

Abstract
Pseudomonas. aeruginosa (PA) is a leading cause of nosocomial pneumonia in patients receiving mechanical ventilation with hyperoxia. Exposure to supraphysiological concentrations of reactive oxygen species during hyperoxia may result in macrophage damage that reduces their ability to phagocytose PA. We tested this hypothesis in cultured macrophage-like RAW 264.7 cells and alveolar macrophages from mice exposed to hyperoxia. Exposure to hyperoxia induced a similarly impaired phagocytosis of both the mucoid and non-mucoid forms of PA in alveolar macrophages and RAW cells. Compromised PA phagocytosis was associated with cytoskeleton disorganization and actin oxidation in hyperoxic macrophages. To test whether moderate concentrations of O2 limit the loss of phagocytic function induced by ≥ 95% O2, mice and RAW cells were exposed to 65% O2. Interestingly, although the resulting lung injury/cell proliferation was not significant, exposure to 65% O2 resulted in a marked reduction in PA phagocytosis that was comparable to that of ≥95% O2. Treatment with antioxidants, even post hyperoxic exposure, preserved actin cytoskeleton organization and phagocytosis of PA. These data suggest that hyperoxia reduces macrophage phagocytosis through effects on actin functions which can be preserved by antioxidant treatment. In addition, administration of moderate rather than higher concentrations of O2 does not improve macrophage phagocytosis of PA.
Keywords: bacterial infection, Pseudomonas aeruginosa, ventilator-associated pneumonia, alveolar macrophages, actin cytoskeleton, oxidation
INTRODUCTION
Nosocomial pneumonia (NP) is a leading cause of morbidity and mortality in the intensive care unit (ICU) [1]. According to the National Nosocomail Infection Survey, 86% of NP is associated with mechanical ventilation with hyperoxia [2]. During an average day in any american ICU, mechanical ventilation is administered to at least 40% of patients. The risk of acquiring ventilator-associated pneumonia (VAP) for ICU patients is estimated to be 1% per day [3]. A postmortem study has shown that pneumonia was present in up to 67% of patients that received mechanical ventilation [4]. Gram-negative aerobic organisms are the predominant causes of VAP. Among these, Pseudomonas aeruginosa (PA) is associated with 21% of the total NP cases [2].
Although PA is one of the leading virulent pathogens in ventilated patients [5], the mechanism(s) for the increased susceptibility of these individuals to infection with this organism remains unclear. The inhalation of supraphysiological oxygen concentrations for prolonged or even transient periods of time results in acute lung injury, which is characterized by the injury and death of pulmonary cells [6–11]. The role of hyperoxia as a potential cause of lung cell injury has been extensively characterized in alveolar epithelial [12–17] and endothelial cells [18, 19]. However, only a few studies have examined it’s impact on alveolar macrophages [20–23]. Alveolar macrophages provide the first line of defense against inhaled bacterial pathogens [24] and are exposed to 13% oxygen under normal physiological conditions [22]. Under conditions of supplemental oxygen therapy, alveolar macrophages, can be exposed to ≥ 95% oxygen. Alveolar macrophages isolated from hyperoxia-exposed animals and macrophages exposed to hyperoxia in vitro exhibit impaired chemotaxis, adherence and phagocytosis of paraffinoil-droplets and Klebsiella [25–29].
Prolonged exposure to hyperoxia can induce cell damage, in part, through the accumulation of increased levels of ROS, mainly superoxide ions (O2−•) [30]. The production of ROS is normally balanced by an elaborate antioxidant defense system [31, 32]. Under hyperoxic conditions, however, excessive ROS production perturbs this balance, devastating the antioxidant protection. We hypothesized that exposure to hyperoxia results in ROS damage that impairs alveolar macrophage phagocytosis of PA and that macrophage function can be preserved by the administration of antioxidant agents that either generally enhance capacity to buffer cellular oxidant status or scavenge superoxide ions (O2−•).
Here, we report that exposure to 95% O2 significantly impairs macrophage phagocytosis of both the mucoid and non-mucoid forms of PA. This impairment of PA phagocytosis is correlated with a severe lung injury and cell proliferation arrest. Interestingly, exposure to 65% O2, a relatively moderate concentration of oxygen, also induced a significant reduction in PA phagocytosis, comparable to what observed at ≥ 95% O2, even though the effects on lung injury and cell proliferation are negligible. One particular feature of this impairment is the disorganization of actin cytoskeleton. Administration of the antioxidants, either superoxide dismutase or procysteine, preserved macrophage phagocytosis of PA as well as actin cytoskeleton organization.
MATERIALS AND METHODS
Cell Culture and Reagents
Mouse macrophage-like RAW 264.7 cells (ATCC, Manassas, VA) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% glutamine, 100 units/ml penicillin/streptomycin (Life technologies Inc., Grand Island, NY) and maintained at 37°C in 5% CO2/95% room air. Hyperoxic exposure was performed in sealed humidified chambers flushed with 95% O2/5% CO2 or 65% O2/5% CO2/30%N2 as previously described [12]. A subgroup of macrophages were treated with superoxide dismutase (SOD1) or procysteine (Cayman, Ann Arbor, MI) either pre- or post- hyperoxic exposure. The number of viable cells and cell proliferation were assessed by 3-(4,5,-dimethyl thiazolyl-2)-2,5-diphenyl tetrazolium bromide assay (MTT) and trypan blue exclusion analyses. All the chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Phagocytosis Assay
PAO1, a motile, piliated and nonmucoid strain of PA, was grown to log phase in Lenox medium (Becton-Dickson Inc., Sparks, MD). CF1, a mucoid clinical isolate of PA from patients with cystic fibrosis, was maintained in a synthetic medium [33]. Procedures to determine phagocytosis were performed as described with minor modifications [29]. After exposure, macrophages were incubated with PA or FITC-labeled latex minibeads (Polysciences, Warrington, PA) at ≈100:1 at 37°C for 60 min. Macrophages were then washed with chilled (4°C) Phosphate Buffered Saline (PBS). Phagocytosis of PA was visually assessed using an immunofluoresent microscope or by colony forming unit assay. To visualize the uptake of PA, macrophages were fixed with 4% paraformaldehyde for 10 minutes, washed with PBS and stained with either 5μg/ml of 4-6-diamidine-2-phenylindoledihydrochloride (DAPI) or Texas Red X-phalloidin (Molecular Probes, Eugene, OR) in 1% bovine serum albumin. The slides were analyzed using an epifluorescence microscope (Nikon, Melville, NY) and a BIO-RAD MRC 600 confocal scanning microscope. The internalization of PA was confirmed by z-series analysis on the confocal microscope. Uptake of PA was determined by counting at least 50 macrophages/slide in triplicate from a minimum of two independent experiments for each group. For the colony forming unit assay, macrophages were lysed in water and viable bacteria enumerated by serial dilution and culture on nutrient agar plates (Becton-Dickinson, Inc).
Exposure to Hyperoxia in vivo and Isolation of Alveolar Macrophages
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research. Male C57BL/6 mice (6–8 weeks), purchased from Jackson Laboratories (Bar Harbor, Maine), were exposed to >99 or 65% for 72 h as described O2 previously and then sacrificed [7]. The lungs were lavaged with 2 separate 1-ml aliquots of sterile saline. The resulting bronchoalveolar lavage fluid (BALF) was pooled, centrifuged and the cellular pellet resuspended and plated in RPMI 1640 medium. After 3 hours, non-adherent cells were removed.
Statistical Analysis
All experiments were performed independently at least twice. The data are presented as mean ± standard error, and analyzed for statistical significance using the one-way analysis of variance (ANOVA). A P value of ≥0.05 was considered significant.
RESULTS
Hyperoxia reduces the ability of macrophages to phagocytose PA and latex beads
To test whether hyperoxia affects the ability of macrophages to phagocytose PA, RAW 264.7 cells were exposed to 95% O2, and then incubated with PAO1, a non-mucoid strain of PA expressing green fluorescent protein. To visualize the internalization of PAO1, RAW 264.7 cells were stained with phalloidin, a reagent that labels the actin cytoskeleton, and analyzed by confocal microscopy. Exposure to 95% O2 for 24 h reduced the uptake of PAO1 significantly to 67±5.6% of room air (RA) control (Fig. 1A, from approximately 9 bacteria/cell to 6 bacteria/cell). The uptake of PAO1 was further reduced to 33±8.6% of the RA control after 48h (data not shown). The effect of hyperoxia on RAW cells to uptake of a FITC-labeled clinical mucoid isolate of PA (CF1) was also examined. The phagocytosis of CF1 was reduced to 59% of RA control in RAW 264.7 cells exposed to 95% O2 for 24 h (Fig. 1B). To determine whether the phagocytosed PA were viable, the number of viable PA was determined by colony forming unit (CFU) analysis in macrophage cell lysates. Compared to the room air controls, the relative colony forming unit (RCFU) for PAO1 was moderately lower in RAW ells following exposure to 95% O2 for 24 h (Fig. 2A). The reduction in RCFU was significantly greater after a 48 h hyperoxic exposure (Fig. 2A). Hyperoxia-reduced RCFU was also observed in RAW 264.7 cells exposed to mucoid CF1. Fig. 2B shows that similar to the results of phagocytic analysis (Fig. 1B), fewer live mucoid CF1 were detected in hyperoxia-exposed macrophages compared to those remaining at room air (RA). A comparison of the results presented in Fig. 2A and Fig. 2B indicates a significant difference in the PAO1 RCFU versus that of the mucoid CF1 strain (90±5% vs 69±3%, respectively; p<0.05) following 24 h of hyperoxia. This difference in CFU became insignificant after a 48 h exposure to 95% O2.
Fig. 1. Reduced Uptake of P. aeruginosa by Macrophages in Hyperoxia.

RAW 264.7 cells were exposed to 95% O2 or to room air (RA) for 24 h, and then incubated for 1 h with either (A) PAO1, a nonmucoid strain of P. aeruginosa expressing green fluorescent protein, or (B) CF1, a FITC-labeled mucoid isolate of P. aeruginosa. RAW 264.7 cells were stained with phalloidin to visualize the macrophages in the field. At least 50 macrophages per slide, 6 slides from 3 independent experiments were counted and the numbers of bacteria/cell were graphed in the lower panels. *, p<0.05 compared to RA controls. Bar, 10 μm.
Fig. 2. Hyperoxia Reduced Viable P. aeruginosa in Macrophages.

RAW 264.7 cells were exposed to 95% O2 or to room air (RA) for indicated time periods, and then incubated for 1 h with either (A) PAO1, a non-mucoid strain of P. aeruginosa expressing green fluorescent protein, or (B) CF1, a FITC-labeled mucoid clinical isolate of P. aeruginosa. The number of viable phagocytosed P. aeruginosa was determined by colony forming unit (CFU) analysis in macrophage cell lysates. The numbers of CFU were normalized to the macrophage number and expressed as the percentage of the number obtained prior to the exposures (which was designated as 100%). *, p<0.05, compared to the room air control; #, p<0.05 compared to 24 h exposure to 95% O2 and PAO1 RCFU.
To assess whether the reduced phagocytosis (Fig. 1A–B) and RCFU of PA (Fig. 2A–B) in hyperoxic RAW 264.7 cells results from increased killing of bacteria due to higher levels of ROS generated in hyperoxic macrophages, the phagocytic activity of RAW 264.7 cells was assessed using FITC-labeled inert latex minibeads. Fig. 3 shows that RAW 264.7 cells exposed to 95% O2 for 24 h also exhibited significantly impaired phagocytosis of latex minibeads compared with macrophages that remained at RA (36.1 ± 0.66% vs 100, n=6, p<0.05)
Fig. 3. Reduced Uptake of Latex Beads in Hyperoxia.
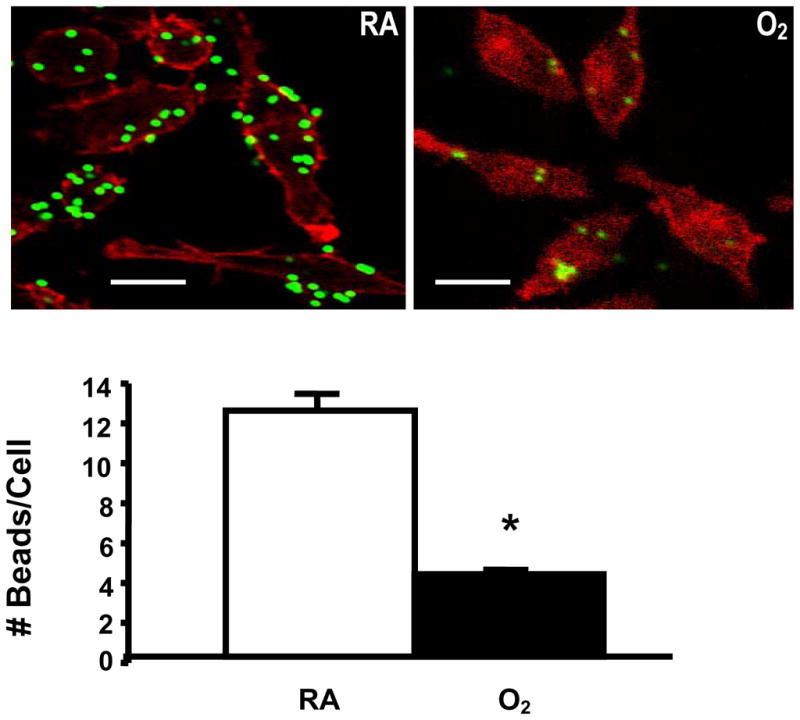
RAW 264.7 cells were exposed to room air (RA) or 95% O2 for 24 h, and then incubated with FITC-labeled latex minibeads for 1 h. RAW 264.7 cells were stained with phalloidin to visualize the macrophages in the field. At least 50 macrophages per slide, 6 slides from 3 independent experiments were counted and the number of beads/macrophage were expressed in the bar graph. *, p<0.05 compared to RA controls. Bar, 10 μm.
Reduced phagocytosis of P. aeruginosa is associated with cytoskeleton disorganization in hyperoxic macrophages
Phagocytosis is driven by a finely controlled rearrangement of the actin cytoskeleton [34]. Hyperoxia affects actin polymerization and distribution in macrophages with impaired phagocytosis of Klebsiella pneumonia (K. pneumonia) [29]. We examined the possibility that hyperoxia may reduce macrophage phagocytosis of PA through effects on actin. Phalloidin staining of RAW 264.7 cells revealed that exposure to hyperoxia resulted in dramatic cytoskeleton disorganization with increased stress fiber number and thickness, depleted cortical actin and large masses of disordered, polymerized cytoplasmic actin. (Fig. 4A). Normoxic macrophages typically exhibited the ability to form phagocytic cups around PA (Fig. 4A, inset). On the other hand, formation of phagocytic cup around PA was sparse in hyperoxic macrophages, although more actin polymerization was observed in these cells (Fig. 4B, inset). Actin polymerization/remodeling is the driving force for particle internalization during phagocytosis. To test whether actin polymerization may directly attenuate PA phagocytosis, RAW 264.7 cells were exposed to Jasplakinolide (JP), a microfilament polymerizing agent [35], stained with phalloidin and then examined for uptake of PAO1. RAW 264.7 cells exposed to 0.5μM and 1μM JP for 1 h exhibited significantly decreased phagocytosis compared to RAW 264.7 cells exposed to vehicle control (0.1% dimethyl sulfoxide) (54±3.8% for 0.5μM JP and 28±2.9% for 1μM JP, Fig. 4B). At these concentrations, JP was not cytotoxic to RAW 264.7 cells (data not shown). Actin polymerization, redistribution and general disorder may therefore contribute to the impairment of PA phagocytosis.
Fig. 4. Hyperoxia-Reduced Phagocytosis of P. aeruginosa is Associated with Disorganization of Actin Cytoskeleton and Oxidation of Actin.

RAW 264.7 cells were exposed to room air (RA) or 95% O2 for 24 h, then incubated with PAO1 for 1 h, and stained with phalloidin (A). Hyperoxic cells (O2) exhibited altered morphology: increased stress fiber formation and thickness, disordered polymeric cytoplasmic actin (A), an absence of phagocytic cups surrounding PAO1 (A, insets) and a loss of cortical actin at margins of macrophages. Bar, 10 μm. B. Equal amounts of proteins were immunoprecipitated with anti-actin antibodies from RAW cells cultured in room air or 95% O2, derivatized with DNPH. The resulting proteins were resolved on SDS-PAGE and probed with anti-DNP antibodies and HRP-conjugated secondary antibodies (left Panel). Control samples without derivatization (Der) were compared with those with derivatization as negative controls for nonspecific binding. Arrows indicate actin. Blots were then stripped and probed with anti-actin antibodies (right Panel, a representative of three independent experiments).
Alveolar macrophages isolated from mice exposed to hyperoxia exhibited reduced phagocytosis of PA ex vivo
Alveolar macrophages (AM) play an essential role in pulmonary defense against bacterial infection [24]. To establish whether our in vitro observations in RAW 264.7 cells are reflective of the events in hyperoxic lungs, AM were assessed for their ability to phagocytose PAO1 ex vivo. AM isolated from C57BL/6 mice exposed to ≥99% O2 for 3 days have a markedly reduced ability for PAO1 uptake compared to the AM from mice that remained at room air (50.89± 2.03%, n=6, p= 0.0001, Fig. 5A) and RCFU counts (53.2 ± 3.6% vs 100 ± 5.4%, n = 6, p<0.05, Fig. 5B). These data indicate that AM exposed to ≥99% O2 in vivo have similarly impaired phagocytosis of PA as RAW 264.7 cells exposed to hyperoxia in vitro. Examination of AM viability by tryphan blue exclusion analysis indicated that > 95% of alveolar macrophages isolated from mice in the hyperoxic group was viable (data not shown). These findings are comparable to those obtained from RAW 264.7 cell exposed to hyperoxia in vitro.
Fig. 5. Disorganization of Actin Cytoskeleton Reduced Phagocytosis of P. aeruginosa.

RAW 264.7 cells were exposed to different concentrations of Jasplakinolide (JP), 0.5 or 1 μM, for 1 h, then incubated with PAO1 for 1 h and stained with phalloidin (B). At least 50 macrophages per slide, 6 slides from 3 independent experiments, were counted and the number of PAO1/macrophage was expressed in the bar graph (A). *, p<0.05, compared to medium control (Con); **, p< 0.05, compared to samples exposed to 0.5 μM JP. Macrophages treated with JP exhibited altered morphology: increased stress fiber formation and thickness, disordered polymeric cytoplasmic actin (B), and a loss of cortical actin at margins of macrophages. Bar, 10 μm.
Macrophages exposed to moderate concentration of oxygen exhibited significant reduced phagocytosis of PA
Extremely high concentrations of oxygen (≥95% O2) are administrated to patients with severe respiratory distress. However, more often patients receive the lowest concentration of oxygen needed [36], in an effort to limit pulmonary oxygen toxicity. To test whether a reduction in supplemental oxygen concentration also limits ROS damage to pulmonary host defenses, we examined whether exposure to moderate hyperoxia (65%) O2 can attenuate ≥95% O2-induced impairment of phagocytosis of PA. In contrast to exposure ≥95% O2, exposure to 65% O2 does not cause significant lung damage in CB57/6L mice (Fig. 6D) or marked cell proliferation arrest in RAW 264.7 cells (Fig. 6B). Intriguingly, exposure to 65% O2 significantly reduced PAO1 uptake by AM from hyperoxic CB57/6L mice (Fig. 6C) and hyperoxic RAW 264.7 cells (Fig. 6A). Similar results were obtained when the number of viable phagocytosed PA were determined by colony forming unit analysis in both hyperoxic alveolar macrophages and RAW 264.7 cells (data not shown). Surprisingly, the reduction in phagocytosis of PA-induced by exposure to 65% O2, was comparable to that observed at ≥95% O2, if not greater (Fig. 6A). In addition, similar alteration of actin cytoskeleton occurred in macrophages exposed to 65% O2 as those to 95% O2 (Fig. 6A, inset).
Fig. 6. Exposure to in vivo Hyperoxia Reduced Phagocytosis of P. aeruginosa in Alveolar Macrophages.

C57BL/6 mice were exposed to >99% O2 for 3 d (O2) or remained in room air (RA). Alveolar macrophages were isolated from BALF, incubated for 1 h with P. aeruginosa, and assessed for the number of phagocytosed P. aeruginosa/macrophage as described in Fig. 1(A) and for live P. aeruginosa by CFU analysis as described in materials and methods (B). Insets in panel A illustrates the disorganized actin cytoskeleton in hyperoxic alveolar macrophages. *, p<0.05, compared to the room air control (n=6 in each group). Bar, 10 μm.
Macrophages exposed to hyperoxia retain proinflammatory responses upon LPs stimulation
Macrophages are responsible for the production of proinflammatory mediators such as TNF-α during the initiation of the inflammatory cascade [37, 38]. To determine if the impaired phagocytosis of PA is linked to changes in secreting proinflammatory cytokines, we investigated whether exposure to hyperoxia affects TNF-α production by RAW 264.7 cells upon exposure to LPS. The level of TNF-α was not detectable in the media of room air control cells or cells treated with hyperoxia alone (Fig. 7). LPS induced substantial accumulation of TNF-α in the media of RAW 264.7 cells cultured in the room air (Fig. 7). Prior exposure to hyperoxia did not significantly alter the ability of RAW 264.7 cells to secrete TNF-α in response to LPS (Fig. 7).
Fig. 7. Exposure to Moderate Hyperoxia Reduced Uptake of P. aeruginosa by Alveolar Macrophages and RAW 264.7 Cells Without Causing Lung Damage or Cell Proliferation Arrest.

C57BL/6 mice & RAW 264.7 cells were exposed to either ≥ 95% or 65% O2 or remained at room air (RA). Both (A–B) RAW 264.7 cells & (C–D) alveolar macrophages isolated from BALF were assessed for phagocytosis of P. aeruginosa as described in Figs.1& 4. The number of bacteria/macrophage was presented in the bar graphs. The number of viable RAW 264.7 cells was assessed by tetrazolium compound (MTT) analysis (B) and mouse lung injury was measured by wet/dry lung ratio (D). *, p<0.05, compared to the room air control; #, p<0.05 compared to those exposure to 95% O2. Bar, 10 μm.
Antioxidants attenuate impairment of P. aeruginosa phagocytosis and disorganization of actin cytoskeleton induced by hyperoxia
Superoxide dismutases (SOD) are antioxidant enzymes that scavenge superoxide radicals. To examine whether the reduced antibacterial function of macrophages in hyperoxia can be restored through reduction of the levels of superoxide, RAW 264.7 cells were pre-treated with SOD-1. Fig. 8 shows that pretreatment with SOD-1 preserves the uptake of PA by RAW 264.7 cells. This effect is not due to an increase in nonspecific binding of PA to RAW cells in the presence of SOD-1, because there was no increase in PA uptake by RAW 264.7 cells that were exposed to room air and treated with SOD-1 (Fig. 8, Panel labeled with SOD/RA).
Fig. 8. Macrophages Exposed to Prolonged Hyperoxia Respond to Endotoxin Stimulation with Production of TNF-α.

RAW 264.7 cells were exposed to 95% O2 or room air (RA) for 48h, and then incubated with 500ng/ml of Endotoxin for 18 hours prior to harvesting of supernatant and measurement of TNF-α with a TNF-α kit (R&D Systems, Minneapolis, MN). TNF-α was not detectable in the media of control cells or cells exposed to O2 alone. Values are means ± SE of >2 separate experiments, n=4.
Procysteine, L-2-oxothiazolidine-4-carboxylate, is a thiol-related cysteine precursor [39, 40] that can increase intracellular levels of the antioxidant glutathione [41, 42]. To test whether it can preserve macrophage phagocytosis of PA, RAW 264.7 cells were treated with 1 mM procysteine, either before or 24 h post hyperoxic exposure. Fig. 9 shows that procysteine pre-treatment markedly enhanced macrophage ability to phagocytose PAO1 (Fig. 9A) as well as mucoid CF1 in macrophages exposed to hyperoxia for 24 h (Fig. 9B). Significant improvements in the ability to phagocytose both PAO1 and CF1 were observed in macrophages exposed to hyperoxia for 48 h (113±9.5% vs 38±7.3% (p<0.05) and 83 ±7.3% vs 38±4.7% (p<0.05) for PAO1 and CF1, respectively). Intriguingly, when macrophages were treated with procysteine 24 h post hyperoxic exposure, the ability to phagocytose both PAO1 and CF1 was also significantly increased (Fig. 9). The degree of improvement in the phagocytosis of PA resulting from pre or post-treatment with procysteine was not significant (Fig. 9). To elucidate the mechanisms of procysteine-rescued phagocytosis of PA, we assessed the organization of the actin cytoskeleton in hyperoxic macrophages in the presence or absence of the treatment. Accompanied with the rescuing phagocytosis of PA, treatment of hyperoxic macrophages with procysteine preserved the normal organization of the actin cytoskeleton (Fig. 10C).
Fig. 9. SOD Preserves P. aeruginosa Uptake by Macrophages in Hyperoxia.

RAW 264.7 cells were exposed to 95% O2 for 24 h in the presence or absence of 250 U/ml superoxide dismutase-1. After hyperoxic exposure, RAW 264.7 cells were incubated with PAO1 for 1 h and stained with phalloidin to visualize the organization of the actin cytoskeleton. PAO1 was pseudocolored as white. At least 50 macrophages per slide were counted and expressed as the number of bacteria/macrophage. Macrophages treated with superoxide dismutase-1, and remained at room air were used as controls. *, p<0.05, compared to the hyperoxic sample without addition of SOD1. Bar, 10 μm.
Fig. 10. Procysteine Preserves Phagocytosis of P. aeruginosa in Hyperoxia.

RAW 264.7 cells were exposed to 95% O2 in the presence (Pre: added 24 h prior to; Post, added 24 h after the start of hyperoxic exposure) of 1 mM procysteine. After hyperoxic exposure, RAW 264.7 cells were incubated with either non-mucoid PAO1 (A) or mucoid CF1 (B) for 1 h, and then assayed for Colony Forming Unit. The numbers of colonies were normalized to the macrophage number and expressed as the percentage of the number (which is designated as 100%) prior to the exposures (either hyperoxia or room air). *, p<0.05, compared to the hyperoxic control without procysteine treatments.
DISCUSSION
Our results indicate that exposure to hyperoxia in either an in vitro cell culture or an in vivo animal model system significantly impairs the ability of macrophages to phagocytose mucoid and non-mucoid strains of PA, a common cause of noscomial pneumonia. Interestingly, exposure of 65% O2, a moderate concentration of oxygen which does not lead to significant lung injury or marked inhibition of cell proliferation, also drastically reduces phagocytosis of PA. Exposure to hyperoxia profoundly alters actin cytoskeleton organization. The ability of macrophages to phagocytose PA in hyperoxia is preserved by antioxidant treatment, which is effective, even when administered 24h after the exposure to hyperoxia. Antioxidant treatment is associated with conservation of normal actin-cytoskeleton organization. Overall, these results suggest that hyperoxia-attenuated phagocytosis of PA is mediated by ROS effects on the actin cytoskeleton.
Reduced phagocytosis of P. aeruginosa is not due to decreased cell viability in macrophages exposed to hyperoxia
Exposure to hyperoxia, regardless whether the concentration of oxygen is ≥ 95% or 65% O2, induced impairment of phagocytosis of PA by macrophages. However, this impairment is not due to the loss of cell viability or cell death. First, few Trypan blue positive signals, an indicator for loss of membrane integrity/cell death, were detected in either RAW 264.7 cells exposed to hyperoxia or alveolar macrophages (AM) isolated from hyperoxic lungs (<5%) at a similar level as those obtained from room air controls. This finding is consistent with previous studies which indicate that exposure to hyperoxia does not decrease viability of macrophages [21, 22, 25, 43, 44]. In addition, RAW 264.7 cells, exposure to 65% O2, could still proliferate (Fig. 6B) even though their phagocytic ability was significantly suppressed (Fig. 6). Furthermore, the ability of hyperoxic RAW 264.7 cells to proliferate was restored when they were re-cultured in room air (data not shown), although exposure of RAW cells to ≥ 95% O2 for up to 48 hours did induce cell proliferation arrest (Fig. 6B). The blunted sensitivity to hyperoxia-induced loss of cell viability is also reflected in the proinflammatory responses [43, 44]. Results in Fig. 7 indicate that hyperoxia does not abolish the ability of macrophages cells to release TNF-α in response to LPS. Other reports also indicate that hyperoxic macrophages retain ability to produce nitric oxide, in response to LPS and interferon-γ [29, 45]. Cell death or loss of cell viability is not therefore a likely explanation for hyperoxia-induced impairment of PA phagocytosis.
Actin cytoskeleton alterations are critical to P. aeruginosa phagocytosis under hyperoxic conditions
The internalization of bacteria by phagocytes is characterized by an actin-dependent extension of the plasma membrane around the particle [34]. This is followed by a secondary activities, such as the release of ROS, proteases and inflammatory cytokines from the phagocyte [46]. Oxidant-induced cell damage is thought to involve the cytoskeleton, with the actin system appearing to be most sensitive to oxidative attack [47]. Hyperoxia has been shown to reduce macrophage phagocytosis of K. pneumonia through effects on the actin [29]. Staining of actin cytoskeleton by phalloidin indicates that hyperoxia causes the broad spectrum of dramatic changes that we collectively describe as “actin disorganization” (Figs. 4 & 5).
Normoxic macrophages exhibited a normal cortical distribution of polymerized actin whereas hyperoxic macrophages exhibited increased levels of polymerized actin with reduced levels of cortical actin. In addition, results shown in Fig. 4 suggest that the distribution/rearrangement of acting cytoskeleton in hyperoxic macrophages does not easily accommodate phagocytic cup formation and particle engulfment. The lack of cortical actin in hyperoxic cells appears to be particularly prohibitive to the internalization of PA. The cellular location of polymerized actin appears to be critical for the impairment of phagocytosis. The large quantities of disordered polymeric actin in the central region of hyperoxic cells may also be obstructive to successful engulfment of PA. Overall, our results suggest that hyperoxia causes impairment of phagocytosis of PA by interfering with the process of directional actin polymerization/remodeling that drives bacterial internalization. Results with Jasplakinolide (Fig. 4B), an agent that increases actin polymerization, indicate that inappropriate actin polymerization may indeed be attributed to the impairment of macrophage phagocytosis of PA. Jasplakinolide has also been shown to have similar effects on uptaking Klebsiella [29].
The mechanism(s) by which hyperoxia induces these effects on actin remains to be fully elucidated. The actin cytoskeleton response to oxidative stress has been studied in numerous different cell types [48–50]. ROS, as well as a variety of other intracellular signals, can mediate the actin cytoskeleton reorganization in preparation for processes such as adhesion under physiological conditions [51, 52]. However, exposure to severe oxidative stress, especially those induce glutathione depletion can cause the formation of inter or intra-molecular disulfide that lead to cross-linking between actin filaments, resulting in excessive polymerization of actins and formation of more stress fibers [52]. In addition, elevated levels of ROS during hyperoxia may lead to the excessive oxidation and glutathionylation of actin’s regulatory proteins [47], causing regulatory dysfunction in normal actin polymerization/remodeling. Therefore, reduction of oxidative stress may be essential for preservation of both normal actin cytoskeleton organization and phagocytic function during hyperoxia.
Antioxidant treatment preserves phagocytosis of P. aeruginosa during hyperoxia
Changes in the structure of the actin cytoskeleton are generally presumed to involve the oxidation of critical protein sulfhydryl residues [53, 54]. Reactive sulfhydryls are located on proteins in all cellular compartments as well as low molecular weight thiols [44]. Thiol moieties are part of the elaborate system of antioxidant defenses that respond to excessive ROS. Procysteine, is a thiol-related compound that has been shown to be associated with increased levels of glutathionine (GSH), the most important antioxidant in mammalian cells [42, 55]. Since exposure to hyperoxia reduces the intracellular level of GSH [56, 57], supplementation with procysteine was expected to enhance GSH levels and reduce ROS induced macrophage damage. Results, shown in Figs. 9&10, demonstrate that procysteine can prevent the hyperoxia-induced alteration of actin cytoskeleton and conserve the phagocytic functions in macrophages. Hyperoxia-induced changes in the actin cytoskeleton which impact phagocytosis of PA may therefore depend, in part, on the oxidation of reactive sulfhydryls. Cysteine 374 (Cys374) at the actin C-terminal segment is the main GSH reactive sulphydryl [47, 52, 58]. Recent studies in fibroblasts have shown that in vivo actin oxidation takes place between Cys374 and glutathionine; this modification was shown to be essential for cytoskeleton organization [52]. Inhibition of this actin glutathionylation, through either GSH depletion or expression of Cys374 redox insensitive mutant profoundly affected fibroblast cytoskeleton assembly. In this respect, actin glutathionylation can be viewed as a protective mechanism against further S-oxidation, permanent protein modification and ultimately irreversible cytoskeleton rearrangements. Interestingly, the oxidation of actin has been demonstrated in hyperoxic macrophages with reduced phagocytosis of K. pneumonia [29]. The superoxide radical is likely to significantly contribute to the reduction in phagocytosis of PA, as our results (Fig. 8) show that SOD-1 has a protective effect on antibacterial function. Even though prolonged exposure to hyperoxia does not suppress the level of SOD expression in macrophages [59], excessive levels of superoxide generated during hyperoxia may require high levels of SOD. In addition to rescuing macrophage function resulting from oxidative stress during hyperoxia, administration of antioxidants may also be useful in reducing ROS generated by bacteria themselves that are harmful to the host defense system [60, 61].
In summary, our study indicates that both moderate and high concentrations of hyperoxia impair macrophage phagocytosis of PA. The administration of moderate, rather than higher, concentrations of supplemental hyperoxia may limit pulmonary oxygen toxicity but the risk for reduced macrophage clearance of PA remains elevated. The effects of hyperoxia on macrophage phagocytosis of PA, however, can be attenuated by antioxidant supplementation. Our current effort is aimed at evaluating the efficacy of antioxidant treatments in the reduction of bacterial burden and in the ultimate improvement of the survival of infected hyperoxic animals. Successful completion of this project will no doubt highlight the possible benefits of antioxidant therapy in maintaining the phagocyte function(s) required for the prevention of nosocomial infection. This may lead to an improvement in the clinical outcomes of ventilated patients, especially those with acute respiratory distresses suffering high morbidity and mortality.
Fig. 11. Procysteine Inhibits Hyperoxia-Suppressed Uptake of PAO1 and Disorganization of the Actin Cytoskeleton in Macrophages.

RAW 264.7 cells were exposed to room air (A), or exposed to 95% O2 for 48 h without (B) or with addition of 1 mM procysteine after 24 h hyperoxia (C). Macrophages were then incubated with PAO1 (green) for 1 h, stained with phalloidin (red) to visualize the organization of actin cytoskeleton in the field. Bar, 10 μm.
Acknowledgments
Authors would like to thank Drs. Sadis Matalon, H. Hank Simms, Jeffrey Kazzaz, Marc Symons, Mike S. Niederman and Jonathan Davis for insightful discussion and invaluable suggestions on this project.
Authors would like to thank Drs. Sadis Matalon, H. Hank Simms, Jeffrey Kazzaz, Marc Symons, Sanna M. Goyert, Mike S. Niederman, Jonathan Davis, and Nicole Palma for insightful discussion and invaluable suggestions on this project/manuscript; Dr. Alice Prince for PAO1. This work was supported by the ALANYS, the Research Funds from the St John’s University College of Pharmacy, the North Shore University Hospital/the Feinstein Institute for Medical Research at the North Shore-Long Island Jewish Health System, and the Winthrop Research Fund.
ABBREVIATIONS
- O2
molecular Oxygen
- O2−•
superoxide ions
- PA
Pseudomonas. Aeruginosa
- K. pneumonia
Klebsiella pneumonia
- ROS
reactive oxygen species
- DAPI
4-6-diamidine-2-phenylindoledihydrochloride
- O2−•
superoxide ions
- TNF-α
tumor necrosis factor-α
- SOD-1
superoxide dismutase-1
- RA
room air
- CFU
colony forming units
- RCFU
relative colony forming units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mehta RM, Niederman MS. Nosocomial pneumonia in the intensive care unit: controversies and dilemmas. J Intensive Care Med. 2003;18:175–188. doi: 10.1177/0885066603254249. [DOI] [PubMed] [Google Scholar]
- 2.Richards MJ, Edwards JR, Culver DH, Gaynes RP. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit Care Med. 1999;27:887–892. doi: 10.1097/00003246-199905000-00020. [DOI] [PubMed] [Google Scholar]
- 3.Intensive Care Antimicrobial Resistance Epidemiology (ICARE) Surveillance Report, data summary from January 1996 through December 1997: A report from the National Nosocomial Infections Surveillance (NNIS) System. Am J Infect Control. 1999;27:279–284. doi: 10.1053/ic.1999.v27.a98878. [DOI] [PubMed] [Google Scholar]
- 4.Marquette CH, Copin MC, Wallet F, Neviere R, Saulnier F, Mathieu D, Durocher A, Ramon P, Tonnel AB. Diagnostic tests for pneumonia in ventilated patients: prospective evaluation of diagnostic accuracy using histology as a diagnostic gold standard. Am J Respir Crit Care Med. 1995;151:1878–1888. doi: 10.1164/ajrccm.151.6.7767535. [DOI] [PubMed] [Google Scholar]
- 5.Garau J, Gomez L. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis. 2003;16:135–143. doi: 10.1097/00001432-200304000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Mantell LL, Horowitz S, Davis JM, Kazzaz JA. Hyperoxia-induced cell death in the lung--the correlation of apoptosis, necrosis, and inflammation. Ann N Y Acad Sci. 1999;887:171–180. doi: 10.1111/j.1749-6632.1999.tb07931.x. [DOI] [PubMed] [Google Scholar]
- 7.Mantell LL, Kazzaz JA, Xu J, Palaia TA, Piedboeuf B, Hall S, Rhodes GC, Niu G, Fein AF, Horowitz S. Unscheduled apoptosis during acute inflammatory lung injury. Cell Death Differ. 1997;4:600–607. doi: 10.1038/sj.cdd.4400278. [DOI] [PubMed] [Google Scholar]
- 8.Crapo JD. Morphologic changes in pulmonary oxygen toxicity. Annu Rev Physiol. 1986;48:721–731. doi: 10.1146/annurev.ph.48.030186.003445. [DOI] [PubMed] [Google Scholar]
- 9.Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis. 1980;122:123–143. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- 10.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA. Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest. 1998;101:1970–1982. doi: 10.1172/JCI1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barazzone C, White CW. Mechanisms of cell injury and death in hyperoxia: role of cytokines and Bcl-2 family proteins. Am J Respir Cell Mol Biol. 2000;22:517–519. doi: 10.1165/ajrcmb.22.5.f180. [DOI] [PubMed] [Google Scholar]
- 12.O’Reilly MA, Staversky RJ, Finkelstein JN, Keng PC. Activation of the G2 cell cycle checkpoint enhances survival of epithelial cells exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2003;284:L368–375. doi: 10.1152/ajplung.00299.2002. [DOI] [PubMed] [Google Scholar]
- 13.Lee PJ, Alam J, Wiegand GW, Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci U S A. 1996;93:10393–10398. doi: 10.1073/pnas.93.19.10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogunlesi F, Cho C, McGrath-Morrow SA. The effect of glutamine on A549 cells exposed to moderate hyperoxia. Biochim Biophys Acta. 2004;1688:112–120. doi: 10.1016/j.bbadis.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Ray P, Devaux Y, Stolz DB, Yarlagadda M, Watkins SC, Lu Y, Chen L, Yang XF, Ray A. Inducible expression of keratinocyte growth factor (KGF) in mice inhibits lung epithelial cell death induced by hyperoxia. Proc Natl Acad Sci U S A. 2003;100:6098–6103. doi: 10.1073/pnas.1031851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Arita Y, Koo HC, Davis JM, Kazzaz JA. Inhibition of c-Jun N-terminal kinase pathway improves cell viability in response to oxidant injury. Am J Respir Cell Mol Biol. 2003;29:779–783. doi: 10.1165/rcmb.2003-0087RC. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang LY, White CW. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L573–584. doi: 10.1152/ajplung.00410.2001. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad S, Ahmad A, Gerasimovskaya E, Stenmark KR, Allen CB, White CW. Hypoxia protects human lung microvascular endothelial and epithelial-like cells against oxygen toxicity: role of phosphatidylinositol 3-kinase. Am J Respir Cell Mol Biol. 2003;28:179–187. doi: 10.1165/rcmb.2002-0004OC. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad S, Ahmad A, Ghosh M, Leslie CC, White CW. Extracellular ATP-mediated signaling for survival in hyperoxia-induced oxidative stress. J Biol Chem. 2004;279:16317–16325. doi: 10.1074/jbc.M313890200. [DOI] [PubMed] [Google Scholar]
- 20.Petrache I, Choi ME, Otterbein LE, Chin BY, Mantell LL, Horowitz S, Choi AM. Mitogen-activated protein kinase pathway mediates hyperoxia-induced apoptosis in cultured macrophage cells. Am J Physiol. 1999;277:L589–595. doi: 10.1152/ajplung.1999.277.3.L589. [DOI] [PubMed] [Google Scholar]
- 21.Nyunoya T, Powers LS, Yarovinsky TO, Butler NS, Monick MM, Hunninghake GW. Hyperoxia induces macrophage cell cycle arrest by adhesion-dependent induction of p21Cip1 and activation of the retinoblastoma protein. J Biol Chem. 2003;278:36099–36106. doi: 10.1074/jbc.M304370200. [DOI] [PubMed] [Google Scholar]
- 22.Nyunoya T, Monick MM, Powers LS, Yarovinsky TO, Hunninghake GW. Macrophages survive hyperoxia via prolonged ERK activation due to phosphatase down-regulation. J Biol Chem. 2005;280:26295–26302. doi: 10.1074/jbc.M500185200. [DOI] [PubMed] [Google Scholar]
- 23.Buccellato LJ, Tso M, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004;279:6753–6760. doi: 10.1074/jbc.M310145200. [DOI] [PubMed] [Google Scholar]
- 24.Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J, Sawa T. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun. 1998;66:3164–3169. doi: 10.1128/iai.66.7.3164-3169.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baleeiro CE, Wilcoxen SE, Morris SB, Standiford TJ, Paine R., 3rd Sublethal hyperoxia impairs pulmonary innate immunity. J Immunol. 2003;171:955–963. doi: 10.4049/jimmunol.171.2.955. [DOI] [PubMed] [Google Scholar]
- 26.Rister M. Effects of hyperoxia on phagocytosis. Blut. 1982;45:157–166. doi: 10.1007/BF00320800. [DOI] [PubMed] [Google Scholar]
- 27.Raffin TA, Simon LM, Braun D, Theodore J, Robin ED. Impairment of phagocytosis by moderate hyperoxia (40 to 60 per cent oxygen) in lung macrophages. Lab Invest. 1980;42:622–626. [PubMed] [Google Scholar]
- 28.Crowell RE, Hallin G, Heaphy E, Mold C. Hyperoxic suppression of Fc-gamma receptor-mediated phagocytosis by isolated murine pulmonary macrophages. Am J Respir Cell Mol Biol. 1995;12:190–195. doi: 10.1165/ajrcmb.12.2.7865216. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly PJ, Hickman-Davis JM, Davis IC, Matalon S. Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am J Respir Cell Mol Biol. 2003;28:443–450. doi: 10.1165/rcmb.2002-0153OC. [DOI] [PubMed] [Google Scholar]
- 30.Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 31.Michiels C, Raes M, Toussaint O, Remacle J. Importance of Se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radic Biol Med. 1994;17:235–248. doi: 10.1016/0891-5849(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 32.Pietarinen P, Raivio K, Devlin RB, Crapo JD, Chang LY, Kinnula VL. Catalase and glutathione reductase protection of human alveolar macrophages during oxidant exposure in vitro. Am J Respir Cell Mol Biol. 1995;13:434–441. doi: 10.1165/ajrcmb.13.4.7546773. [DOI] [PubMed] [Google Scholar]
- 33.Terry JM, Pina SE, Mattingly SJ. Role of energy metabolism in conversion of nonmucoid Pseudomonas aeruginosa to the mucoid phenotype. Infect Immun. 1992;60:1329–1335. doi: 10.1128/iai.60.4.1329-1335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan G. Differences in the mode of phagocytosis with Fc and C3 receptors in macrophages. Scand J Immunol. 1977;6:797–807. doi: 10.1111/j.1365-3083.1977.tb02153.x. [DOI] [PubMed] [Google Scholar]
- 35.Isowa N, Liu M. Role of LPS-induced microfilament depolymerization in MIP-2 production from rat pneumocytes. Am J Physiol Lung Cell Mol Physiol. 2001;280:L762–770. doi: 10.1152/ajplung.2001.280.4.L762. [DOI] [PubMed] [Google Scholar]
- 36.ACCP-NHLBI National Conference on Oxygen Therapy. Chest. 1984;86:234–247. doi: 10.1378/chest.86.2.234. [DOI] [PubMed] [Google Scholar]
- 37.Standiford TJ, Wilkowski JM, Sisson TH, Hattori N, Mehrad B, Bucknell KA, Moore TA. Intrapulmonary tumor necrosis factor gene therapy increases bacterial clearance and survival in murine gram-negative pneumonia. Hum Gene Ther. 1999;10:899–909. doi: 10.1089/10430349950018300. [DOI] [PubMed] [Google Scholar]
- 38.Chen GH, Reddy RC, Newstead MW, Tateda K, Kyasapura BL, Standiford TJ. Intrapulmonary TNF gene therapy reverses sepsis-induced suppression of lung antibacterial host defense. J Immunol. 2000;165:6496–6503. doi: 10.4049/jimmunol.165.11.6496. [DOI] [PubMed] [Google Scholar]
- 39.Fidelus RK, Tsan MF. Enhancement of intracellular glutathione promotes lymphocyte activation by mitogen. Cell Immunol. 1986;97:155–163. doi: 10.1016/0008-8749(86)90385-0. [DOI] [PubMed] [Google Scholar]
- 40.Kalayjian RC, Skowron G, Emgushov RT, Chance M, Spell SA, Borum PR, Webb LS, Mayer KH, Jackson JB, Yen-Lieberman B, et al. A phase I/II trial of intravenous L-2-oxothiazolidine-4-carboxylic acid (procysteine) in asymptomatic HIV-infected subjects. J Acquir Immune Defic Syndr. 1994;7:369–374. [PubMed] [Google Scholar]
- 41.Brown LA, Harris FL, Guidot DM. Chronic ethanol ingestion potentiates TNF-alpha-mediated oxidative stress and apoptosis in rat type II cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L377–386. doi: 10.1152/ajplung.2001.281.2.L377. [DOI] [PubMed] [Google Scholar]
- 42.Lee YC, Lee KS, Park SJ, Park HS, Lim JS, Park KH, Im MJ, Choi IW, Lee HK, Kim UH. Blockade of airway hyperresponsiveness and inflammation in a murine model of asthma by a prodrug of cysteine, L-2-oxothiazolidine-4-carboxylic acid. Faseb J. 2004;18:1917–1919. doi: 10.1096/fj.04-2212fje. [DOI] [PubMed] [Google Scholar]
- 43.Danielson DS, Heagy W, Nieman KM, West MA. Relative hyperoxia augments lipopolysaccharide-stimulated cytokine secretion by murine macrophages. Surgery. 2003;133:538–546. doi: 10.1067/msy.2003.148. [DOI] [PubMed] [Google Scholar]
- 44.O’Brien-Ladner AR, Nelson ME, Cowley BD, Jr, Bailey K, Wesselius LJ. Hyperoxia amplifies TNF-alpha production in LPS-stimulated human alveolar macrophages. Am J Respir Cell Mol Biol. 1995;12:275–279. doi: 10.1165/ajrcmb.12.3.7873193. [DOI] [PubMed] [Google Scholar]
- 45.Pepperl S, Dorger M, Ringel F, Kupatt C, Krombach F. Hyperoxia upregulates the NO pathway in alveolar macrophages in vitro: role of AP-1 and NF-kappaB. Am J Physiol Lung Cell Mol Physiol. 2001;280:L905–913. doi: 10.1152/ajplung.2001.280.5.L905. [DOI] [PubMed] [Google Scholar]
- 46.Bautista AP. Free radicals, chemokines, and cell injury in HIV-1 and SIV infections and alcoholic hepatitis. Free Radic Biol Med. 2001;31:1527–1532. doi: 10.1016/s0891-5849(01)00745-6. [DOI] [PubMed] [Google Scholar]
- 47.Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001;31:1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- 48.Mirabelli F, Salis A, Marinoni V, Finardi G, Bellomo G, Thor H, Orrenius S. Menadione-induced bleb formation in hepatocytes is associated with the oxidation of thiol groups in actin. Arch Biochem Biophys. 1988;264:261–269. doi: 10.1016/0003-9861(88)90593-0. [DOI] [PubMed] [Google Scholar]
- 49.Mirabelli F, Salis A, Vairetti M, Bellomo G, Thor H, Orrenius S. Cytoskeletal alterations in human platelets exposed to oxidative stress are mediated by oxidative and Ca2+-dependent mechanisms. Arch Biochem Biophys. 1989;270:478–488. doi: 10.1016/0003-9861(89)90529-8. [DOI] [PubMed] [Google Scholar]
- 50.Bellomo G, Mirabelli F, Vairetti M, Iosi F, Malorni W. Cytoskeleton as a target in menadione-induced oxidative stress in cultured mammalian cells. I. Biochemical and immunocytochemical features. J Cell Physiol. 1990;143:118–128. doi: 10.1002/jcp.1041430116. [DOI] [PubMed] [Google Scholar]
- 51.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–944. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J Biol Chem. 2006 doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- 53.Milzani A, DalleDonne I, Colombo R. Prolonged oxidative stress on actin. Arch Biochem Biophys. 1997;339:267–274. doi: 10.1006/abbi.1996.9847. [DOI] [PubMed] [Google Scholar]
- 54.Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, DalleDonne I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000;9:1774–1782. doi: 10.1110/ps.9.9.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 56.Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 57.Housset B, Hurbain I, Masliah J, Laghsal A, Chaumette-Demaugre MT, Karam H, Derenne J. Toxic effects of oxygen on cultured alveolar epithelial cells, lung fibroblasts and alveolar macrophages. Eur Respir J. 1991;4:1066–1075. [PubMed] [Google Scholar]
- 58.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 59.Aerts C, Wallaert B, Gosset P, Voisin C. Relationship between oxygen-induced alveolar macrophage injury and cell antioxidant defence. J Appl Toxicol. 1995;15:53–58. doi: 10.1002/jat.2550150112. [DOI] [PubMed] [Google Scholar]
- 60.Bolm M, Chhatwal GS, Jansen WT. Bacterial resistance of daf-2 mutants. Science. 2004;303:1976. doi: 10.1126/science.303.5666.1976a. author reply 1976. [DOI] [PubMed] [Google Scholar]
- 61.Day BJ, van Heeckeren AM, Min E, Velsor LW. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect Immun. 2004;72:2045–2051. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]