexed with its cognate ssDNA-binding proteA common step in genetic recombination. Proc Nat Acad Sci USA. 2002;99:15327–15332. doi: 10.1073/pnas.252633399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witte G, Urbanke C, Curth U. DNA polymerase III chi subunit ties single-stranded DNA binding protein to the bacterial replication machinery. Nucleic Acids Res. 2003;31:4434–4440. doi: 10.1093/nar/gkg498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadman CJ, McGlynn P. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004;32:6378–6387. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shereda RD, Bernstein DA, Keck JL. A central role for SSB in Escherichia coli RecQ DNA helicase function. J Biol Chem. 2007;282:19247–19258. doi: 10.1074/jbc.M608011200. [DOI] [PubMed] [Google Scholar]
- 19.Buss JA, Kimura Y, Bianco PR. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 2008;36:7029–7042. doi: 10.1093/nar/gkn795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suski C, Marians KJ. Resolution of converging replication forks by RecQ and topoisomerase III. Molecular Cell. 2008;30:779–789. doi: 10.1016/j.molcel.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelman Z, Yuzhakov A, Andjelkovic J, O'Donnell M. Devoted to the lagging strand-the subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998;17:2436–2449. doi: 10.1093/emboj/17.8.2436. PMID: 9545254 {Medline} [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curth U, Genschel J, Urbanke C, Greipel J. In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res. 1996;24:2706–2711. doi: 10.1093/nar/24.14.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dillingham M, Tibbles K, Hunter J, Bell J, Kowalczykowski S, Webb M. Fluorescent single-stranded DNA binding protein as a probe for sensitive, real-time assays of helicase activity. Biophys J. 2008;95:3330–3339. doi: 10.1529/biophysj.108.133512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy R, Kozlov A, Lohman T, Ha T. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009;461:1092–1097. doi: 10.1038/nature08442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ. Independent positioning and action of Escherichia coli replisomes in live cells. Cell. 2008;133:90–102. doi: 10.1016/j.cell.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kremers G, Goedhart J, van Munster E, Gadella TJ. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry. 2006;45:6570–6580. doi: 10.1021/bi0516273. [DOI] [PubMed] [Google Scholar]
- 27.Shaner N, Steinbach P, Tsien R. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 28.Giepmans B, Adams S, Ellisman M, Tsien R. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 29.Zacharias D, Tsien R. Molecular biology and mutation of green fluorescent protein. Methods Biochem Anal. 2006;47:83–120. [PubMed] [Google Scholar]
- 30.Kinebuchi T, Shindo H, Nagai H, Shimamoto N, Shimizu M. Functional domains of Escherichia coli single-stranded DNA binding protein as assessed by analyses of the deletion mutants. Biochemistry. 1997;36:6732–6738. doi: 10.1021/bi961647s. PMID: 9184154 {Medline} [DOI] [PubMed] [Google Scholar]
- 31.Miyawaki A, Tsien R. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 32.Lohman TM, Overman LB. Two binding modes in Escherichia coli single strand binding protein-single stranded DNA complexes. Modulation by NaCl concentration. J Biol Chem. 1985;260:3594–3603. [PubMed] [Google Scholar]
- 33.Kozlov A, Lohman T. Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry. 2002;41:6032–6044. doi: 10.1021/bi020122z. [DOI] [PubMed] [Google Scholar]
- 34.Roman LJ, Kowalczykowski SC. Characterization of the helicase activity of the Escherichia coli RecBCD enzyme using a novel helicase assay. Biochemistry. 1989;28:2863–2873. doi: 10.1021/bi00433a018. [DOI] [PubMed] [Google Scholar]
- 35.Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bianco PR. DNA helicases. In: Lovett ST, editor. EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. Washington, DC: ASM Press; 2010. [Google Scholar]
- 37.Slocum SL, Buss JA, Kimura Y, Bianco PR. Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J Mol Biol. 2007;367:647–664. doi: 10.1016/j.jmb.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, Baskin RJ. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature. 2001;409:374–378. doi: 10.1038/35053131. [DOI] [PubMed] [Google Scholar]
- 39.Bianco PR, Bradfield JJ, Castanza LR, Donnelly AN. Rad54 oligomers translocate and cross-bridge double-stranded DNA to stimulate synapsis. J Mol Biol. 2007;374:618–640. doi: 10.1016/j.jmb.2007.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brewer LR, Corzett M, Balhorn R. Protamine-induced condensation and decondensation of the same DNA molecule. Science. 1999;286:120–123. doi: 10.1126/science.286.5437.120. [DOI] [PubMed] [Google Scholar]
- 41.Roman LJ, Eggleston AK, Kowalczykowski SC. Processivity of the DNA helicase activity of Escherichia coli recBCD enzyme. J Biol Chem. 1992;267:4207–4214. [PubMed] [Google Scholar]
- 42.Dillingham MS, Spies M, Kowalczykowski SC. RecBCD enzyme is a bipolar DNA helicase. Nature. 2003;423:893–897. doi: 10.1038/nature01673. [DOI] [PubMed] [Google Scholar]
- 43.Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, Kowalczykowski SC. A molecular throttle: the recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell. 2003;114:647–654. doi: 10.1016/s0092-8674(03)00681-0. [DOI] [PubMed] [Google Scholar]
- 44.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO Journal. 2007;26:4239–4251. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu D, Windsor M, Gellman S, Keck J. Peptide inhibitors identify roles for SSB C-terminal residues in SSB/exonuclease I complex formation. Biochemistry. 2009;48:6764–6771. doi: 10.1021/bi900361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- 47.Lohman TM, Green JM, Beyer RS. Large-scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under lambda PL control. Biochemistry. 1986;25:21–25. doi: 10.1021/bi00349a004. [DOI] [PubMed] [Google Scholar]
- 48.Gill S, von Hippel P. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. PMID: 2610349 {Medline} [DOI] [PubMed] [Google Scholar]
- 49.Biosciences G. Gel Filtration. Principles and methods. Uppsala, Sweden: GE Healthcare Bio-Sciences AB; 2002. [Google Scholar]
- 50.Dziegielewska B, Beerman TA, Bianco PR. Inhibition of RecBCD enzyme by antineoplastic DNA alkylating agents. J Mol Biol. 2006;361:898–919. doi: 10.1016/j.jmb.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 51.Pezza RJ, Camerini-Otero RD, Bianco PR. Hop2-mnd1 condenses DNA to stimulate the synapsis phase of DNA strand exchange. Biophys J. 2010;99:3763–3772. doi: 10.1016/j.bpj.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neuman K, Abbondanzieri E, Landick R, Gelles J, Block S. Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell. 2003;115:437–447. doi: 10.1016/s0092-8674(03)00845-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.