

Abstract
Background
Acute lung injury is common during sepsis. Whereas gaseous exchange often can be supported adequately, death results frequently from cardio-circulatory depression, the mechanisms of which remain unclear. The aim of this study was to determine whether cardio-circulatory dysfunction during sepsis results from release of macrophage migration inhibitory factor (MIF) by the lung.
Methods
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) in adult Sprague-Dawley rats. Macrophage MIF was measured in the plasma sampled from the right ventricle (pre-lung) and left atrium (post-lung).
Results
The concentration of macrophage MIF in each of the post-lung samples was higher than in the corresponding pre-lung sample at 6 h (p = 0.015; paired t-test), 20 h (p = 0.008), and 30 h (p = 0.026) after the induction of sepsis. Next, rats that underwent CLP were treated with either saline (control) or our specific MIF inhibitor, (S,R)-3-(4-hydroxyphenyl)-4,5-dehy-dro-5-isoxazole acetic acid methyl ester (ISO-1). Echocardiography revealed that ISO-1 significantly improved the left ventricular end-diastolic volume index (p = 0.02), stroke volume index (p = 0.01), and cardiac index (p = 0.02) at 30 h after the induction of sepsis.
Conclusions
The lung appears to release significant amounts of macrophage MIF into the systemic circulation during late sepsis. Inhibition of MIF in a clinically relevant time frame blocked polymicrobial peritonitis-induced cardio-circulatory dysfunction. Inhibition of MIF may be a useful strategy to prevent cardio-circulatory deterioration associated with late sepsis.
Sepsis is a progressive systemic response to infection that can result in shock, profound myocardial depression, and cardiopulmonary collapse [1–4]. Prior to the systemic circulatory deterioration, respiratory insufficiency may be persistent and necessitate ventilatory support in as many as 85% of patients [5]. Of the individuals requiring mechanical ventilation, approximately one-half progress to the acute respiratory distress syndrome (ARDS) [5]. However, most patients who die while receiving mechanical ventilation during sepsis do not succumb to respiratory insufficiency but rather to multiple organ dysfunction or refractory sepsis [6]. This often results in cardiac collapse [7–9] that is characterized by low cardiac output in the setting of inappropriate vasodilatation [10,11].
Macrophage migration inhibitory factor (MIF) is a pro-inflammatory mediator that plays a crucial role in sepsis [12–14], as well as being a cardiac depressant [15,16]. The factor was identified as a peptide released by the pituitary gland in response to endotoxemia in vivo [17] and exists as a homotrimer [18,19]. Between each two adjacent subunits lies a hydrophobic cavity that confers the ability to catalyze the tautomerization of substrates such as D,L-dopachrome methyl esters into their corresponding indole derivatives. Whereas tautomerase activity is an ancient phenomenon, there is no evidence that modern species use it in synthetic pathways. However, blocking of this active site inhibits MIF induction of arachidonic acid release, MIF glucocorticoid-regulating activity [20], and MIF-induced ERK-1/2 MAP kinase activation, p53-dependent apoptosis, and proliferation of serum-starved cells [21].
Calandra et al. reported macrophages to be peripheral sources of MIF in response to inflammatory stimuli in vivo [22]. They also detected MIF protein in macrophage-rich organs, suggesting that the MIF in various tissues may be at least partly macrophage-derived. However, they examined normal tissues, and did not identify the lung as an MIF-rich organ [22]. Subsequently, Donnelly et al. [23] demonstrated that MIF is present in increased concentrations in the lungs of individuals with ARDS, and that alveolar macrophages are a source of the protein. Those investigators also showed that MIF augments, and anti-MIF antibodies attenuate, the secretion of the pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α) and interleukin-8 (IL-8) from alveolar macrophages. Recently, we reported increased concentrations of MIF in the cardiopulmonary circulation 30 h after the induction of sepsis [24]. We observed that fluorescently tagged MIF, instilled into the lung via the trachea, could translocate to the systemic circulation in late sepsis. Chagnon et al. showed that MIF can have a deleterious effect on cardiac function in endotoxemic mice [16]. However, the source of the MIF in that model and the more clinically relevant model of septic peritonitis remains undefined. We believe that crucial to understanding the role of MIF in the cardiac dysfunction and lethality associated with sepsis is the identification of its site of production and release. Therefore, we investigated the lung as a major source of MIF release into the systemic circulation during sepsis. Using our specific MIF antagonist, we defined the role of MIF in cardio-circulatory function in the late phase of polymicrobial sepsis.
MATERIALS AND METHODS
Synthesis and use of (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1)
The MIF antagonist ISO-1 was synthesized as described previously [14]. Briefly, to a solution of 4-hydroxybenzaldoxime (1 g, 5.85 mmol) in dimethylformamide (DMF) (120 mL) was added N-chlorosuccinimide (1.95 g, 14.6 mmol). The reaction mixture was stirred for 4 h at room temperature, affording the chloro-oxime. To this solution, methyl-3-butenoate (0.93 mL, 8.78 mmol) was added, followed by the dropwise addition of triethylamine (1.23 mL, 8.78 mmol) in DMF (12 mL). The reaction mixture was heated to 40 °C and stirred for 48 h. The solvent was removed in vacuo and the residue was taken up in EtOAc. The EtOAc extract was washed with water, dried (anhydrous Na2SO4), and concentrated in vacuo. The residue was purified by flash column chromatography (hexane/EtOAc 4/3) to afford ISO-1 (550 mg, 40%). The ISO-1 was diluted for use with a minimal amount of dimethylsulfoxide (DMSO) and further diluted with saline (0.9% w/v) so that the final concentration of DMSO was 5%, which also was included in the saline control (Saline) [14].
Animal model of sepsis
All experimentation was approved by the Institutional Animal Care and Use Committee at The Feinstein Institute for Medical Research. Sprague-Dawley rats were maintained under standard laboratory conditions. The rats were fasted overnight but allowed water ad libitum. On the day of surgery, with the animal under isoflurane anesthesia (2.0%), the anterior abdominal wall was shaved. A midline incision was made, and the cecum was ligated with double 3-0 silk just distal to the ileocecal valve tightly enough to stop blood inflow completely and then punctured twice with an 18-gauge needle. Approximately 2 mm of fecal material were extruded. The cecum was placed back in the abdomen, and the peritoneal wall was closed with 4-0 monofilament polypropylene sutures. The animals were resuscitated with 0.9% NaCl (3 mL/100 g body weight). The model had a mortality rate of approximately 43% at 30 h.
Experimental design and sample treatment
In the first set of experiments, rats (319 ± 37 g), both normal cage controls and at various times after cecal ligation and puncture (CLP) (6 h, 20 h, and 30 h), were anesthetized, and blood samples were collected from the left atrium and right ventricle after opening the thorax. First, blood from the left atrium was collected, then that from the right ventricle, into a 1-mL heparinized syringe using a 25-ga needle. The sampling from both chambers was completed within one min in the absence of arrest. The samples were centrifuged (956 × g, 5 min.), and the plasma was stored at −70 °C until assayed. The animals were euthanized and postmortem bronchoalveolar lavage (BAL) was performed by instilling and withdrawing sterile physiological saline (8 mL at 4°C) through a tracheal cannula. This procedure was repeated twice, and all BAL fluid was pooled. The BAL fluid was centrifuged (106 × g, 5 min.), and the supernatant portions were stored at −70 °C until assayed. Total cell numbers were counted using a hemocytometer, and slides were prepared and stained for differential counts. For organ collection, blood was flushed from the lung with cold saline (approximately 30 mL) instilled into the right ventricle using a 20-ga needle. The organ samples were harvested, frozen immediately in liquid nitrogen, and stored at −70 °C for MIF protein analysis.
In the second set of experiments, rats (281 ± 15 g) underwent CLP and were then treated with either MIF antagonist ISO-1 or saline. The ISO-1 (16 mg/rat in a total of 300 microliters) or an equal volume of saline was given intraperitoneally at 6 h, 18 h, and 24 h after CLP. The dose of ISO-1 was derived from our earlier rodent studies [14] to be approximately one-half the maximal dose required to achieve a four-fold increase in the survival rate after 14 days. Transthoracic echocardiography of each rat was performed under isoflurane anesthesia (1.5%) both before CLP as well as 30 h afterward.
Measurement of cytokines
The concentrations of MIF and macrophage inhibitory protein-2 (MIP-2) in the plasma and BAL supernatant liquids were measured by an enzyme-linked immunosorbent assay (Chemicon International, Temecula, CA, and Biosource International, Inc. Camarillo, CA, respectively) according to the manufacturers’ instructions.
Protein extraction and Western blotting
Lung and heart tissues were thawed and homogenized on ice in Tris-buffered saline (TBS; 50 mM Tris and 150 mM NaCl, pH 7.5) containing 1% octylphenolpoly(ethyleneglycol ether)(Nonidet) 40 (Roche Diagnostics Corp., Indianapolis, IN), 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate (SDS), 2 mM ethylene-diaminetetraacetic acid (EDTA), and 1 mM phenylmethylsulfonyl fluoride (PMSF) and centrifuged. The lysate protein concentration was quantified using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Protein was diluted two parts sample to one part Laemmli sample buffer (Bio-Rad) and resolved on an 18% SDS polyacrylamide gel (Ready Gel, Bio-Rad) under reducing conditions. Pre-stained SDS-PAGE standards (PageRuler™ Prestained Protein Ladder, Fermentas, Inc., Hanover, MD) were run with each gel to determine the approximate molecular weight of the detected bands. Recombinant mouse MIF (from Christine Metz) was used as a positive control. The separated proteins were transferred onto Immobilon filters (Immobilon™ Transfer Membranes, Millipore, Bedford, MA). The MIF was detected with the polyclonal anti-MIF antibody R102 (from Christine Metz) [25] and visualized with a peroxidase-conjugated donkey anti-rabbit-IgG antibody and enhanced chemiluminescence (ECL; Amersham Pharamacia, Piscataway, NJ). Equivalency of loading of Western blots was assessed using a goat polyclonal anti-β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The lysates obtained from the tissues were compared with those of each time point. The amount of specific protein present was quantified by densitometry supported by Quantity One 4.2.2 software (Bio-Rad) and expressed in relative units. Densitometric measurements were standardized relative to known quantities of rmMIF electrophoresed and Western blotted as a control.
Echocardiography
Transthoracic echocardiography was performed in the same rats under isoflurane anesthesia (1.5%) before CLP, as well as 30 h afterward, using a 7-MHz transducer (Acuson Sequoia System; Siemens Medical Solutions USA, Inc., Malvern, PA) to obtain a two-dimensional short-axis image at the level of the papillary muscles. The imaging plane-guided left ventricle (LV) M-mode tracings were obtained for measurement of heart rate (HR), LV end-diastolic dimension, and LV end-systolic dimension. The ejection fraction (EF), % fractional shortening (%FS), end-diastolic volume (EDV), end-systolic volume (ESV), and stroke volume (SV) were calculated automatically. Cardiac output (CO), cardiac index (CI), end-diastolic volume index (EDVI), and stroke volume index (SVI) were calculated with HR, SV, EDV, and weight using the following formulae: CO = SV × HR, CI = CO/weight (kg), EDVI = EDV/weight (kg), and SVI = SV/weight (kg). Data represent the average of three cardiac cycles from at least three separate scans. The same qualified technician performed all echocardiograms. The LV internal dimensions were measured according to the recommendations of the American Society for Echocardiography leading-edge method from three consecutive cardiac cycles [26]. Echocardiographic parameters at 30 h after CLP were compared with those measured pre-CLP in each rat, and indexed as a percentage of baseline values in each study.
Statistics
Unless otherwise indicated, data are expressed as the mean ± standard deviation (SD). Two groups were compared using Student’s t-test. Multiple groups were compared using analysis of variance (ANOVA) with a post hoc Dunn’s or Tukey analysis. Changes of MIF in plasma over time were assessed using a repeated measures ANOVA.
RESULTS
Cell counts and protein concentration in BAL
The effect of CLP within the alveolar space was assessed (Table 1). Lungs harvested from rats at 0 h, 6 h, 20 h, or 30 h after CLP were lavaged. Leukocytosis was present 30 h after surgery, which increased the total cell count (p < 0.05; 30 h vs. 0 h, 6 h, or 20 h) and neutrophil count (p < 0.05; 30 h vs. 0 h). However, there was no significant increase in protein concentration, suggesting absence of severe pulmonary injury during this time.
Table 1.
Cell Counts AND Protein Concentration IN Bronchoalveolar Lavage Fluid AFTER Cecal Ligation AND Puncturea
| 0 h | 6 h | 20 h | 30 h | |
|---|---|---|---|---|
| Total cell counts (× 105) | 3.34 ± 0.52 | 4.39 ± 0.51 | 3.16 ± 0.89 | 7.76 ± 0.88b |
| Total neutrophil counts | 497 ± 361 | 5,260 ± 3,620 | 5,400 ± 3,700 | 75,400 ± 52,000c |
| Protein (mcg/mL) | 65.8 ± 5.71 | 70.8 ± 10.04 | 92.1 ± 14.94 | 57.3 ± 6.47 |
Values are group means ± SE; n = 6 for 0 h and 6 h, n = 4 for 20 h and 30 h.
P < 0.05 vs. 0 h, 6 h, or 20 h (one-way ANOVA and Tukey test).
P < 0.05 vs. 0 h (Kruskal-Wallis one-way ANOVA on ranks and Dunn’s method).
Accumulation of MIF in the lung
Macrophage MIF accumulated in both the air spaces and the parenchyma of the lung. There were significant increases in the concentration of MIF and the α-chemokine MIP-2 in the alveoli at 20 h and 30 h after CLP (Fig. 1). These two cytokines showed similar accumulation patterns, with both increasing by 6 h and peaking by 30 h after CLP. In addition, MIF was increased significantly within the lung tissue by 20 h after the induction of sepsis (Fig. 2).
FIG. 1.

Concentrations of MIF and MIP-2 in BAL liquid at 0 h, 6 h, 20 h, and 30 h after CLP. (A) Sepsis induced increases in MIF at both 20 h and 30 h (p < 0.05 compared with 0 h; one-way ANOVA and Tukey test; n = 7, 6, 4, and 6 for 0 h, 6 h, 20 h, and 30 h, respectively). (B) Sepsis also induced increases in MIP-2 at both 20 h and 30 h (p < 0.05 compared with 0 h; Kruskal-Wallis one-way ANOVA and Dunn’s method; n = 7, 6, 10, and 15 for 0 h, 6 h, 20 h, and 30 h, respectively).
FIG. 2.

Expression of MIF in lung after CLP. The lungs were homogenized, and MIF was identified by Western blot (n = 6 per group except at 20 h, with 4 rats). Band densities of MIF:β-actin were assessed. The MIF protein was increased significantly 20 h after CLP (p < 0.05; one-way ANOVA and Tukey test).
Release of lung-derived MIF into the pulmonary circulation
The concentration of MIF in each of the post-lung samples at 6 h, 20 h, and 30 h after CLP was significantly higher than in the corresponding pre-lung samples, with mean increases of 416.1 ± 501.8%, 116.8 ± 47.2%, and 84.8 ± 35.7 % (p = 0.015, 0.008, and 0.026, respectively, by paired t-test) (Fig. 3). The MIF concentration in post-lung samples remained higher than the pre-lung values over the next 24 h (p< 0.003) (Fig. 4A). In contrast to MIF, MIP-2 showed no concentration gradient between pre- and post-lung samples during sepsis (Fig. 4B).
FIG. 3.

Macrophage migration inhibitory factor is released into pulmonary circulation after CLP. Blood was collected from right ventricle (RV) as pre-lung sample and left atrium (LA) as post-lung sample. Concentration of MIF in each of the post-lung samples at 6 h and 20 h after CLP was higher than in the corresponding pre-lung samples, with a mean increase of 416.1 ± 501.8% at 6 h, 116.8 ± 47.2% at 20 h, and 84.8 ± 35.7%. Paired t-test analysis showed that differences were significant at 6 h (p = 0.015), 20 h (p = 0.008), and 30 h (p = 0.026).
FIG. 4.

Time course of MIF and MIP2 concentration in blood sampled pre- or post-lung. Blood was collected from right ventricle (RV; ●) as pre-lung sample and left atrium (LA; ■) as post-lung sample, and concentrations of MIF and MIP2 were measured (n = 7, 6, 4, and 6 for 0 h, 6 h, 20 h, and 30 h, respectively). (A) MIF concentration shows significant correlation between time after induction of sepsis and source of blood sample (p < 0.003). Pairwise comparisons found no difference between MIF concentrations in RV and LA at 0 h. However, there was significant difference at 6 h (p < 0.002), 20 h (p < 0.002), and 30 h (p < 0.003), when MIF was significantly higher in post-lung than pre-lung sample. There was no significant change in MIF in pre-lung samples over time. However, MIF was significantly higher in post-lung samples at 20 h (p < 0.0004) and 30 h (p < 0.0005) than at 0 h. *Significant difference between pre- and post lung value (p < 0.05). (B) MIP2 concentration in blood peaked at 6 h after CLP, with no concentration difference between pre-and post-lung blood samples.
Changes in MIF accumulation in heart during sepsis
Consistent with the findings of Garner et al. in their endotoxemia studies [15], we found a decrease in the amount of MIF within the cardiac tissue following the induction of sepsis, with recovery of normal concentrations by 20 h after CLP. Thus, the mean MIF:β-actin protein ratio in the heart at 6 h post CLP was slightly lower than that in the control animals, although the difference was not statistically significant. However, there was a statistically significant increase of MIF in the heart between 6 h and 20 h after CLP (Fig. 5).
FIG. 5.
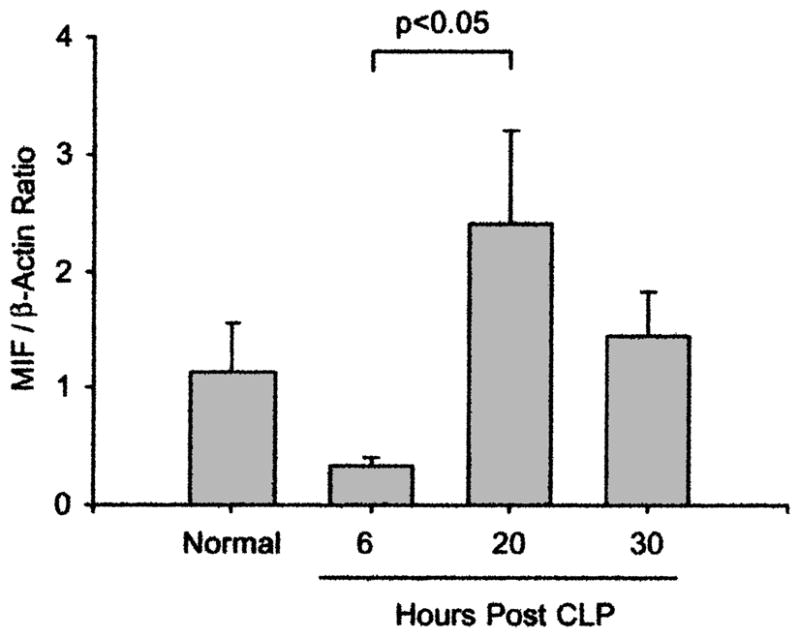
MIF, constitutively expressed in heart, is decreased 6 h after induction of sepsis. MIF:β-actin protein ratios in heart after CLP are shown. Hearts were homogenized, and amount of MIF was measured by Western blot (n = 6 per group except at 20 h, with 4 rats). Band densities of MIF: β-actin were calculated. The MIF:β-actin protein ratios in heart are not significantly increased at 20 h after CLP compared with normal values. However, concentration at 20 h is significantly higher than at 6 h (p < 0.05; Kruskal-Wallis one-way ANOVA and Dunn’s method).
Echocardiography
Serial M-mode echocardiographic analyses were performed in the same animals before (baseline, pre) and 30 h after (post) CLP. The initial measurement of HR was made within 15 min of the induction of anesthesia, following stabilization of the animal. At 30 h after induction of sepsis, in animals that had received only saline, there was a significant increase (p ≤ 0.05) in the mean HR (pre 370 ± 26; post 432 ± 27 beats/min), and decreases in mean EDVI (pre 1.96 ± 0.25; post 1.13 ± 0.51 mL/kg), SVI (pre 1.72 ± 0.3; post. 1.00 ± 0.42 mL/kg), and CI (pre 59.8 ± 12.4; post 40.2 ± 17.5 mL/min./kg). However, there were no significant changes in EF (pre 88.6 ± 4.31%; post 87.5 ± 7.32%) or %FS (pre 54.6 ± 5.37%; post 53.7 ± 10.22%).
The ISO-1 inhibitor binds to the MIF tautomerase active site, thereby reducing its pro-inflammatory activities such as induction of NF-κB activation and TNF-α release [4] and arachidonic acid release [21], and it significantly improves survival in sepsis [14]. Therefore, we examined whether ISO-1 treatment could prevent the changes in HR, EDVI, SVI, and CI. Treatment of the rats with ISO-1 significantly improved EDV, SVI, and CI but had no significant effect on HR (Fig. 6).
FIG. 6.

Inhibition of MIF improves cardiac function during sepsis. Changes in echocardiographic parameters (EF, HR; %FS, SVI, EDVI, CI) between pre and 30 h post-CLP in individual rats treated with ISO-1 (n = 8) or vehicle (saline) alone (n = 6) were assessed. Paired t-test revealed that with sepsis, there was significant (p < 0.05) increase in mean HR and decreases in mean EDVI, SVI, and CI, although there were no significant changes in EF or %FS. Treatment with ISO-1 significantly improved SVI, EDVI, and CI, with statistically significant differences between ISO-1 and saline-treated groups of p = 0.01, 0.02, and 0.02, respectively.
DISCUSSION
Macrophage MIF is a cardiac depressant factor shown previously to play a crucial role in the pathogenesis of sepsis [12,14]. Our study demonstrates that during sepsis, the lung accumulates MIF both in the parenchymal tissue and in the alveolar spaces, and releases it to the systemic circulation. In our previous study, we examined the release of MIF from the heart/lung block by sampling blood from the aorta and inferior vena cava at single time point. In our current study, by collecting blood immediately pre- and post-lung, we defined more clearly that MIF is released from the lung and that release occurs over a long period. In addition, because MIF is released from the lung by 6 h post-sepsis, we administered our small-molecule MIF antagonist, ISO-1, starting at this time. Thus, ISO-1 was administered 6 h, 18 h, and 24 h after CLP, and the compound protected against polymicrobial peritonitis-induced cardio-circulatory dysfunction 30 h after the induction of sepsis. The study demonstrates that the strategy of MIF blockade for the prevention of cardio-circulatory deterioration is reasonable, and can be employed in a clinically relevant time frame. Indeed, our recent study in mice indicates that the initial administration may be delayed at least until 24 h post-sepsis and still have substantial beneficial effects [14].
We first determined that the lung is a major source of plasma MIF during sepsis by collecting blood from the left atrium (post-lung) and from the right ventricle (pre-lung). These results show that the lung can release MIF into the pulmonary circulation during the late phases of sepsis. The lung contains many cell types, including macrophages and endothelial and epithelial cells, all of which have the ability to produce and release MIF [22,23,27–29]. In addition, inflammatory-activated neutrophils, which also have MIF-deriving ability [30,31], migrate into the alveolar spaces during sepsis, particularly that associated with acute lung injury [32]. Although the normal lung may not be rich in MIF [22], our data show clearly that lung is a source of this cytokine during sepsis. However, whereas Donnelly et al. showed the potential role of alveolar macrophages in the production of MIF during ARDS [23], the actual cell type responsible for the pulmonary-derived MIF in late sepsis remains to be defined.
In times of increased pulmonary vascular permeability, as is seen frequently in sepsis, inflammatory mediators such as TNF-α, IL-1, IL-6, and IL-8 can leak into the blood stream, where they contribute to the inflammatory milieu [6,33–35]. There also is evidence that lung damage (particularly from injurious ventilator strategies) is accompanied by pulmonary elaboration of these inflammatory mediators into the blood stream [36–38]. However, we did not use mechanical ventilation in our rat model, and leukocytosis in the absence of increased protein concentrations suggests that the lung can release MIF even in the absence of overt lung injury. In addition, although MIF and MIP-2 showed similar accumulation patterns in the BAL fluids, the MIF and MIP-2 concentrations in plasma were dissimilar; only MIF was higher in the post-lung than the pre-lung samples and in post- and pre-lung blood samples. These results further suggest that MIF release from the lung to the pulmonary circulation may not be secondary to leakage of the protein from the alveolar space into the pulmonary blood vessels. Thus, whereas it is clear that MIF is released into the pulmonary circulation during sepsis, further studies are needed to define the mechanism of this release.
Although the lung is an important source of MIF during sepsis, increasing the plasma concentration as much as 416% in a single pass, our data show that by the time the plasma returns to the right ventricle, the MIF concentration has returned to its pre-lung value, indicating that the protein is being cleared from the plasma at sites and by mechanisms that remain to be defined. In an earlier study [24], we collected blood from a catheter placed in the inferior vena cava below the diaphragm and advanced to a position adjacent to the right atrium (pre-lung) and a catheter in the carotid artery that was advanced to a position in the aorta adjacent to the LV (post-heart). In that study, at 30 h post-CLP, the MIF concentration in the post-heart/lung sample was only 25 ± 16% higher than that in the pre-lung sample. In the current study, blood collected from the right ventricle and left atrium showed that at 30 h post-CLP, the MIF concentration was approximately 85 ± 36% higher in the post-lung sample than in the pre-lung sample. This finding suggests that the heart removes some MIF from the plasma, although because the data were obtained in separate experiments, this hypothesis needs further investigation. Nevertheless, the blood leaving the lung perfuses the heart immediately. Therefore, we examined the effect of this MIF on the cardio-circulatory system.
Using echocardiography, we observed that the MIF inhibitor, ISO-1, can protect the cardio-circulatory system in late-phase sepsis. The inhibitor is the lead compound obtained through a strategy aimed at designing MIF antagonist drugs by targeting the tautomerase catalytic site [39]. Inhibition of this catalytic site is associated with loss of MIF pro-inflammatory properties both in vivo and in vitro, establishing a role for the catalytic active site of MIF in inflammatory activities [20]. We have shown that MIF inhibition by ISO-1 dramatically improves survival in sepsis [14]. In the present study, CI, SVI, and EDVI were preserved in the late phase of sepsis by MIF blockade with ISO-1. The late phase of sepsis is characterized by a hypodynamic circulation: Low cardiac output and depressed tissue perfusion. Cardiac index and SV are dependent on several factors, including contractility, preload, and afterload [40]. The maintenance of EDVI suggests preservation of both cardiac preload and afterload, which would account for the preservation of CI and SVI. Furthermore, Garner et al. [15] showed that recombinant MIF induces rapid cardiac depression in Langendorff perfusion studies, with significant decreases in both systolic and diastolic performance. This suggests that the MIF effects in our model are not secondary to changes in loading alone. In addition, we demonstrated down-regulation of cardiomyocyte function in a mouse CLP model using stress-activated signaling pathway activation (MAPK phosphorylation) as a marker of potential contractile dysfunction [24]. Chagnon et al. [16] demonstrated that cardiomyocyte apoptosis is associated with sepsis-induced cardiac dysfunction. This conclusion was supported by their study using both upstream (caspase-3, cytochrome c) and downstream (oligo-nucleosomal fraction) markers. In addition, cardiomyocyte apoptosis can have a profound affect on cardiac contractility during sepsis [16]. Further studies will be needed to define whether MIF blockade can protect against these changes in contractility during sepsis. This may be possible using the approach of Kumar et al. [41], who showed that human endotoxemia is associated with depression of load-independent contractility indices such as end-systolic myocardial wall stress (σes)/ESVI, which is one of the most load-independent of the contractility indices.
Although studies using lipopolysaccharide to induce sepsis demonstrate cardiac dysfunction and the protective effect of the MIF-blocking strategy [15,16], some investigators reported difficulty measuring cardiac contractility in the late phase of polymicrobial sepsis [42,43], a more clinically relevant model than endotoxemia alone. Kovacs et al. [42] suggested that the rat CLP model is not appropriate to examine the myocardial depression of clinical sepsis. Many of their echocardiographic parameters are similar to our own (no significant change of %FS between the normal condition and sepsis, and decreased EDVI [end-diastolic dimension according to Kovacs et al.]). However, the anesthesia used by Kovacs et al. was peritoneal ketamine, which lowers the HR considerably. Using normal mice, Roth et al. [44] recommended inhaled isoflurane as the anesthetic of choice if the model is known to cause cardiac depression. We followed this recommendation, using 1.5% isoflurane for our current study, and have shown an increase in HR, which suggests that cardiac function was not suppressed by the anesthesia. In addition, an advantage of our method was the use of the noninvasive technique of echocardiography. This method allowed us to examine cardiovascular function in the same animal at different time points.
In summary, we have shown that during sepsis, the lung accumulates MIF in both the parenchyma and the air spaces and releases it to the systemic circulation. Treatment with the MIF antagonist ISO-1 dramatically inhibits polymicrobial peritonitis-induced cardio-circulatory dysfunction 30 h after CLP. This result demonstrates that blocking MIF released from the lung to prevent cardio-circulatory deterioration can be achieved in a clinically relevant time frame, and provides the basis for new strategies for the treatment of sepsis.
Acknowledgments
We thank Dr. Hal A. Skopicki for his insightful comments and input on the manuscript. This work was supported by NIH R01 GM 060197 and HL 081655 (EJM).
Footnotes
Recipient of Surgical Infection Society New Member Award.
References
- 1.Parker MM, Shelhamer JH, Bacharach SL, et al. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 2.Bone RC. The pathogenesis of sepsis. Ann Intern Med. 1991;115:457–469. doi: 10.7326/0003-4819-115-6-457. [DOI] [PubMed] [Google Scholar]
- 3.Parrillo JE, Parker MM, Natanson C, et al. Septic shock in humans: Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 4.Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530–538. doi: 10.1007/s00134-003-1662-x. [DOI] [PubMed] [Google Scholar]
- 5.Bernard GR, Wheeler AP, Russell JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 6.Happel KI, Nelson S, Summer W. The lung in sepsis: Fueling the fire. Am J Med Sci. 2004;328:230–237. doi: 10.1097/00000441-200410000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Zilberberg MD, Epstein SK. Acute lung injury in the medical ICU: Comorbid conditions, age, etiology, and hospital outcome. Am J Respir Crit Care Med. 1998;157:1159–1164. doi: 10.1164/ajrccm.157.4.9704088. [DOI] [PubMed] [Google Scholar]
- 8.Monchi M, Bellenfant F, Cariou A, et al. Early predictive factors of survival in the acute respiratory distress syndrome: A multivariate analysis. Am J Respir Crit Care Med. 1998;158:1076–1081. doi: 10.1164/ajrccm.158.4.9802009. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y, Song Z, Zhou X, et al. A 12-month clinical survey of incidence and outcome of acute respiratory distress syndrome in Shanghai intensive care units. Intensive Care Med. 2004;30:2197–2203. doi: 10.1007/s00134-004-2479-y. [DOI] [PubMed] [Google Scholar]
- 10.Vincent JL, Gris P, Coffernils M, et al. Myocardial depression characterizes the fatal course of septic shock. Surgery. 1992;111:660–667. [PubMed] [Google Scholar]
- 11.Vincent JL, Reuse C, Frank N, et al. Right ventricular dysfunction in septic shock: Assessment by measurements of right ventricular ejection fraction using the thermodilution technique. Acta Anaesthesiol Scand. 1989;33:34–38. doi: 10.1111/j.1399-6576.1989.tb02856.x. [DOI] [PubMed] [Google Scholar]
- 12.Bozza M, Satoskar AR, Lin G, et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calandra T, Echtenacher B, Roy DL, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 14.Al-Abed Y, Dabideen D, Aljabari B, et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J Biol Chem. 2005;280:36541–36544. doi: 10.1074/jbc.C500243200. [DOI] [PubMed] [Google Scholar]
- 15.Garner LB, Willis MS, Carlson DL, et al. Macrophage migration inhibitory factor is a cardiac-derived myocardial depressant factor. Am J Physiol Heart Circ Physiol. 2003;285:H2500–H2509. doi: 10.1152/ajpheart.00432.2003. [DOI] [PubMed] [Google Scholar]
- 16.Chagnon F, Metz CN, Bucala R, Lesur O. Endotoxin-induced myocardial dysfunction: Effects of macrophage migration inhibitory factor neutralization. Circ Res. 2005;96:1095–1102. doi: 10.1161/01.RES.0000168327.22888.4d. [DOI] [PubMed] [Google Scholar]
- 17.Bernhagen J, Calandra T, Mitchell RA, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto H, Suzuki M, Nakagawa A, et al. Crystallization of rat liver macrophage migration inhibitory factor for MAD analysis. J Struct Biol. 1995;115:331–334. doi: 10.1006/jsbi.1995.1057. [DOI] [PubMed] [Google Scholar]
- 19.Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-Å resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci USA. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lubetsky JB, Dios A, Han J, et al. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 21.Dios A, Mitchell RA, Aljabari B, et al. Inhibition of MIF bioactivity by rational design of pharmacological inhibitors of MIF tautomerase activity. J Med Chem. 2002;45:2410–2416. doi: 10.1021/jm010534q. [DOI] [PubMed] [Google Scholar]
- 22.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly SC, Haslett C, Reid PT, et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3:320–323. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 24.Lin X, Sakuragi T, Metz CN, et al. Macrophage migration inhibitory factor within the alveolar spaces induces changes in the heart during late experimental sepsis. Shock. 2005;26:556–563. doi: 10.1097/01.shk.0000183238.70374.a8. [DOI] [PubMed] [Google Scholar]
- 25.Fingerle-Rowson G, Koch P, Bikoff R, et al. Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol. 2003;162:47–56. doi: 10.1016/S0002-9440(10)63797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiller NB, Shah PM, Crawford M, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 27.Nishihira J, Koyama Y, Mizue Y. Identification of macrophage migration inhibitory factor (MIF) in human vascular endothelial cells and its induction by lipopolysaccharide. Cytokine. 1998;10:199–205. doi: 10.1006/cyto.1997.0276. [DOI] [PubMed] [Google Scholar]
- 28.Arndt U, Wennemuth G, Barth P, et al. Release of macrophage migration inhibitory factor and CXCL8/interleukin-8 from lung epithelial cells rendered necrotic by influenza A virus infection. J Virol. 2002;76:9298–9306. doi: 10.1128/JVI.76.18.9298-9306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makita H, Nishimura M, Miyamoto K, et al. Effect of anti-macrophage migration inhibitory factor antibody on lipopolysaccharide-induced pulmonary neutrophil accumulation. Am J Respir Crit Care Med. 1998;158:573–579. doi: 10.1164/ajrccm.158.2.9707086. [DOI] [PubMed] [Google Scholar]
- 30.Riedemann NC, Guo RF, Gao H, et al. Regulatory role of C5a on macrophage migration inhibitory factor release from neutrophils. J Immunol. 2004;173:1355–1359. doi: 10.4049/jimmunol.173.2.1355. [DOI] [PubMed] [Google Scholar]
- 31.Lin X, Yang H, Sakuragi T, et al. α-Chemokine receptor blockade reduces high mobility group box 1 (HMGB1) protein induced lung inflammation and injury and improves survival in sepsis. Am J Physiol Lung Cell Mol Physiol. 2005;I289:L583–L590. doi: 10.1152/ajplung.00091.2005. [DOI] [PubMed] [Google Scholar]
- 32.Warshawski FJ, Sibbald WJ, Driedger AA, Cheung H. Abnormal neutrophil-pulmonary interaction in the adult respiratory distress syndrome: Qualitative and quantitative assessment of pulmonary neutrophil kinetics in humans with in vivo 111indium neutrophil scintigraphy. Am Rev Respir Dis. 1986;133:797–804. [PubMed] [Google Scholar]
- 33.Tutor JD, Mason CM, Dobard E, et al. Loss of compartmentalization of alveolar tumor necrosis factor after lung injury. Am J Respir Crit Care Med. 1994;149:1107–1111. doi: 10.1164/ajrccm.149.5.8173748. [DOI] [PubMed] [Google Scholar]
- 34.Schutte H, Lohmeyer J, Rosseau S, et al. Bron-choalveolar and systemic cytokine profiles in patients with ARDS, severe pneumonia and cardiogenic pulmonary oedema. Eur Respir J. 1996;9:1858–1867. doi: 10.1183/09031936.96.09091858. [DOI] [PubMed] [Google Scholar]
- 35.Monton C, Torres A, El-Ebiary M, et al. Cytokine expression in severe pneumonia: A bronchoalveolar lavage study. Crit Care Med. 1999;27:1745–1753. doi: 10.1097/00003246-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- 37.von Bethmann AN, Brasch F, Nusing R, et al. Hyper-ventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med. 1998;157:263–272. doi: 10.1164/ajrccm.157.1.9608052. [DOI] [PubMed] [Google Scholar]
- 38.Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109–116. doi: 10.1164/ajrccm.160.1.9803046. [DOI] [PubMed] [Google Scholar]
- 39.Cvetkovic I, Al-Abed Y, Miljkovic D, et al. Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology. 2005;146:2942–2451. doi: 10.1210/en.2004-1393. [DOI] [PubMed] [Google Scholar]
- 40.Carabello BA. Evolution of the study of left ventricular function: Everything old is new again. Circulation. 2002;105:2701–2703. doi: 10.1161/01.cir.0000021240.86593.9d. [DOI] [PubMed] [Google Scholar]
- 41.Kumar A, Bunnell E, Lynn M, et al. Experimental human endotoxemia is associated with depression of load-independent contractility indices: Prevention by the lipid A analogue E5531. Chest. 2004;126:860–867. doi: 10.1378/chest.126.3.860. [DOI] [PubMed] [Google Scholar]
- 42.Kovacs A, Courtois MR, Barzilai B, et al. Reversal of hypocalcemia and decreased afterload in sepsis: Effect on myocardial systolic and diastolic function. Am J Respir Crit Care Med. 1998;158:1990–1998. doi: 10.1164/ajrccm.158.6.9804114. [DOI] [PubMed] [Google Scholar]
- 43.Zhou M, Wang P, Chaudry IH. Cardiac contractility and structure are not significantly compromised even during the late, hypodynamic stage of sepsis. Shock. 1998;9:352–358. doi: 10.1097/00024382-199805000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Roth DM, Swaney JS, Dalton ND, et al. Impact of anesthesia on cardiac function during echocardiography in mice. Am J Physiol Heart Circ Physiol. 2002;282:H2134–140. doi: 10.1152/ajpheart.00845.2001. [DOI] [PubMed] [Google Scholar]