

Abstract
Axonal degeneration is a hallmark of many debilitating neurological disorders and is thought to be regulated by mechanisms distinct from those governing cell body death. Recently, caspase 6 activation via amyloid precursor protein (APP) cleavage and activation of DR6 was discovered to induce axon degeneration after NGF withdrawal. We tested whether this pathway is involved in axonal degeneration caused by withdrawal of other trophic support, axotomy or vincristine exposure. Neurturin deprivation, like NGF withdrawal activated this APP/DR6/caspase 6 pathway and resulted in axonal degeneration, however, APP cleavage and caspase 6 activation were not involved in axonal degeneration induced by mechanical or toxic insults. However, loss of surface APP (sAPP) and caspase 6 activation were observed during axonal degeneration induced by dynactin 1(Dctn1) dysfunction, which disrupts axonal transport. Mutations in Dctn1 are associated with motor neuron disease and frontal temporal dementia, thus suggesting that the APP/caspase 6 pathway could be important in specific types of disease-associated axonal degeneration. The NGF deprivation paradigm, with its defined molecular pathway, was used to examine the context of Nmnat-mediated axonal protection. We found that although Nmnat blocks axonal degeneration after trophic factor withdrawal, it did not prevent loss of axon sAPP or caspase 6 activation within the axon, suggesting it acts downstream of caspase 6. These results indicate that diverse insults induce axonal degeneration via multiple pathways and that these degeneration signals converge on a common, Nmnat-sensitive program that is uniquely involved in axonal, but not cell body, degeneration.
Introduction
Why is axonal degeneration a common element of a diverse set of neurological disorders? A favorite hypothesis is that a wide range of neuronal insults triggers a general axonal self-destruction program. In support of this hypothesis, overexpression of Wlds, or its component Nmnat, blocks degeneration in response to many distinct stimuli (Mack et al., 2001; Ferri et al., 2003; Araki et al., 2004; Sasaki et al., 2006, 2009a,b; Yahata et al., 2009). The intrinsic pathways that drive degeneration are poorly understood, however calcium influx, regulated protein degradation, and JNK activation by DLK have all been implicated (Coleman, 2005; Luo and O'Leary, 2005; Miller et al., 2009).
A general molecular pathway leading from the initial insult to the effectors of axonal breakdown has remained elusive. Although axonal destruction shares morphological features with apoptosis, most studies indicate that it is a caspase-independent process as manipulation of the mitochondrial apoptotic machinery or caspase inhibitors fail to block axonal degeneration (Sagot et al., 1995; Finn et al., 2000; Watts et al., 2003; Whitmore et al., 2003). Recently, however a candidate pathway for axonal degeneration following NGF withdrawal that involves activation of caspase 6 was identified (Nikolaev et al., 2009). In this paradigm, removal of nerve growth factor (NGF) initiates a cascade of events whereby axonal APP is cleaved and the shed N-terminal APP fragment activates the tumor necrosis factor (TNF) receptor DR6. The activation of DR6 leads to caspase 6 activation and eventual axonal destruction.
Defects in axonal transport and insufficient trophic support are thought to be involved in many neurological disorders (De Vos et al., 2008). Dctn1 is a component of the dynactin complex that interacts with dynein to facilitate retrograde transport of cargoes from the axon terminal to the neuronal cell body. Missense mutations in DCTN1, which encodes dynactin 1, are linked to slowly progressing motor neuron diseases, ALS, ALS with FTD, and Perry syndrome (Puls et al., 2003, 2005; Münch et al., 2004, 2005; Wider and Wszolek, 2008; Farrer et al., 2009; Vilariño-Güell et al., 2009). Patients with these mutations also manifest mild sensory neuropathy (Puls et al., 2005). Mutation of DCTN1 is thought to cause neuronal dysfunction via disruption of retrograde axonal transport that results in decreased availability of trophic factors in the cell body. Because of the potential involvement in trophic factor deprivation, we hypothesized that the APP/DR6/caspase 6 pathway could play a role in neurological disorders caused by defective axonal transport. Furthermore, this pathway could also be involved in axonal degeneration due to mechanical injury, as blocking APP cleavage reduces axonal degeneration, neuronal loss and cognitive deficits resulting from traumatic brain injury (Loane et al., 2009). The identification of this APP-dependent pathway is an important advance in our understanding of the mechanism of axonal degeneration, however it remains unclear whether APP processing is a common initiating step in axonal degeneration or instead in one of multiple stimulus-dependent axonal degeneration programs (for review, see Kim and Tsai, 2009; Wiedemann, 2009).
Materials and Methods
Reagents.
The following primary antibodies were used: Anti-amyloid precursor protein (1:100; MAB348, Millipore), Active Caspase-6 Polyclonal Antibody (1:100, BioVision), anti-N-APP polyclonal antibody (20 μg/ml, Fisher Scientific), Neurofilament mouse monoclonal (1:1000, Sigma-Aldrich). The secondary antibodies used were Alexa Fluor 488- and 594-conjugated anti-mouse and anti-rabbit antibodies (1:500; Invitrogen). BACE inhibitor (10 μm; OM99-2) and cell-permeable caspase 6 inhibitor (VEID-CHO) were purchased from Calbiochem. The Apo Logix carboxyfluorescein caspase 6 detection kit was purchased from Cell Technology Inc. All other reagents were from Sigma-Aldrich unless otherwise noted.
Culture of SCG and DRGs.
Tissue culture plates were coated with poly-d-lysine and laminin (Invitrogen) and neurons were cultured as previously described (Sasaki et al., 2009a). Briefly, DRGs were collected from CD1 mouse (Charles River) embryos at embryonic day 12.5, whereas SCGs were collected from P0 Pups. DRGs were treated with 0.05% trypsin and 0.02% EDTA at 37°C for 15 min. SCGs were dissociated with collagenase (1 mg/ml) at 37°C for 30 min which was followed by trypsinization for 15 min at 37°C. After dissociation, DRGs and SCGs were triturated by passing repeatedly through a 1 ml pipette tip and resuspended in 500 μl of Neurobasal medium (Invitrogen) containing 0.2% B27 (Invitrogen) and 50 ng/ml NGF (2.5S; Harlan Bioproducts). After brief centrifugation (2000 × g), the cell pellet was resuspended in 50 μl of complete medium and neurons were plated using drop culture method by placing (2 μl) of cell suspension slightly below the center of each 24 well and incubated at 37°C with 5% CO2 for 1 h. After cell attachment, 500 μl of complete medium containing 1 μm 5-fluoro-2-deoxyuridine, and 1 μm uridine was added to each well.
Lentivirus-mediated gene knockdown.
Oligonucleotides were designed to target proteins using parameters set by Genescript on its siRNA design website (Genescript) (Wang and Mu, 2004). Fifty-nine base pair oligonucleotides were engineered containing both forward and reverse sequences for the 19 bp siRNA target sequences linked together with a hairpin loop, and ligated into AgeI-BamHI cut pFUHIV downstream of the U6 promoter (Chen et al., 2006). The lentiviral-expressing siRNAs were produced and the efficiency of knockdown was ascertained by measuring the Dctn1 mRNA levels. The target sequences used were siRNA #1 GCAGTGTGGACGTGTATAA, siRNA #2 GAAGATCCGAAGGCGGATG and siRNA #3 CCAGAGACTTTTGATTTCA.
Construction of lentiviral expression plasmids.
mCherry was obtained from Dr. Roger Tsien (University of California, San Diego, La Jolla, CA) (Sharner et al., 2004). The cytNmnat1-mCherry fusion gene and Bcl-xl cDNA (kindly provided by S. J. Weintraub, Washington University, St. Louis, MO) were cloned into a lentiviral expression plasmid (Lois et al., 2004).
Trophic factor deprivation.
For NGF withdrawal experiments, SCG neurons were grown in NGF-containing medium (50 ng/ml) cultured for 5 d in vitro (DIV5). The NGF-containing medium was removed and neurons were washed with NGF-depleted medium twice. Neurons were then grown in NGF-depleted neurobasal/B27 medium containing goat anti-NGF antibody (1:1000). For Neurturin deprivation experiments, SCG neurons were established and grown for 2 d in NGF-containing medium. The neurons were washed with Neurobasal medium containing 0.2% B27 to remove NGF and then cultured in Neurobasal medium containing 0.2% B27 and Neurturin (40 ng/ml). After 3 d, the neurons were washed twice with neurobasal containing 0.2% B27 without Neurturin and then cultured in Neurturin-free medium.
Inhibitor and APP-blocking antibody treatment.
In all the experiments, inhibitors (caspase 6 inhibitor; 25 μm, BACE inhibitor; 10 μm) and APP-blocking antibody (anti-APP; 20 μg/ml, Fisher Scientific) were added to the medium at the time of NGF withdrawal and axotomy or vincristine treatment.
Axotomy and vincristine treatment.
Axons were severed by a micro-scalpel (Fine Science Tools) after DIV5. Vincristine was added after DIV5 at a concentration of 0.04 μm in NGF-supplemented medium.
Lentiviral infection of DRG neurons.
For infection of DRG neurons, lentivirus (105–106 infectious units) was added at DIV1 and 4 d later (to allow for adequate transgene expression) experiments were performed. Viral infection and transgene expression were examined via fluorescent microscopy of the EGFP or mCherry reporter.
Dctn1 siRNA studies.
DRG neurons (DIV1) were infected with lentivirus-expressing Dctn1 or Luciferase siRNAs. To test the effects of Bcl-xl and cytNmnat1 overexpression, cells were simultaneously infected with lentiviruses expressing Dctn1 siRNA, along with viruses expressing Bcl-xl or cytNmnat1. After infection, cultures were maintained for an additional 7 d in the presence of 50 ng/ml NGF. To monitor neuronal cell death, ethidium homodimer (100 nm final) was added, cells were fixed after 30 min, and fluorescent (dead) DRG neurons were counted. Axonal degeneration was assessed as described below.
NGF internalization assay.
Lyophilized NGF was fluorescently labeled using a Cy3 labeling kit as per manufacturer's instructions (GE Healthcare Biosciences). DRG neurons were plated in the inner chamber of compartmentalized chambers, maintained for 14 d, and then infected with lentiviruses expressing either Dctn1 or Luciferase siRNA. Seven days postinfection NGF was removed from both the inner (cell body) and the outer (axonal) chambers and the neurons were incubated overnight in medium lacking NGF. Cy3-NGF (100 ng/ml final) was added to the axonal compartment and incubated at 37°C for 4 h. The percentage of Cy3-positive neurons (indicating internalized NGF) was determined using fluorescence microscopy.
Immunohistochemistry.
After fixation, cells were washed with PBS and blocked with 4% normal donkey serum in TBST (Tris-buffered saline plus 0.1% Triton X-100) (1 h, 37°C). Cells were incubated in primary antibody (4°C overnight) and antibody binding was visualized with Alexa Fluor 488- or 594-conjugated anti-mouse and anti-rabbit secondary antibodies (25°C, 1 h). To visualize APP on the axonal surface, cells were incubated with MAB348 (1:100) without permeabilization. Cells were incubated in primary antibody (4°C overnight) and antibody binding was visualized with Alexa Fluor 594-conjugated anti-mouse secondary antibody (25°C, 1 h). For quantification of sAPP loss, axons showing at least four patches of decreased staining (i.e., areas of sAPP loss) were counted from 4 random fields for each condition. Each experiment was repeated three times and the percentage of axons bearing >4 patches of APP loss was determined.
Quantification of axonal degeneration.
Axonal degeneration was quantified as described previously (Sasaki et al., 2009a). Briefly, after axonal severing, phase contrast images were taken using an inverted microscope with a 20× objective (Eclipse TE 300; Nikon). MetaMorph software (Molecular Devices) was used to image 3–4 random fields of distal axons from each well with 40 ms exposure time by using CCD camera (Cool SNAP ES; Nikon). After brightness and background intensity adjustment by the auto-level function in Adobe Photoshop, images were analyzed by NIH ImageJ. The total axonal area was determined by the total number of detected pixels after binarization of the image. Nondegenerated axons showed continuous tracts, while degenerating axons were fragmented, beaded and showed up as aggregates. Degenerated axons were detected using the particle analyzer of ImageJ as small particles, while healthy axonal areas were represented as large continuous areas. The degeneration index is the ratio of fragmented axon area to total axon area. Sixteen fields were evaluated for each condition and each experiment was repeated three times.
Results
N-terminal APP fragmentation is not a universal initiator of axon degeneration
To test the generality of the APP/caspase 6 pathway for axon degeneration, we used three distinct triggers of axonal degeneration in dorsal root ganglion (DRG) and superior cervical ganglion (SCG) neurons: NGF withdrawal, axotomy, and treatment with the chemotherapeutic agent vincristine. If cleavage of APP is the initial trigger for axonal degeneration, then blocking APP cleavage with a BACE inhibitor or using anti-APP antibodies to block its interaction with DR6 should block axonal degeneration initiated by each of these insults. We treated DIV5 neurons with the BACE inhibitor OM99-2 (10 μm) or anti-N-terminal amyloid precursor protein (N-APP) antibody (20 μg/ml) at the time of axonal injury. Axonal degeneration was monitored over 24 h using phase-contrast microscopy and a degeneration index (D. I.) was calculated (see Materials and Methods). Axonal degeneration increases dramatically after NGF deprivation (Fig. 1B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Consistent with previous findings, inhibiting APP cleavage with a BACE inhibitor or a function-blocking antibody against the shed N-APP fragment blocked axonal degeneration triggered by NGF withdrawal (Fig. 1A,B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (Nikolaev et al., 2009). In contrast, neither the BACE inhibitor nor the N-APP function-blocking antibody protected axons following axotomy or vincristine treatment (Fig. 1A,C,D; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Hence, APP shedding is necessary for axonal degeneration following trophic factor withdrawal but not for degeneration induced by mechanical or chemical insults.
Figure 1.
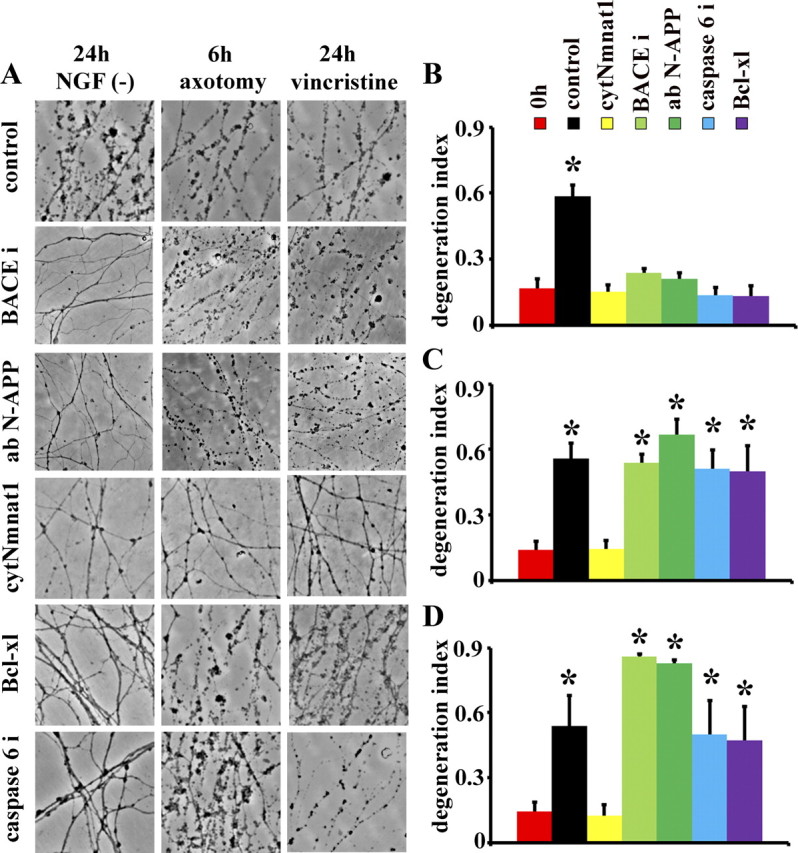
Distinct pathways of axonal degeneration activated by trophic factor deprivation, axotomy and vincristine intoxication. A, Dorsal root ganglia (DRG) were cultured for 5 d and axonal degeneration was induced by NGF withdrawal [NGF (-)], axotomy or vincristine. Treatment with the BACE inhibitor (BACEi; OM99-2), which inhibits APP cleavage, or anti-N-terminal APP-blocking antibody (ab N-APP) blocked axonal degeneration caused by NGF withdrawal, but not degeneration caused by axotomy or vincristine. Similarly, caspase 6 inhibitor (Caspase 6i; zVEID-FMK) and overexpression of Bcl-xl delayed axonal degeneration caused by NGF deprivation only. In contrast, expression of cytNmnat1 prevented axonal degeneration in all paradigms tested. B–D, Image analysis software was used to provide a quantitative analysis of axonal degeneration (i.e., degeneration index) after NGF deprivation (B), axotomy (C), and vincristine administration (D). These analyses highlight the fact that BACE inhibitor, N-APP-blocking antibody, caspase 6 inhibitor and Bcl-xl only block axonal degeneration stimulated by NGF withdrawal, whereas cytNmnat1 protected axons after all insults tested. Degeneration indices before treatment insults are indicated as 0 h. Significantly different (*p < 0.001, Student's t test) from neurons at time 0 (0 h NGF withdrawal).
Apoptotic pathways are not involved in axonal degeneration caused by traumatic or toxin damage
While APP may be unnecessary for axonal degeneration following mechanical and neurotoxic insults, apoptotic pathways involving activation of caspase 6 may still be a common downstream effector of diverse stimulus-dependent degeneration pathways. We used two methods to test the role of caspase 6 in axonal degeneration. First, we used Bcl-xl overexpression to block activation of the mitochondrial apoptotic signaling pathway because it is downstream of N-APP-stimulated activation of the DR6 receptor. Second, we evaluated the pharmacological inhibition of caspase 6. We also tested the effects of overexpression of cytNmnat1, a cytosolic mutant of Nmnat1 (nicotinamide mononucleotide adenylyltransferase 1) that provides robust axonal protection after axotomy (Sasaki et al., 2006). We found that axonal degeneration following NGF withdrawal is blocked by Bcl-xl, cytNmnat1 or caspase 6 inhibitors (Fig. 1A,B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material), although the protection afforded by caspase 6 inhibition was for a shorter time even with repeated daily additions of the inhibitor (supplemental Fig. S2, available at www.jneurosci.org as supplemental material, data not shown). In contrast to the protection by cytNmnat1, axonal destruction initiated by axotomy or vincristine intoxication was not affected by caspase 6 inhibition or Bcl-xl overexpression (Fig. 1A,C,D). These experiments demonstrate that loss of axonal sAPP, stimulation of apoptotic signaling, and caspase 6 activation are not involved in axonal degeneration caused by mechanical or toxic insults and imply that multiple pathways are involved in triggering axonal degeneration.
While the APP/caspase pathway is selectively involved following trophic factor withdrawal, there are also likely to be shared components in the pathways leading to axonal degeneration. For example, Wlds and its functional component Nmnat effectively block axonal degeneration caused by both mechanical and chemical injuries and Wlds preserves NGF-deprived axons (Deckwerth and Johnson, 1994; Mack et al., 2001; Araki et al., 2004; Sasaki et al., 2006, 2009a,b), suggesting these injuries activate a common downstream axonal execution pathway. To test this hypothesis we overexpressed a cytoplasmically targeted variant of Nmnat, cytNmnat1, that provides robust axonal protection (Sasaki et al., 2009b), in SCG neurons. In the presence of cytNmnat1, axons are robustly protected from degeneration after NGF deprivation, axotomy and vincristine treatment (Fig. 1A–C). As previously reported for wlds SCG neurons (Deckwerth and Johnson, 1994), cytNmnat1 preserved both axonal integrity and metabolic activity after NGF deprivation (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These findings suggest that Nmnat-mediated axonal protection occurs via the inhibition of a common axon degeneration pathway.
cytNmnat1 does not block loss of axonal sAPP or caspase 6 activation after NGF withdrawal
Axonal degeneration after NGF deprivation requires APP cleavage, N-APP fragment induced downstream signaling through the DR6 receptor, and subsequent caspase 6 activation (Nikolaev et al., 2009). We therefore investigated whether N-APP shedding occurs in response to other axonal degeneration insults by examining the surface loss of APP on axons after axotomy and vincristine treatment. As previously reported (Nikolaev et al., 2009), NGF depletion induces the loss of axonal surface immunoreactivity for N-APP in patches (Fig. 2A); however, surface APP was not lost when axonal degeneration was induced by axotomy or vincristine (Fig. 2B,C). These results are consistent with the lack of axonal protection by N-APP-blocking antibodies or BACE inhibitors after mechanical or chemical injury (Fig. 1).
Figure 2.

Loss of axonal sAPP is not involved in axonal degeneration induced by mechanical or toxin damage. A–C, Immunofluorescent microscopic analysis using an antibody to the APP N terminus to examine the “patchy” loss of surface APP (sAPP) on axons of neurons deprived of NGF (A), following axotomy (B), or treated with vincristine (C). Note the loss of sAPP following NGF withdrawal but not after mechanical or toxic insults, and that the loss of sAPP after NGF withdrawal persists despite the expression of cytNmnat1 or Bcl-xl. Quantification of the percentage of axons with decreased sAPP is displayed for each condition (see Materials and Methods). D, Conditioned medium was subjected to Western blot analysis for cleaved N-terminal APP fragment using anti-N-APP antibody. The presence of shed N-terminal APP fragments was detected after NGF deprivation even under conditions where the axons were protected due to overexpression of Bcl-xl or cytNmnat1. Significantly different (*p < 0.001, Student's t test) from neurons grown in the presence of NGF.
Caspase 6 activation is downstream of N-APP generation (Nikolaev et al., 2009) and caspase 6 inhibition effectively blocks axonal degeneration after NGF withdrawal. While caspase 6 inhibitors are ineffective in preserving axons after mechanical or chemical injury, caspase 6 activation could be one of several independent mechanisms that drive axonal degeneration induced by these insults. We stained for activated caspase 6 after injury, and while we observed a punctate staining pattern of activated caspase 6 in NGF-deprived axons, we did not detect caspase 6 activation after axotomy or vincristine treatment (Fig. 3). Together, these results lead us to conclude that loss of surface APP and caspase 6 activation play essential roles in axonal demise induced by NGF withdrawal but that it is not involved in mechanical or toxin-mediated axonal damage.
Figure 3.

Nmnat1 protects axons in the presence of activated caspase 6 and axonal degeneration induced by axotomy and vincristine does not involve caspase 6 activation. Immunofluorescence detection of cleaved caspase 6 in SCG axons (top, microtubule staining; bottom, cleaved caspase 6 staining) reveals that caspase 6 is activated within 20 h after NGF deprivation. The activation of caspase 6 is blocked by expression of Bcl-xl but not by the expression of cytNmnat1, despite the fact that cytNmnat1 effectively prevents axonal fragmentation after NGF withdrawal. Immunofluorescence analysis of cleaved caspase 6 in SCG neurons revealed that caspase 6 is not activated after axotomy or vincristine treatment.
The preservation of NGF-deprived axons by cytNmnat1 provided an opportunity to investigate where Nmnat functions to protect axons doomed by activation of the APP/caspase 6 pathway. We first determined whether surface APP loss in SCG neurons expressing either cytNmnat1 or Bcl-xl occurred after NGF withdrawal. Even though both molecules provided robust axonal protection, neither protein blocked axonal sAPP loss as determined by immunostaining (Fig. 2A) or Western blot analysis of the conditioned media (Fig. 2D), indicating that they both function downstream of N-APP generation. Next, we examined the effects of cytNmnat1 and Bcl-xl overexpression on caspase 6 activation in NGF-deprived axons. We observed robust axonal caspase 6 activation in control neurons, but not in those expressing Bcl-xl, indicating that the apoptotic cascade was blocked as expected (Fig. 3). Surprisingly, cytNmnat1 did not block activation of caspase 6 despite the robust protection of these axons (Fig. 3). These results demonstrate that Nmnat exerts its axonal protective effects at a step that lies downstream (or parallel) to the loss of axonal sAPP and caspase 6 activation invoked by NGF-deprivation.
Nmnat-mediated protection is restricted to axons
Along with axonal degeneration, NGF withdrawal causes extensive cell death. Interestingly, neurons from transgenic mice overexpressing Bcl-2 are protected from both axon and cell soma loss after NGF deprivation (Greenlund et al., 1995). Since both Nmnat1 and Bcl-xl protect axons from degeneration after trophic factor withdrawal, we investigated whether cytNmnat1 and Bcl-xl can also protect against NGF-deprived neuronal death. Cell death was monitored by phase-contrast microscopy (Fig. 4) and ethidium homodimer uptake (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The percentage of dead neurons increased until essentially all soma were degenerated (neuronal death) by 72 h after NGF-deprivation. Neurons expressing Bcl-xl showed minimal somal or axonal degeneration; however, cell size was remarkably smaller than those maintained with NGF (Fig. 4A; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). In contrast, neurons expressing cytNmnat1 had somal degeneration similar to control neurons despite robust axonal protection. Further investigation revealed that soma degeneration followed a similar time course in control and cytNmnat1-expressing neurons with 47 ± 8% of control and 54 ± 13% of Nmnat-expressing neuronal cell bodies remaining refractile and excluding ethidium homodimers (intact) after 24 h and only 3% at 72 h after NGF withdrawal (Fig. 4B; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In contrast, the cell bodies of neurons expressing Bcl-xl were protected from degeneration with 68.7 ± 6.8% of cell bodies excluding ethidium homodimer after 72 h of NGF withdrawal (Fig. 4B; supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 4.
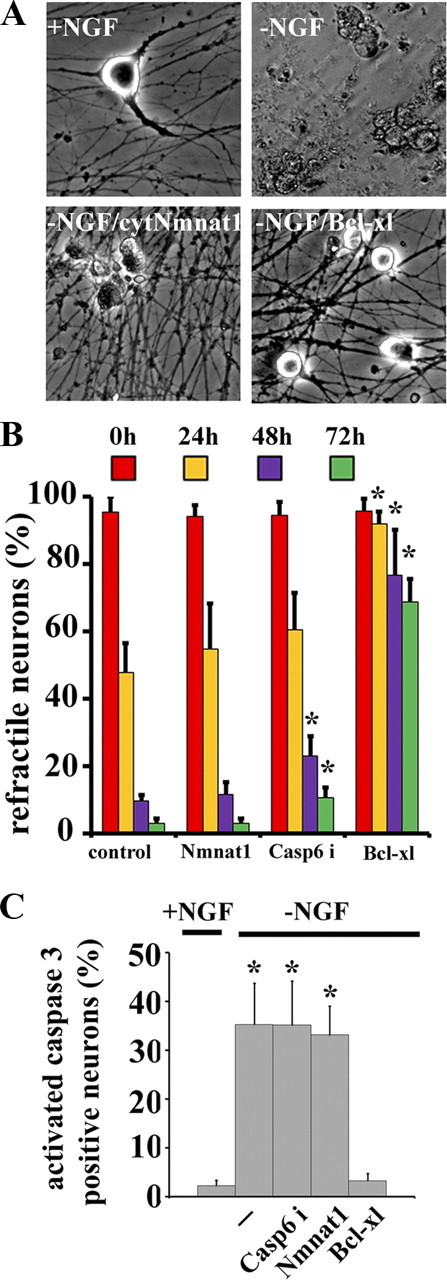
Bcl-xl inhibits both axonal and somal degeneration after NGF deprivation whereas cytNmnat1 specifically inhibits axon degeneration. A, Phase contrast microscopy of DRG soma cultured in the presence of NGF or 72 h after NGF deprivation. In the absence of NGF, extensive cell soma degeneration was observed in control (−NGF) and cytNmnat1-expressing neurons (−NGF/cytNmnat1). In contrast, Bcl-xl-expressing neuronal soma (−NGF/Bcl-xl) remained refractile (alive) but were smaller than neurons grown in NGF. B, Quantitative analysis of refractile soma after NGF deprivation showed that most soma (>90%) degenerated 72 h after NGF withdrawal in control, cytNmnat1-expressing, and caspase 6 inhibitor (caspase 6i)-treated DRG neurons. The majority (68%) of Bcl-xl-expressing neuronal soma remained intact at 72 h after NGF deprivation. Significantly different (*p < 0.001, Student's t test) from control neurons after NGF withdrawal. C, Quantification of activated caspase 3-positive neurons demonstrated that neither caspase 6 inhibitors nor cytNmnat1 expression blocked caspase 3 activation in the cell soma; however Bcl-xl expression completely blocked caspase 3 activation after NGF deprivation. Significantly different (*p < 0.001, Student's t test) from neurons supplemented with NGF. A total of 1100 neurons from each condition were counted and experiments were repeated 3 times.
We further investigated the mechanism of cell soma protection by examining caspase 3 activation, a marker of apoptotic cell death. Within 24 h after NGF withdrawal, activated caspase 3 was detected using cleaved caspase 3-specific antibody in control neurons, those treated with a caspase 6 inhibitor, and neurons expressing cytNmnat1. However, no activated caspase 3 was detected in neurons expressing Bcl-xl (Fig. 4C). Bcl-xl protects both the axon and cell body by inhibiting activation of caspases, whereas Nmnat mediates only axonal protection via a mechanism that is independent of caspase activation.
To investigate whether the APP/DR6/caspase 6 pathway is specific for neurotrophins that signal through Trk receptors or is a more general phenomenon linked to loss of trophic support, we examined the responses of SCG neurons to Neurturin deprivation. We chose to examine Neurturin because it also supports SCG neuron growth and survival (Kotzbauer et al., 1996) as it signals through a different receptor, namely a receptor complex containing the Ret tyrosine kinase receptor (Baloh et al., 2000). SCG neuronal cultures were established in NGF and then switched to Neurturin. After growing for 3 d in Neurturin, Neurturin was withdrawn and neuronal death and axonal degeneration were examined. We found that Neurturin withdrawal caused loss of surface APP (Fig. 5A) (patchy APP staining was observed in only 6 ± 2% axons in control vs 87 ± 6% after Neurturin deprivation). Similar to the effects of NGF withdrawal, Neurturin deprivation caused caspase 6 activation in axons (Fig. 5B). In addition, inhibition of caspase 6 as well as overexpression of cytNmnat1 or Bcl-xl blocked axon degeneration induced by Neurturin deprivation (Fig. 5C). These results indicate that caspase 6 is an important driver of axon degeneration resulting from withdrawal of different classes of neurotrophic factors acting through unique receptors.
Figure 5.
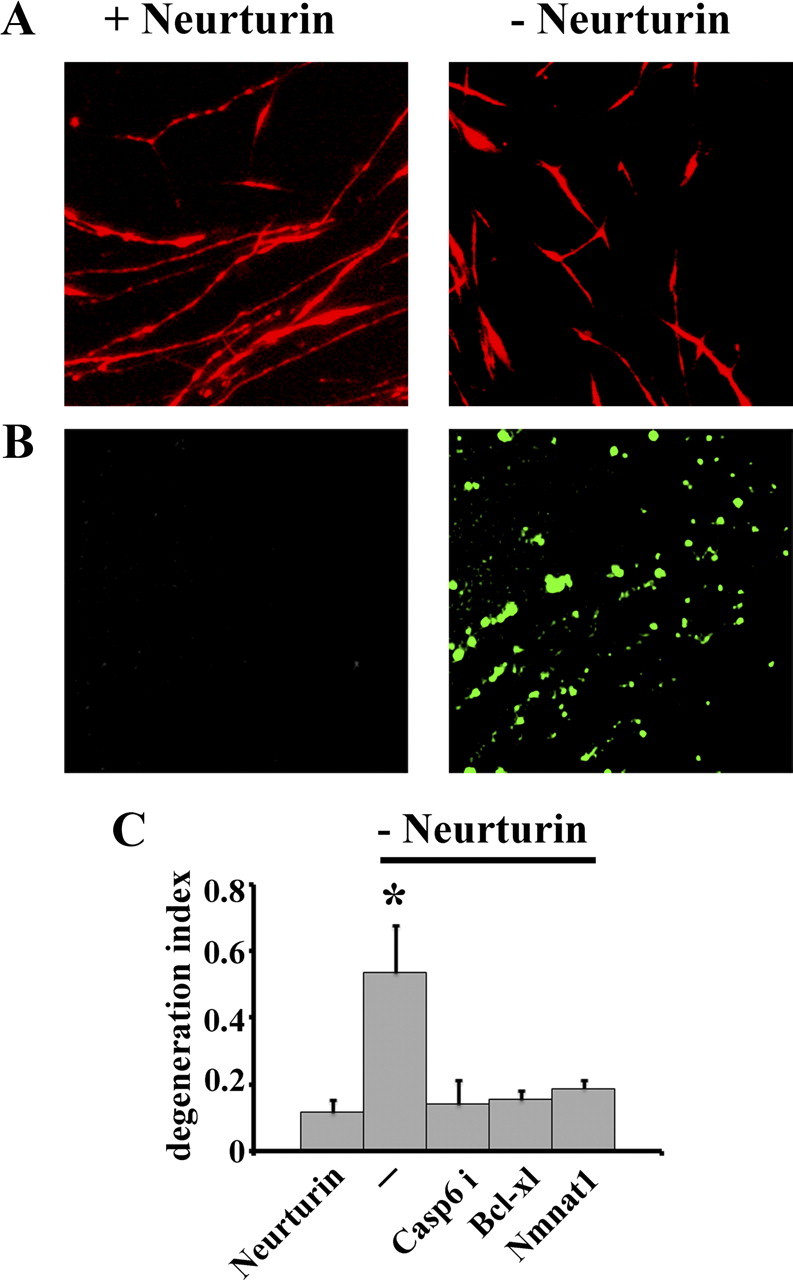
Neurturin deprivation results in loss of surface N-terminal APP and caspase 6 activation. A, Immunofluorescent microscopic analysis using an antibody to the APP N terminus demonstrated the patchy loss of surface APP (sAPP) on axons of SCG neurons deprived of Neurturin. B, Active caspase 6 was detected in SCG axons via visualization with carboxyfluorescein within 20 h after Neurturin withdrawal. C, Quantitative analysis of axonal degeneration in SCG neurons deprived of Neurturin. Note that treatment with caspase 6 inhibitor (VEID-CHO) or expression of Bcl-xl or cytNmnat1 protects against axonal degeneration induced by Neurturin withdrawal. *Significantly different (p<0.001, Student's t test) from neurons grown in the presence of Neurturin.
Axonal degeneration caused by Dctn1 dysfunction can be inhibited by Nmnat or caspase 6 inhibition
The APP/DR6/caspase 6 signaling pathway plays an important role in NGF deprivation-mediated axonal degeneration. Mutations that disrupt retrograde axonal transport are often associated with neurological disorders in which axonal degeneration is an important component. Dctn1 is a component of the dynactin complex that associates with dynein to drive retrograde axonal transport of multiple cargoes along the microtubule lattice (LaMonte et al., 2002; Schroer, 2004). Mutations in DCTN1 are associated with motor neuron degenerative disorders, ALS associated with frontotemporal dementia (FTD), and Perry syndrome, which has symptoms of Parkinsonism (Puls et al., 2003, 2005; Münch et al., 2004, 2005; Wider and Wszolek, 2008; Farrer et al., 2009; Vilariño-Güell et al., 2009). These mutations impair the transport of trophic factor back to the neuronal cell body (Laird et al., 2008), leading to speculation that neurodegeneration in these patients is due to lack of trophic factors in the soma (Puls et al., 2005).
Disruption of dynein function leads to decreased NGF transport and increased death in cultured DRG neurons (Wu et al., 2007). We therefore tested whether depletion of Dctn1 induces axonal degeneration via activation of the APP/DR6/caspase 6 pathway. DRG neurons were infected with lentiviruses expressing Dctn1 siRNA constructs. We constructed many and extensively used three different Dctn1 siRNAs in these studies. The efficiency of knockdown for these three siRNAs was quantified using real-time RT-PCR analysis. We found that Dctn1–1 and Dctn1-2 siRNAs reduced Dctn1 mRNA levels by 96% and 90%, respectively; whereas Dctn-3 siRNA resulted in only a 15% reduction in Dctn1 mRNA (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). DRG neurons were infected with lentiviruses expressing each of these siRNAs constructs a time when they are critically dependent on NGF (DIV1). DRG neurons infected with Dctn1-1 or Dctn-2 siRNA showed extensive axon and soma degeneration, whereas those infected with Dctn1-3 siRNA were indistinguishable from neurons expressing a Luciferase siRNA (Fig. 6A–D). Bcl-xl overexpression blocked both axonal and somal degeneration (Fig. 6A–D), whereas cytNmnat1 blocked only axonal degeneration in Dctn1-deficient neurons. These results were similar to those obtained with neurons after NGF withdrawal, suggesting that the mechanism of neuronal damage caused by Dctn1 knockdown and trophic factor deprivation is similar.
Figure 6.

Apoptotic pathways play important roles in axonal degeneration and cell death caused by Dctn1 knock down. A, Phase contrast microscopy showing axonal degeneration 7 d after DRG neurons were infected with lentiviruses expressing Luciferase (Luc) or Dctn1 siRNA. Extensive axonal degeneration occurred after Dctn1 knockdown that was blocked by Bcl-xl expression, caspase 6 inhibitor (caspase 6i) treatment, or cytNmnat1 expression. Axons of Luciferase siRNA-expressing neurons remained intact. B, Ethidium homodimer staining overlying phase contrast images of DRG neurons show extensive cell death in Dctn1 knockdown neurons compared with those expressing Luciferase siRNA. Bcl-xl expression blocked cell death after Dctn1 knockdown; however cytNmnat1 expression or caspase 6 inhibition failed to block neuronal death. C, Quantitative analysis of axonal degeneration (i.e., degeneration index) displayed in A demonstrated the protection afforded by Bcl-xl, caspase 6 inhibitor and cytNmnat1. D, Quantification of ethidium homodimer-positive neurons showed that only Bcl-xl expression blocks neuronal cell death caused by Dctn1 knockdown. The number of dead DRG neurons in each condition was scored and the percentage of dead neurons is displayed. Significantly different (*p < 0.001, Student's t test) from neurons expressing Luciferase siRNA.
To assess whether degeneration induced by Dctn1 knockdown is due to NGF transport deficits, we measured NGF internalization (see Materials and Methods) and found that depletion of Dctn1 decreases the amount of NGF that is internalized (Fig. 7A,B). Indeed, the magnitude of the NGF transport defect with each Dctn siRNA was directly correlated with the amount of Dctn1 knockdown and the extent of axon and soma damage. The axonal degeneration, along with the NGF trafficking defects, observed in neurons deficient in Dctn1 prompted us to examine the APP/DR6/caspase 6 pathway in these neurons. We found that, like NGF withdrawal, Dctn1 knockdown initiated loss of surface APP that could be blocked by BACE inhibition (Fig. 8A,B). However, in contrast to axon degeneration after NGF withdrawal, BACE inhibition or APP-blocking antibody did not block Dctn1-induced axonal degeneration (Fig. 8C). This is particularly intriguing as caspase 6 inhibition was effective in blocking axonal degeneration in Dctn1-deficient neurons. Moreover, Bcl-xl overexpression blocked both axon degeneration and cell death in this paradigm (Fig. 6). The surprising dichotomy between blockade of APP surface loss and caspase 6 inhibition prompted us to examine caspase 6 activation in neurons expressing Dctn1 siRNA. We found that caspase 6 was activated in neurons lacking Dctn1 even in the presence of BACE inhibitors (Fig. 8D), indicating that caspase 6 can be activated independently of APP cleavage. Together, these results suggest that deficits in axonal transport cause axonal degeneration via mechanisms that are similar to those invoked by trophic factor deprivation as they both involve caspase 6 activation. However, since caspase 6 is activated independent of loss of surface APP in Dctn-deficient neurons, it appears that multiple pathways are capable of activating this enzyme and inducing axonal degeneration.
Figure 7.
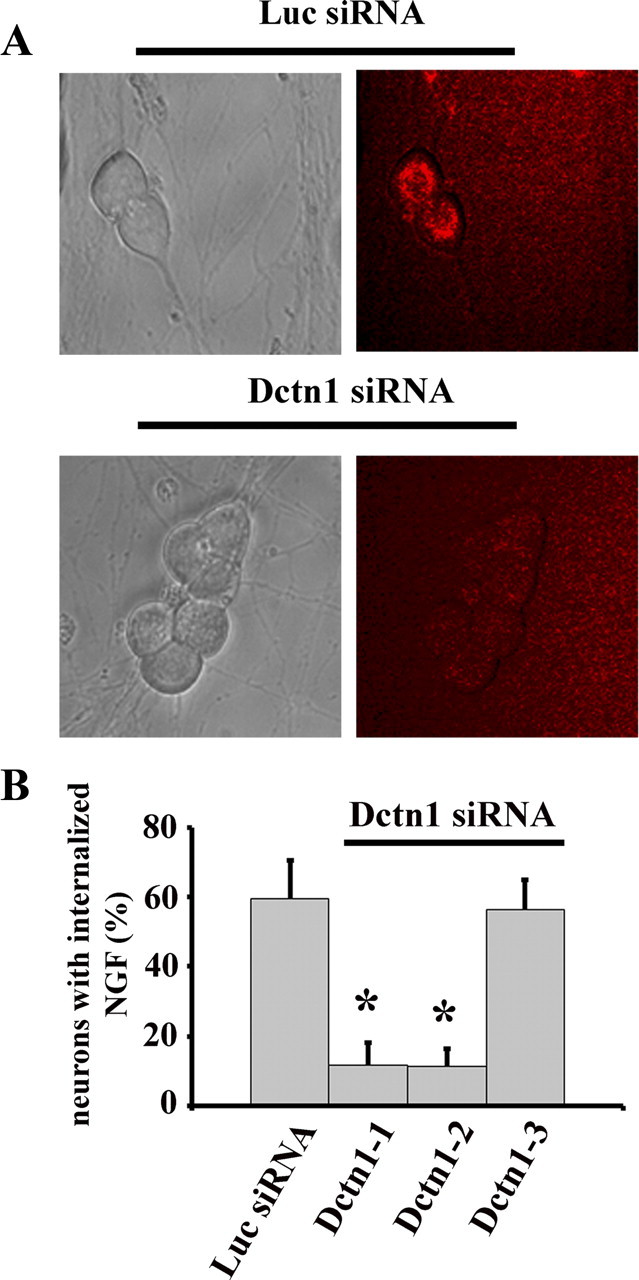
NGF internalization is blocked by Dctn 1 knockdown. A, Fluorescent microscopy of NGF trafficking using Cy3 labeled NGF. DRG neurons (DIV14) were infected with lentiviruses expressing either Luciferase (Luc) or Dctn1 siRNA for 7 d. Neurons were deprived of NGF for 16 h then Cy3 labeled NGF was added to the medium. After 4 h, Cy3 labeled NGF appeared in the cell bodies of control neurons (Luc siRNA); however, no Cy3 NGF was observed in the neuronal soma after Dctn1 knockdown (Dctn1 siRNA). B, The percentage of neurons containing Cy3-labeled NGF in the cell body was determined. Note the normal level of internalized NGF in neurons expressing Dctn1-3 siRNA that produces poor knockdown and no axonal degeneration. A total of 400 neurons were analyzed and the experiments were repeated 3 times. Significantly different (*p < 0.001, Student's t test) from Luciferase siRNA infected neurons.
Figure 8.
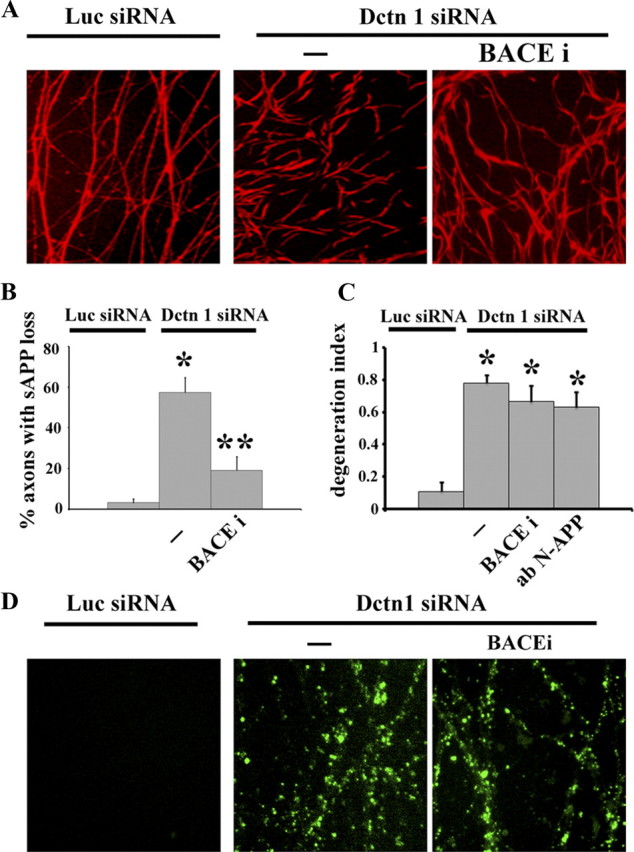
APP-regulated pathways are not involved in axonal degeneration or caspase 6 activation in Dctn1 deficient neurons. A, Immunofluorescent microscopic analysis using an antibody to the APP N terminus demonstrated the characteristic patchy loss of sAPP on axons from neurons expressing Dctn1 siRNA. Compare to the continuous staining observed on axons from neurons expressing Luciferase (Luc) siRNA. The Dctn1 siRNA-induced loss of sAPP was blocked by treatment with BACE inhibitor (BACEi; OM99-2). B, Quantification of the percentage of axons with decreased sAPP is displayed for each condition (see Materials and Methods). Significantly different from Dctn1 siRNA-expressing neurons (**p < 0.001). C, Axonal degeneration after Dctn1 knockdown was not blocked by BACE inhibition or anti-N-terminal APP-blocking antibody (ab N-APP). Significantly different from Luciferase siRNA-expressing neurons (*p < 0.001). D, Fluorescent microscopic analysis of caspase 6 activation in axons was performed using DRG neurons infected with lentivirus expressing either Luciferase (Luc) siRNA or Dctn1 siRNA. Activation of caspase 6 was detected in Dctn1 siRNA-expressing DRG neurons (Dctn 1 siRNA) using carboxyfluorescein visualization. No caspase 6 activity (fluorescence) was observed in Luciferase siRNA-expressing axons. Dctn1 knockdown induced strong fluorescence indicating caspase 6 activation. Inhibition of APP cleavage by BACE inhibitor did not block activation of caspase 6 after Dctn1 knockdown.
Discussion
Axonal degeneration is a hallmark of many debilitating neurological disorders. Identifying the mechanisms that drive such degeneration may lead to treatments that block or delay axon loss. The recent identification of the APP/DR6/caspase 6 pathway (Nikolaev et al., 2009) was a benchmark in the field because it suggested rational targets for novel therapeutic interventions. Because this pathway is well defined, it provided an opportunity to examine whether it represents a common mechanism, activated in response to multiple insults, to promote axonal degeneration. We inhibited this pathway at multiple levels: APP cleavage, APP function, apoptotic pathway activation, or caspase 6 activation; and discovered that while each of these manipulations block axonal degeneration following trophic factor withdrawal (NGF or Neurturin), none of them provide axonal protection following axotomy or vincristine treatment. This implies that multiple stimulus-dependent pathways can invoke the cascade of events required to bring about axon demise.
Apoptotic pathways appear to play important roles in axonal degeneration triggered by trophic factor withdrawal (e.g., NGF or Neurturin). This is supported by our observation that Bcl-xl blocks caspase 6 activation and axonal degeneration in neurons deprived of trophic support. NGF deprivation stimulates parallel pathways to induce the apoptotic process and caspase activation. One is dependent on Bim induction and is responsible for Bax translocation to mitochondria, whereas the other involves activation of Jnk pathways (Putcha et al., 2001). Moreover, NGF deprivation activates glycogen synthase kinase 3 (GSK3) at the axon tip, which initiates an apoptotic signal that travels back to the neuronal cell body to induce Jun-mediated neuronal apoptosis (Mok et al., 2009). Another GSK3-mediated process that promotes apoptosis is the phosphorylation of Bax and its subsequent translocation into mitochondria (Linseman et al., 2004). Axonal retrograde death-promoting signals that lead to apoptotic pathway activation have also been observed in mouse models of ALS (Perlson et al., 2009). While blocking APP cleavage is clearly sufficient to suppress axonal degeneration after trophic factor deprivation, the mechanism of apoptotic pathway activation downstream of APP cleavage remains to be elucidated.
APP cleavage and activation of apoptotic pathways occurs after loss of BDNF, NT3, and NGF-mediated Trk receptor activation (Nikolaev et al., 2009). We found that withdrawal of Neurturin, which signals through the Ret receptor tyrosine kinase, also stimulated APP/caspase6 axon degeneration signaling events. The difference between Trk and Ret mediated signaling is substantial (Klesse and Parada, 1999; Airaksinen and Saarma, 2002; Huang and Reichardt, 2003), yet the loss of signaling after NGF or Neurturin deprivation appears to stimulate common axonal degeneration signals. These results suggest that common adaptor molecules downstream of Trk and Ret receptors (e.g., Shc, GRB2, or PI3 kinase) may play key roles in the regulation of APP processing and axon stability.
The dynein/dynactin1 complex facilitates retrograde axonal transport of signaling molecules, including neurotrophins, from the periphery back to the cell body. Mutations in dynactin 1 (DCTN1) are linked to a number of slowly progressing neurological disorders (Puls et al., 2003, 2005; Münch et al., 2004, 2005; Wider and Wszolek, 2008; Farrer et al., 2009; Vilariño-Güell et al., 2009), and the neuronal damage in these disorders is speculated to involve loss of trophic factor delivery to the neuronal cell body. We found that loss of Dctn1 caused impaired NGF trafficking, surface APP loss, and caspase 6 activation. Although caspase 6 inhibition blocked axonal degeneration caused by Dctn1 depletion, inhibition of APP cleavage by inhibiting BACE did not block the axonal degeneration or caspase 6 activation, suggesting that caspase 6 activation in Dctn1-depleted neurons is independent of APP cleavage. This could occur due to impaired retrograde axonal transport in Dctn1-depleted neurons as this would limit the availability (at the neuronal cell body) of vital factors and signaling molecules associated with signaling endosomes such as P-Trk, Erk1/2 and Erk5 (Delcroix et al., 2003) to induce apoptotic pathway activation. Furthermore, Dctn1 interacts with Kinesin II to regulate transport of organelles including mitochondria (Deacon et al., 2003). Dctn1 deficiency could therefore also disrupt neuronal energy homeostasis, another potential mechanism of caspase 6 activation.
While distinct, stimulus-dependent mechanisms trigger axonal degeneration in response to trophic factor deprivation, mechanical injury, or chemical toxicity; each of these pathways is blocked by expression of Nmnat1, a component of the Wlds protein (Araki et al., 2004; Sasaki et al., 2006, 2009a,b; Yahata et al., 2009). Nmnat is the most potent axonal protection factor known, however, its mechanism of action remains obscure. The requirement for the APP/caspase 6 pathway for axonal degeneration induced by trophic factor deprivation afforded an opportunity to identify where in the cascade Nmnat is exerting its protective effects. In neurons overexpressing cytNmnat1, NGF withdrawal still leads to loss of axonal sAPP and activation of axonal caspase 6, yet axonal degeneration does not occur. These findings are consistent with the prevention of degeneration in caspase-positive dendrites by Wlds protein in Drosophila (Schoenmann et al., 2010). From these observations, three important aspects of axonal degeneration emerge: (1) Nmnat influences a step downstream or parallel to caspase 6 activation; (2) caspase 6 activation does not occur in all axonal injury paradigms; and (3) distinct, injury-specific pathways of axonal degeneration must converge on a common, Nmnat-sensitive axonal degeneration program.
Although cytNmnat1 protects axons from degeneration after loss of trophic support or Dctn1 knockdown, it failed to protect the cell soma. Our results and previous experiments indicate that different signaling cascades are responsible for degeneration of the axon and soma (Nikolaev et al., 2009). For example, after NGF deprivation inhibitors of caspase 6 and caspase 3 selectively inhibit axon or soma degeneration, respectively. Further work to pinpoint the critical substrates of caspase 6 in axons, as well as the steps in axonal degeneration that are modulated by Nmnat will be crucial if we are to exploit its potential for treating the axonopathy accompanying neurological disorders.
Footnotes
This work was supported by National Institutes of Health (NIH) Neuroscience Blueprint Center Core Grant P30 NS057105 to Washington University; the HOPE Center for Neurological Disorders, Washington University ADRC NIA P50 AG05681-25 (A.D.) and NIH Grants NS040745 (J.M.), AG13730 (J.M.), and NS065053 (A.D. and J.M.); Muscular Dystrophy Association Grants 10040 (J.M.); and Craig H. Neilsen Foundation Grant 124030 (J.M.). We thank Kelli Simburger, Amy Strickland, Amber Neilson, and Tim Fahrner for experimental assistance, and members of the Milbrandt laboratory for their comments on this manuscript and helpful discussions.
The authors and Washington University may derive benefit from a licensing agreement with Sirtris Pharmaceuticals, which did not provide any support for this work.
References
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors—implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Chen B, Nelson DM, Sadovsky Y. N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J Biol Chem. 2006;281:2764–2772. doi: 10.1074/jbc.M507330200. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Deacon SW, Serpinskaya AS, Vaughan PS, Lopez Fanarraga M, Vernos I, Vaughan KT, Gelfand VI. Dynactin is required for bidirectional organelle transport. J Cell Biol. 2003;160:297–301. doi: 10.1083/jcb.200210066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilariño-Güell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund LJ, Korsmeyer SJ, Johnson EM., Jr Role of BCL-2 in the survival and function of developing and nature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Kim D, Tsai LH. Bridging physiology and pathology in AD. Cell. 2009;137:997–1000. doi: 10.1016/j.cell.2009.05.042. [DOI] [PubMed] [Google Scholar]
- Klesse LJ, Parada LF. Trks: signal transduction and intracellular pathways. Microsc Res Tech. 1999;45:210–216. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<210::AID-JEMT4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Jr, Milbrandt J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, Griffin J, Price DL, Martin LJ, Wong PC. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med. 2009;4:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok SA, Lund K, Campenot RB. A retrograde apoptotic signal originating in NGF-deprived distal axons of rat sympathetic neurons in compartmented cultures. Cell Res. 2009;19:546–560. doi: 10.1038/cr.2009.11. [DOI] [PubMed] [Google Scholar]
- Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Münch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58:777–780. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Perlson E, Jeong GB, Ross JL, Dixit R, Wallace KE, Kalb RG, Holzbaur EL. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009;29:9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009a;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009b;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, Yaron A. Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci. 2010;30:6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Vilariño-Güell C, Wider C, Soto-Ortolaza AI, Cobb SA, Kachergus JM, Keeling BH, Dachsel JC, Hulihan MM, Dickson DW, Wszolek ZK, Uitti RJ, Graff-Radford NR, Boeve BF, Josephs KA, Miller B, Boylan KB, Gwinn K, Adler CH, Aasly JO, Hentati F, et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology. 2009;72:2024–2028. doi: 10.1212/WNL.0b013e3181a92c4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mu FY. A Web-based design center for vector-based siRNA and siRNA cassette. Bioinformatics. 2004;20:1818–1820. doi: 10.1093/bioinformatics/bth164. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- Wider C, Wszolek ZK. Rapidly progressive familial parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome): a literature review. Parkinsonism Relat Disord. 2008;14:1–7. doi: 10.1016/j.parkreldis.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Wiedemann C. Development and disease: Alzheimer's protein in embryonic pruning. Nat Rev Neurosci. 2009;10:244. [Google Scholar]
- Wu C, Ramirez A, Cui B, Ding J, Delcroix JD, Valletta JS, Liu JJ, Yang Y, Chu S, Mobley WC. A functional dynein–microtubule network is required for NGF signaling through the Rap1/MAPK pathway. Traffic. 2007;8:1503–1520. doi: 10.1111/j.1600-0854.2007.00636.x. [DOI] [PubMed] [Google Scholar]
- Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]