

Abstract
Network-based drug design holds great promise in clinical research as a way to overcome the limitations of traditional approaches in the development of drugs with high efficacy and low toxicity. This novel strategy aims to study how a biochemical network as a whole, rather than its individual components, responds to specific perturbations in different physiological conditions. Proteins exerting little control over normal cells and larger control over altered cells may be considered as good candidates for drug targets. The application of network-based drug design would greatly benefit from using an explicit computational model describing the dynamics of the system under investigation. However, creating a fully characterized kinetic model is not an easy task, even for relatively small networks, as it is still significantly hampered by the lack of data about kinetic mechanisms and parameters values. Here, we propose a Monte Carlo approach to identify the differences between flux control profiles of a metabolic network in different physiological states, when information about the kinetics of the system is partially or totally missing. Based on experimentally accessible information on metabolic phenotypes, we develop a novel method to determine probabilistic differences in the flux control coefficients between the two observable phenotypes. Knowledge of how differences in flux control are distributed among the different enzymatic steps is exploited to identify points of fragility in one of the phenotypes. Using a prototypical cancerous phenotype as an example, we demonstrate how our approach can assist researchers in developing compounds with high efficacy and low toxicity.
Keywords: metabolic control analysis, network-based drug design, Monte Carlo sampling
1. Introduction
The main challenge in drug discovery consists of developing drugs which are both effective and selective, hence non-toxic. In this respect, drug development approaches have often assumed that the enzyme chosen as target plays the role of ‘rate limiting step’ for the biological function of interest [1]. However, single and completely rate-limiting steps barely exist [2] and it is not easy to identify them if they do. Potent inhibitors of essential enzymes often do not show the expected effect at the cellular level [3–5]. The network of biochemical interactions in living cells buffers changes introduced in a single enzymatic step. Likewise, local changes may induce unforeseen adverse side-effects on the whole system [4]. This interconnectedness of biological function usually results in poor predictive power with respect to the requirement of high efficiency and low toxicity for a drug.
With the advent of systems biology, drug discovery has been shifting its focus from a single-molecule to a system-level perspective, and the concept of network-based drug design has been introduced [3–6]. Within this new paradigm, the aim is to study a biochemical network as a whole and identify points of fragility specifically characterizing an altered phenotype (as in a cancer cell) [7,8]. By targeting these points of fragility, a significant response in altered cells is induced. Differential network-based drug design then maximizes the difference in response of target cells versus normal cells [9]. A possible implementation of network-based drug design is based on metabolic control analysis (MCA), where the concept of fragility is expressed in terms of control coefficients [10]. Differential MCA, in particular, has been proposed as a tool to understand how the system responds to specific perturbations under different physiological conditions or in different host cells, hence providing a way to assess both the efficiency and the specificity of a compound designed to target specific enzymes [3,9,11].
However, one fundamental requirement for applying MCA is the availability of a fully characterized kinetic model of the system under study. Creating such a model is not an easy task, even for relatively small metabolic networks [12,13]. In most cases, the detailed enzymatic mechanisms governing the dynamics of the different metabolic steps are unknown and precise knowledge of kinetic parameters under the relevant in vivo conditions is usually not available. The resulting uncertainty in predicting the dynamic behaviour is significant and increases drastically with the size of the system [12]. Randomized sampling of the parameter space represents a way in which such uncertainty can be quantified and probabilistic insights about the dynamical behaviour of the system can be obtained. Such probabilistic approaches have been recently used in a number of different studies, spanning from applications on MCA [14,15], on metabolic engineering [16], to a description of the dynamics in metabolic networks [17–19]. Here we build upon these ideas and present a Monte Carlo approach to identify differences between the control profiles of a metabolic network in two different settings, one representative of a tumour cell and the other of a corresponding normal cell. Our aim is to show how putative targets for drugs operating at the metabolic level may be identified in a probabilistic manner, when only partial knowledge is available with respect to the kinetic properties of the system. In particular, our analysis is based on experimentally accessible information on metabolic phenotypes, such as observable concentrations and fluxes, rather than on detailed knowledge of kinetic parameters. It is demonstrated that a combination of such phenotypic data, together with heuristic assumptions about the properties of typical enzyme-catalysed reactions, already allows for a fast and efficient way to explore the effectiveness of putative drug targets. Our method makes use of biophysical constraints on the metabolic network, as provided by mass-conservation and thermodynamics, and implements an efficient numerical scheme to allow scanning of a large parameter-space, making it applicable to networks of large size. As a proof-of-concept, we apply our methodology to identify the points of fragility characterizing a paradigmatic cancer metabolic phenotype. We demonstrate that our method allows us to identify those enzymes that exert a high differential control upon a given relevant system property, and thus represent suitable sites to specifically target the cancerous phenotype.
2. Methods
2.1. Metabolic control analysis
The dynamic behaviour of a metabolic system, consisting of m metabolites and r reactions, can be described by a set of ordinary differential equations of the form
| 2.1 |
where S is an m-dimensional vector denoting the set of metabolite concentrations and N is the m × r stoichiometry matrix. The r-dimensional vector v = v(S,K) describes the functional form of the reaction rates, which depend on the metabolite concentrations S and the set of kinetic parameters K. The presence of mass-conservation relationships and conserved chemical moieties in the network results in linear dependencies among the rows of N. In this case, one distinguishes between a set of independently variable metabolite concentrations Sind and a set of dependent metabolite concentrations Sdep. To characterize the dynamics of the system it is sufficient to consider the set of independent variables, making use of a reduced stoichiometry matrix N′ consisting of linearly independent rows only. The link between N′ and N is provided by the expression N = L · N′, where L denotes the link matrix [20].
To evaluate the system, each reaction has to be assigned a particular rate equation and each kinetic parameter needs to be assigned a specific quantitative value. Given a set of initial conditions, equation (2.1) can then be solved computationally, using standard methods of numerical integration. Usually, the model gives rise to one or more steady states (v0, S0) that can be compared with experimentally observed values. The response of the system to a perturbation of a kinetic parameter, representing for example the action of a drug, can be quantified in terms of control coefficients, as introduced by MCA [2,21–23]. In particular, given an effector pi which acts directly on the enzymatic step i, the (scaled) concentration control coefficient 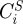 is defined as [20,24]
is defined as [20,24]
 |
2.2 |
and quantifies the ratio between the relative change of the steady-state concentration of metabolite S and the relative change in the catalytic action of enzyme i (induced by an infinitesimal change in the parameter pi). Analogously, the (scaled) flux control coefficient  is defined as
is defined as
 |
2.3 |
and quantifies the response of the system in terms of the relative change in a steady-state flux J. Reder [20] showed that the matrix CS of the concentration control coefficients can be expressed as
| 2.4 |
where J′ denotes the Jacobian of the system with respect to the independent variables, while  and
and 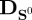 denote square matrices with elements S0 and v0 on the diagonal, respectively, and zero elsewhere.
denote square matrices with elements S0 and v0 on the diagonal, respectively, and zero elsewhere.
For the matrix of flux control coefficients CJ the following expression holds:
 |
2.5 |
Because J′ can be written in terms of the link matrix and the reduced stoichiometry matrix as
| 2.6 |
it follows from equations (2.4)–(2.6) that, once the network topology (as represented by L and N′) and the network ‘phenotype’ (as represented by  and
and  ) are known, the only quantities to be evaluated in order to retrieve the control coefficients are the partial derivatives ∂v/∂S. These, in turn, depend on the functional form of the rate equations governing the dynamics of each enzymatic step, the kinetic parameters and the metabolic state where the derivatives are evaluated. When full kinetic information is available, the control coefficients can be readily calculated using available software packages, such as COPASI [25], or online simulation tools for biochemical models that reside in repositories, such as JWS Online [26].
) are known, the only quantities to be evaluated in order to retrieve the control coefficients are the partial derivatives ∂v/∂S. These, in turn, depend on the functional form of the rate equations governing the dynamics of each enzymatic step, the kinetic parameters and the metabolic state where the derivatives are evaluated. When full kinetic information is available, the control coefficients can be readily calculated using available software packages, such as COPASI [25], or online simulation tools for biochemical models that reside in repositories, such as JWS Online [26].
2.2. Differential metabolic control analysis
To locate suitable drug targets, one aims to identify those enzymes where a perturbation—usually a decrease in enzyme activity induced by an applied inhibitor—elicits a large response in at least one vital aspect of the functioning of the diseased cell, whereas a similar decrease in the activity of the same enzyme is less detrimental for the functioning, or ‘phenotype’, of normal cells. We note that the diseased metabolic phenotype often corresponds to changes in either enzyme concentrations (because of mutations in regulatory elements or signal transduction genes), or availability of external substrates (influx), whereas most Michaelis–Menten constants remain unchanged. Given detailed kinetic models of both phenotypes, the putative action of a drug is applied to both models and the difference in the response is assessed using differential MCA or direct simulations.
The vital aspect of the diseased cell that is targeted may be the production flux of ATP or it could be the growth rate itself. Because it is often a flux, we shall denote it by Jtarget. Our aim is then to locate suitable sites for the action of a drug which results in the maximal differential response in this desired flux between diseased phenotype and normal phenotype. Suitable drug targets are chosen according to the following, alternative or simultaneous, criteria:
-
— Maximal selectivity. We assume that the effect we aim for is the decrease of Jtarget in the diseased cells. In this case, we are interested in enzymes which exert a positive control upon Jtarget in the altered phenotype:
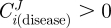 . Independent of whether the effect induced in normal cells consists of a decrease or increase of Jtarget, we require this effect to be lower, in magnitude, than in altered cells. In terms of flux control coefficients of an enzyme i, this criterion can be expressed as follows:
. Independent of whether the effect induced in normal cells consists of a decrease or increase of Jtarget, we require this effect to be lower, in magnitude, than in altered cells. In terms of flux control coefficients of an enzyme i, this criterion can be expressed as follows:
2.7 The higher the value of
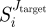 , the stronger the differential response between normal and altered cells. We note that equation (2.7) does not impose any restriction on the sign nor on the value of
, the stronger the differential response between normal and altered cells. We note that equation (2.7) does not impose any restriction on the sign nor on the value of  , provided that its magnitude is smaller than the magnitude of
, provided that its magnitude is smaller than the magnitude of  . Indeed, enzymes with non-negligible negative value of
. Indeed, enzymes with non-negligible negative value of  may be also considered suitable targets as long as the difference in magnitude with
may be also considered suitable targets as long as the difference in magnitude with  allows us to reach an acceptable specificity through an appropriate dosage of an applied inhibitor.
allows us to reach an acceptable specificity through an appropriate dosage of an applied inhibitor. - — Minimal toxicity. To avoid other system properties (not captured by the flux of interest Jtarget) undergoing important changes in normal cells, we require the normal phenotype to be wholly robust against perturbation in the drug target activity. In this respect, we define the toxicity coefficient, which quantifies how far the normal phenotype deviates from its original metabolic state after an inhibition of enzyme i:
where the summation is over all the N exchange fluxes of the system. This definition of toxicity reflects a black-box perspective, where the behaviour of the system is assessed through its inputs and outputs. However, other definitions of toxicity are possible and are examined later in §4.
2.8
2.3. A Monte Carlo strategy
A straightforward implementation of the strategy described above is only rarely applicable, as detailed kinetic models, describing the normal as well as the diseased phenotype, are usually not available. To overcome this limitation, we implement a Monte Carlo strategy which allows us to deal with incomplete knowledge of kinetic parameters and explicitly take into account available phenotypic data. In particular, we assume that for both states of the system the measured metabolic phenotype, characterized by a set of concentrations and fluxes, is known. In this respect, our approach is based on the fact that high-throughput metabolomics and fluxomics studies are now standard techniques in the analysis of cellular metabolism [17,27–32]. We proceed according to the following rationales:
— We require that the map of the metabolic network of interest is known and is the same for both the diseased and the normal phenotype. In addition, we assume that the topology of regulatory interactions is, as far as possible, available.
— For some of the enzymatic steps, information about the detailed functional form of the rate laws may be available. However, in the absence of such information, we employ heuristic assumptions about generic reaction characteristics to describe the dependencies of flux rates with respect to substrates, products and allosteric effectors.
— To obtain a probabilistic understanding on how the control is distributed, the kinetic parameters are sampled randomly from pre-assigned intervals. Each sampled set of parameters is made compliant with the given metabolic phenotypes and additional thermodynamic and biophysical constraints, by rescaling the maximal activity of every reaction step.
— For each sampled set of parameters the differential response in the normal and the disease phenotype is evaluated.
Our workflow is illustrated in figure 1. In the following, each step is described more thoroughly and the details of its implementation are given.
Figure 1.

Workflow of our Monte Carlo approach. The Monte Carlo approach described in the text receives as input the two metabolic states under comparison (where each metabolic state is defined in terms of fluxes and metabolite concentrations at stationary condition). The computation of the control coefficients, following the sampling of the parameter values, is done assuming that the rate equations and the equilibrium constants are known. Where the detailed enzyme mechanism is unknown, heuristic approximate rate equations are used.
2.4. Defining generic reaction characteristics
To evaluate the differential response, each rate equation has to be assigned a specific mathematical form. To describe the kinetics of enzyme catalysed reactions for which the true kinetics is unknown, several possible heuristic approximate rate equations have been proposed in the literature [33–35]. In general, good results are obtained for functional forms that follow generalized Michaelis–Menten equations. Given a reaction converting a set of substrates A into a set of products B
such rate equation can be written as follows:
 |
2.9 |
where
- — freg is a dimensionless prefactor describing the interactions of the enzyme with allosteric regulators. Following the definition given in Liebermeister & Klipp [35], we write
where Pk and Ql denote, respectively, a generic (allosteric) inhibitor and activator, and
2.10  and
and  denote their corresponding binding constants. This equation assumes that the activators and inhibitors are non-competitive.
denote their corresponding binding constants. This equation assumes that the activators and inhibitors are non-competitive. — Vmax is the forward maximal rate of the enzyme and implicitly depends on the enzyme concentration.
—
 is the concentration of reactant Ai divided by its Michaelis constant
is the concentration of reactant Ai divided by its Michaelis constant 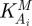 .
.—
 is the concentration of reactant Bj divided by its Michaelis constant
is the concentration of reactant Bj divided by its Michaelis constant  .
.- — Γ is the mass–action ratio, defined as

— Keq denotes the equilibrium constant of the reaction.


The functional form described above captures the generic characteristics of enzyme catalysed reactions, such as reversibility, product inhibition, saturation and a reaction direction that only depends upon the mass–action ratio relative to the equilibrium constant. In the case study presented in this paper, we used equations in the form proposed in Liebermeister & Klipp [35] for every reaction.
2.5. Defining the kinetic parameters
Once the functional form of each rate equation is specified, the equations have to be populated with their respective kinetic constants. Again, we may assume that a (usually small) number of kinetic parameters is available, either by direct experimentation or from data repositories [36,37]. To account for the remaining unknown or uncertain values, we implement a Monte Carlo approach to systematically evaluate the behaviour of the network in a probabilistic manner. The idea is to sample the parameters so that their values comply with known phenotypic data and satisfy basic biophysical and thermodynamic constraints.
Our starting point consists of a metabolic state as characterized by a flux distribution v0 and a set of metabolite concentrations S0. The flux distribution v0 satisfies the stationarity constraint dS0/dt ≡ N · v0 = 0, and the direction of each flux must be consistent with the set of concentrations, relative to its equilibrium constant. Reaction parameters are then assigned or sampled according to the following criteria:
— Equilibrium constants. The equilibrium constants are physico-chemical quantities which reflect the change in standard Gibbs free energy occurring in a reaction. They do not depend on the specific organism or cell type, but may depend on intracellular parameters, such as temperature. While as yet a large-scale detailed experimental quantification of the change in free energy is not available, a number of algorithms exists that allow for a reasonable computational approximation [38–42]. In the case study presented here, the values assigned to the equilibrium constants are obtained from Holzhütter [43].
— Michaelis–Menten constants. The Michaelis–Menten constants are genuine enzyme kinetic parameters and are sampled randomly in intervals
 . Different options are available to specify the interval borders. Here, the interval boundaries are chosen according to the observed metabolite concentrations. We sample the values of the parameters from intervals covering at least two orders of magnitudes around the steady-state concentrations of the corresponding metabolite. In particular, if [S]1 and [S]2 denote the concentration of a metabolite S in the two metabolic states, the affinity, inhibition and activation constants of any enzyme in respect to S are sampled between min{[S]1,[S]2} × 10−α and min{[S]1,[S]2} × 10−β, with α and β representing adjustable factors. To ensure that the sampled values are evenly spread among the different order of magnitudes around [S]1 and [S]2, the sampling is performed using a logarithmic distribution. In the sampling process no distinction is made between parameters relating to regulatory interaction and substrate and product affinities. The results shown in the next section refer to sampling conditions where α = β = 1. The robustness of our results was subsequently tested for different choices of α and β, and different sampling distributions.
. Different options are available to specify the interval borders. Here, the interval boundaries are chosen according to the observed metabolite concentrations. We sample the values of the parameters from intervals covering at least two orders of magnitudes around the steady-state concentrations of the corresponding metabolite. In particular, if [S]1 and [S]2 denote the concentration of a metabolite S in the two metabolic states, the affinity, inhibition and activation constants of any enzyme in respect to S are sampled between min{[S]1,[S]2} × 10−α and min{[S]1,[S]2} × 10−β, with α and β representing adjustable factors. To ensure that the sampled values are evenly spread among the different order of magnitudes around [S]1 and [S]2, the sampling is performed using a logarithmic distribution. In the sampling process no distinction is made between parameters relating to regulatory interaction and substrate and product affinities. The results shown in the next section refer to sampling conditions where α = β = 1. The robustness of our results was subsequently tested for different choices of α and β, and different sampling distributions.— Maximum rates. Given the metabolic state and the parameters defined above, it follows from equation (2.9) that the maximum reaction velocity Vmax is unambiguously determined. In other terms, to make the sampled values of Michaelis–Menten constants compliant with the metabolic state (v0, S0) and the thermodynamics of the system (determined by the equilibrium constants Keq), the maximum reaction velocity is computed by reversing equation (2.9) with respect to Vmax.
2.6. Evaluating the control coefficients
The sampling is performed iteratively. For each sampled set of parameters, the partial derivatives ∂v/∂S are evaluated based on the generalized Michaelis–Menten rate equation described above. Both metabolic states were tested for stability by evaluating their corresponding Jacobian. Parameter sets resulting in Jacobians with positive real part among their spectrum of eigenvalues were discarded, otherwise the control coefficients were evaluated using equations (2.4)–(2.5). We note that the computation only employs basic matrix inversion and multiplication—a procedure which is orders of magnitude faster than numerical integration, making our approach applicable to systems of large size. The process is repeated iteratively until 2.5 × 104 samples are obtained.
2.7. Assessing the suitability of drug target candidates
The differences in the control profiles between diseased and normal phenotype are evaluated according to the criteria of maximal selectivity and minimal toxicity specified above. Because of the probabilistic nature of our approach, these criteria have to be reformulated such that they describe the average, over all the sampling iterations, of the two quantities defined in equations (2.7)–(2.8). As an additional criterion, we evaluate the reliability of the estimated average selectivity. As a quantitative measure of the quality of our prediction, we define the reliability coefficient as the ratio of the average selectivity to the standard deviation of the sampled selectivities:
 |
2.12 |
2.8. Defining the diseased phenotype
To show the applicability of our method on a system of reasonable complexity, we consider a case study to identify suitable sites for drug intervention specifically targeting a cancerous phenotype. As to the best of our knowledge no detailed characterization of a cancer metabolic phenotype (in terms of fluxes and metabolite concentrations) exists, we employ a modelling approach to obtain a set of fluxes and concentrations representative of a generic cancer phenotype.
Our starting point consists of a fully defined metabolic map of human erythrocyte metabolism [43], modified for our purpose (figure 2). Two different sets of maximal activities (Vmax) are used to reproduce the flux patterns characteristic of a normal cell and a paradigmatic cancer cell. In particular, the cancerous metabolic phenotype is obtained by increasing the Vmax of those enzymes which are often overexpressed in cancer, namely the glucose transporter [44,45], hexokinase (HXK) [46–49] and phosphofructokinase (PFK) [46,49,50]. The activity of a fourth enzyme, the glutathione oxidase (GSHox), is also increased in order to achieve a higher flux through the pentose phosphate pathway, as observed by Richardson et al. [51].
Figure 2.

Metabolic map of central carbon metabolism. The metabolic map was derived, in its main features, from Holzhütter's model of erythrocyte metabolism and subsequently enriched with a reaction representing the TCA cycle and a reaction representing the oxidative phosphorylation process. Glucose transporter (GLT), lactate transporter (LCT), phosphoribosylpyrophosphate synthetase (PRPPS) and the tricarboxylic acid (TCA) branch represent the exchange fluxes of the system. ALD, aldolase; TK, transketolase; TPI, triosephosphate isomerase.
We emphasize that the model itself is not used in further analysis and its purpose is solely to obtain a kinetically and thermodynamically consistent set of concentrations and fluxes that represents the common features shared among a wide panel of different cancerous cell lines, in particular a higher uptake of glucose and an enhanced production of lactate [52–54]. Details of model construction and analysis are given in the electronic supplementary material.
3. Results
3.1. Identifying the control profile of the system based on the topology and the metabolic state
The conjecture underlying our probabilistic approach is that some of the control properties of a metabolic system are independent from the precise magnitude of the enzyme parameters. The rationale behind this conjecture is that the signs and magnitudes of the control coefficients are determined to an appreciable extent by the topology of the system as well as the metabolic state under consideration [20,55]. To investigate our conjecture, we distinguish between two extreme scenarios: if the control coefficients are entirely determined by the metabolic map and phenotype, then their value should be independent from the specific choice of the kinetic parameters; on the other hand, if stoichiometry and phenotype had no bearing on control properties at all, then the control coefficients calculated for randomized values of the kinetic parameters should have infinitely wide and flat distributions.
Figure 3 shows the distributions obtained for the control exerted by selected enzymes upon the glucose import flux in the normal phenotype. In some of the cases, the control coefficients are distributed fairly narrowly with respect to the entire probable range of values. For example, the control coefficient of the glucose transporter (GLT, figure 3a) is distributed almost entirely between 0.01 and 0.05. This reinforces an earlier conclusion from a precise calculation that this control coefficient is small and that inhibitors of the glucose transporter are unlikely to be toxic to human erythrocytes [56]. More importantly, it confirms the strength of our conjecture for this case; i.e. within the limits of experimental accuracy [2], the value of this flux control coefficient can be estimated on the basis of the metabolic map, the metabolic phenotype and very limited kinetic information. The other panels of figure 3 show that this phenomenon is not unique. It is also valid for the control exerted on this same flux by other enzymes in the system, also for ones with higher flux control. Figure 3 also shows that the accuracy by which metabolic map and metabolic phenotype determine the flux control coefficient is not always the same. For the phosphofructokinase, for example, the estimated flux control coefficients on glucose import exhibit a broad distribution covering almost the entire interval between zero and one (figure 3b). The other panels in figure 3 are chosen as representative cases where the distribution of the control coefficients over the uptake of glucose is either mainly confined in the negative semiaxes (as opposed to glucose transporter and phosphofructokinase) or is spread evenly around zero, allowing no best guess on the sign of the actual control coefficient. See also the electronic supplementary material for a magnified depiction of the individual distributions.
Figure 3.

Calculated distributions of the control exerted by some enzymes over the glucose uptake flux in the normal phenotype. (a) Glucose transporter (GLT), (b) phosphofructokinase (PFK), (c) transketolase 1 (TK1), (d) phosphoglyceratekinase (PGK), (e) lactate dehydrogenase (LDH), (f) ATPase.
Figure 4 summarizes the distributions of the estimated control coefficients for the entire network. Each position in the matrix corresponds to the control exerted by an enzyme (columns) upon a flux within the network (rows) and shows the (colour-coded) percentage of calculated control coefficients that is positive. If the box is white then the flux control coefficient is positive independent, to a good extent, of the values of the kinetic constants. For example, the figure shows that the control exerted by the first six enzymes on all the first 13 reactions is positive, as indeed might be expected from the network topology shown in figure 2. Likewise, the enzymes 14–18 (corresponding to G6PDH, 6PGD, GSSG, GSHox and EP) mostly exert a negative control on the flux through reaction 3 (PGI), again consistently with our expectations from the network topology. However, other results are less straightforward to interpret, showing the utility of our calculations for complex reaction network. Overall, figure 4 confirms our conjecture that often at least the sign of the control coefficient is, to a good extend, independent from the precise values of kinetic parameters—enabling us to obtain a best guess for putative drug targets in the face of incomplete information.
Figure 4.

Matrix of the flux control coefficient in the normal metabolic state. The matrix of the flux control coefficients in the normal metabolic phenotype is represented as a grey-scale matrix. The entry (j,i) is associated with the statistical control exerted by enzyme i (column index) upon flux j (row index). More in particular, the shade of the entry represents the percentage of calculated control coefficients that is positive. The ends of the colours scale represent the extreme situations in which the distribution lies entirely over positive (white) or negative (black) values. The numbers at the left and the bottom of the matrix refers to the different reaction steps in the system as depicted in figure 2.
3.2. Identifying candidate targets for drug intervention
From the perspective of limiting the survival or proliferation of cells that function as parasites in the human body, a possible strategy consists of inhibiting glycolysis, provided the pathway is phenotypically different in the parasitic versus the host cells [3,5,57,58]. From an MCA perspective, this translates into inhibiting the activity of an enzyme which exerts a major control over (for example) the uptake of glucose in the diseased/parasitic phenotype and a minor control over the same flux in the normal phenotype. The rationale of such an approach is to starve the diseased/parasitic cells without significantly affecting the host. From here on we will refer to this specific strategy to present and comment on our results.
Figure 5 shows the distributions of the selectivity coefficients with respect to the uptake of glucose for the same enzymes of figure 3a,b, in particular, show that the selectivity coefficients of the glucose transporter (GLT) and phosphofruktokinase (PFK) are mainly distributed over negative values, meaning that the control exerted by these enzymes upon the uptake of glucose is higher in the normal phenotype than in the cancer phenotype. Consequently, GLT and PFK cannot be considered good candidate targets (cf. [59]). This result is somewhat expected as the diseased phenotype was taken to be due to overexpression of four enzymes among which GLT and PFK (see §2). In general, however, the overexpression of an enzyme does not necessarily imply a decrease in the control it exerts, as the activity of the enzyme must be considered in the context of the entire network. Other enzymes show the desired selectivity in terms of their control on the target flux between diseased and normal cells. This is the case for phosphoglycerate kinase (PGK, figure 5d) and lactate transporter (LCT, figure 5e), the selectivity coefficients of which are mainly distributed over positive values, although the magnitude of these values differ substantially between the two enzymes.
Figure 5.

Calculated distributions of the selectivity coefficient. The distribution of the selectivity coefficients with respect to the uptake of glucose are shown for the same enzymes as in figure 3.
Because of the probabilistic nature of our approach, the quantities defining the criteria of maximal selectivity and minimal toxicity, i.e. the selectivity coefficient  and the toxicity coefficient Ti, respectively, are taken in their average values
and the toxicity coefficient Ti, respectively, are taken in their average values 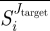 and
and  , where the average is computed over all the sampling iterations. For the statements about enzyme selectivity to be meaningful we also consider the reliability coefficient
, where the average is computed over all the sampling iterations. For the statements about enzyme selectivity to be meaningful we also consider the reliability coefficient  as defined in equation (2.12). We now have three criteria by which to compare potential molecular drug targets. In any actual situation we will need to look at all three criteria simultaneously. To make the weighing of the three criteria as transparent as possible, we introduce the following scaled quantities:
as defined in equation (2.12). We now have three criteria by which to compare potential molecular drug targets. In any actual situation we will need to look at all three criteria simultaneously. To make the weighing of the three criteria as transparent as possible, we introduce the following scaled quantities:
 |
3.1 |
 |
3.2 |
 |
3.3 |
where σi, 1/τi and ρi are the normalized selectivity, the normalized safety and the normalized reliability, respectively. We note that the normalized safety is defined so that it increases with decreasing toxicity.
By restricting our search for putative targets to only those enzymes with positive average selectivity 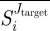 , i.e. enzymes which tend to produce the wanted inhibiting effect on Jtarget in the diseased phenotype, the three normalized criteria are bound between 0 and 1.
, i.e. enzymes which tend to produce the wanted inhibiting effect on Jtarget in the diseased phenotype, the three normalized criteria are bound between 0 and 1.
Figure 6 depicts the values of the normalized selectivity, reliability and safety for all the enzymes with positive average selectivity  . A most interesting result was that one enzyme target was both most selective and most reliable, and was also hardly toxic. This target was phosphoglyceratekinase (PGK). The fact that all three criteria come out with a single enzyme target suggests that that target could indeed be exceptionally valuable. It also suggests that the methodology we have introduced may be useful; a lesser result would have been if the target that was most selective would have been least reliable and most toxic, or if the least toxic target would also have been least selective. However, in general, we expect that no single target is best according to all three criteria introduced above. The more important result therefore is that our methodology leads to a clear separation of the potential targets at least in the dimensions of selectivity and reliability. In this respect the criterion of maximal safety (i.e. minimal toxicity) appeared to be less discriminatory for the enzymes shown.
. A most interesting result was that one enzyme target was both most selective and most reliable, and was also hardly toxic. This target was phosphoglyceratekinase (PGK). The fact that all three criteria come out with a single enzyme target suggests that that target could indeed be exceptionally valuable. It also suggests that the methodology we have introduced may be useful; a lesser result would have been if the target that was most selective would have been least reliable and most toxic, or if the least toxic target would also have been least selective. However, in general, we expect that no single target is best according to all three criteria introduced above. The more important result therefore is that our methodology leads to a clear separation of the potential targets at least in the dimensions of selectivity and reliability. In this respect the criterion of maximal safety (i.e. minimal toxicity) appeared to be less discriminatory for the enzymes shown.
Figure 6.

The normalized selectivity (σi), safety (1/τi) and reliability (ρi), defined in equations (3.1)–(3.3), respectively, are plotted versus each other. Enzymes with negative values of  (see text) are not represented in the graph. The grey plane in (a) represents the set of points in the three-dimensional space sharing the same score, as defined by equation (3.4). Different scores are represented by different planes, parallel to each other. (b–d) show the orthogonal projections of the three-dimensional scatter-plot shown in (a).
(see text) are not represented in the graph. The grey plane in (a) represents the set of points in the three-dimensional space sharing the same score, as defined by equation (3.4). Different scores are represented by different planes, parallel to each other. (b–d) show the orthogonal projections of the three-dimensional scatter-plot shown in (a).
We may wish to classify drug targets with a single score, which takes into account all three criteria (selectivity, reliability and safety), perhaps with different weights. To do so, we define the following quantity:
 |
3.4 |
as the score to be assigned to enzyme i. Equation (3.4) represents a plane in the three-dimensional space defined by σi, ρi and 1/τi. For a minimally required score Z*, one may draw the corresponding plane in figure 6a and require all enzyme targets one wishes to consider for further development to lie above that plane. In figure 6a, we have drawn the plane corresponding to Z* = 1/3 and weight factors wσ = 4, wτ = 2 and wρ = 1. This specific choice for the weight factor values was made to prioritize the maximal selectivity over the minimal toxicity, and the latter over the requirement of minimal uncertainty. Table 1 shows the list of enzyme with positive average selectivity, sorted by decreasing value of Z.
Table 1.
Suitability of the different enzymes as drug targets. The suitability of each enzyme as drug target is evaluated according to the three criteria of selection described in the text. The score is computed through equation (3.4) where wσ = 4, wτ = 2 and wρ = 1 (see text).
| enzyme | score |
|---|---|
| PGK | 0.99 |
| ENO | 0.56 |
| LDH | 0.48 |
| PGM | 0.45 |
| LCT | 0.40 |
| TPI | 0.38 |
| GAPDH | 0.36 |
| ALD | 0.35 |
| 6PGD | 0.32 |
3.3. Robustness of the results
As a probabilistic method, our approach crucially relies on the robustness of the results with respect to different ways of sampling the parameter values. To this end, we repeated the data generation process and the subsequent analysis using different sampling conditions. In particular, we changed the range of values from which the parameters were sampled and the sampling distribution. For each different set of sampling conditions, we ranked the suitability of the different enzyme as drug targets as explained above.
Table 2 summarizes the results obtained for the different sampling conditions, showing PGK as the highest scoring target for all of them. As expected, enzymes with lower scores than PGK did not always keep their position in the ranking list. The lower the score the higher the chance for the enzyme to enter the list in a different position, when the sampling condition was altered. In most of the cases, however, the top six entries of the list remained the same. In particular, PGK always emerged as the best guess as putative target, while enolase (ENO) and phosphoglycerate mutase (PGM) were always found within the top four entries of the list, and lactate dehydrogenase (LDH) was almost always found between the 3rd and the 6th position (see the electronic supplementary material for more details). The use of a linear sampling distribution caused the most different results in respect to the scoring list obtained with a logarithmic distribution, even for the high-scoring enzymes (except for PGK, which was always at the top of the list). This fact can be ascribed to the highly asymmetric sampling of the parameter values in respect to the metabolite concentrations. When using a linear distribution, there is a much higher probability that the sampled value of the Michaelis–Menten constants exceed the concentration of the corresponding metabolites. In our case, where α = β = 1 (see §2 for their definition), for each reaction step statistically at least 90 per cent of the sampled values were larger than the concentration of their corresponding metabolites. This implies a scenario where the saturation level of all the enzymes in respect to all their reactants and modifiers was almost always smaller that 50 per cent. On the other hand, when using a logarithmic distribution, the value of the Michaelis–Menten constants tended to be evenly sampled around the metabolite concentrations, or, more precisely, among the orders of magnitude spanned by the sampling intervals.
Table 2.
Best drug target guesses for different sampling conditions. The first two columns refer to the parameters α and β used to define the intervals from which the parameter values were sampled. The first four rows refer to sampling conditions where the intervals were defined separately for each parameter, as discussed in §2. The borders of this interval were defined as min {[S]1, [S]2} × 10α and max {[S]1, [S]2} × 10β, where {[S]1, [S]2} denotes the set of concentrations of all the metabolites in the two metabolic states.
| α | β | sampling function | best guess |
|---|---|---|---|
| 1 | 1 | logarithmic | PGK |
| 1 | 2 | logarithmic | PGK |
| 2 | 1 | logarithmic | PGK |
| 2 | 2 | logarithmic | PGK |
| 1 | 1 | linear | PGK |
| 1a | 1a | logarithmic | PGK |
asampling performed on the same interval for all the parameters.
3.4. Comparison with the dynamic model
To assess the significance of our result, we compared the insights gained through our statistical approach with the results obtainable directly through a dynamic simulation. The dynamic model used to retrieve the cancer metabolic state (see §2 and electronic supplementary material) was used as workbench for this assessment. The two sets of parameters characterizing the two metabolic states (normal and cancer) were both modified by decreasing the activity (Vmax) of PGK from 12.9 to 6.5 mM h−1, simulating the addition of a non-competitive inhibitor. Figure 7 shows how some of the fluxes of the metabolic system changed in the two phenotypes in response to this perturbation. Table 3 shows the relative changes recorded once the system reached the new steady state after the perturbation.
Figure 7.

Effects induced on the principal fluxes of the system by decreasing the activity of PGK. The plots show the effect of decreasing the activity of PGK from 12.9 to 6.5 mM h−1 in the two phenotypes (normal, thin solid line; disease, thick solid line). Six main fluxes are considered: (a) glucose uptake, (b) PRPP, (c) lactate production, (d) flux entering the TCA cycle, (e) ATPase and (f) flux through the oxidative phosphorylation process.
Table 3.
Relative changes in the principal fluxes of the system after decreasing the activity of PGK. The relative changes reported are the result of decreasing the activity of PGK from 12.9 to 6.5 mM s−1.
| metabolic branch/process | normal state (%) | cancer state (%) |
|---|---|---|
| glucose uptake | −7 | −44 |
| lactate production | −61 | −82 |
| TCA cycle | −1 | −8 |
| phosphoribosylpyrophosphate synthetase (PRPPS) | +9 | +13.0 |
| ATPase | −2 | −12 |
| oxidative phosphorylation | −1 | −8 |
4. Discussion
Despite a surge in the perspectives of using mechanistic mathematical modelling in clinical development [60–63], parametric uncertainty remains a challenge to mechanistic approaches in medicine [13]. However, contrary to the scarcity of kinetic data, the comprehensive quantification of all concentrations and fluxes within a metabolic system is, at least in principle, experimentally feasible. The question we addressed in this paper is whether the knowledge of a metabolic phenotype only—expressed in terms of fluxes and metabolite concentrations at steady-state—allows for a probabilistic understanding of how the control properties of a biochemical system are distributed among the different enzymatic steps and metabolic processes. We developed a Monte Carlo approach which aims to provide researchers with a probabilistic description of how the control properties of a metabolic network differ between two fully characterized metabolic phenotypes, when only minimal knowledge of the system is available with respect to its kinetic parameters. Our goal was to locate points of fragility in a diseased/pathogen phenotype which can be considered for drug interventions with maximal effectiveness and minimal toxicity. In particular, enzymes which exert a major control over a certain property of interest in a diseased/pathogen phenotype, and a minor control over the same property in the normal/host phenotype, represent good putative targets. In our method, the control profiles characterizing the two phenotypes under comparison are determined for sampled values of the unknown affinity, inhibition and activation constants. Our Monte Carlo approach provides us with a statistical understanding of how the control is differentially distributed between the two metabolic states, where the word ‘statistical’ should be interpreted in the sense of uncertainty rather than in the sense of population dispersion.
The system under study was a simplified reconstruction of the central carbon metabolism, with two metabolic states that were representative of a normal and a paradigmatic cancerous phenotype. The results presented in this work refer to a clinical strategy aimed to starve specifically cancer cells, i.e. to locate enzymes which exhibit a high differential control upon the uptake of glucose, denoted as Jtarget, between the two phenotypes.
In the development of new drugs, pharmaceutical companies tend to give priority to the maximal effect that a compound can induce in the disease cells. Among the palette of compounds which pass this filter, a second screening is performed to look for compounds which leave the normal phenotype unaltered as much as possible. Our approach is different. To assess the suitability of the enzymes as possible targets for an anti-cancer drug, three different criteria were considered in this paper: maximal selectivity, maximal safety (i.e. minimal toxicity) and maximal reliability. The first criterion was formulated in such a way as to encompass both the requirements of high effectiveness and high selectivity with respect to the specific property one wants to affect in the diseased cells. High values of the selectivity coefficient correspond to a high (positive) control over Jtarget in the diseased cell, and a relatively small control over the same flux in the normal cells. In particular, we introduced the selectivity coefficient, as defined in equation (2.7), to quantify the differential response of the system to inhibition of enzyme i. Alternative definitions than equation (2.7) may also be considered. For example, Bakker et al. [3] defined it as the ratio between the control over Jtarget in the parasitic/diseased cell over the same control in the host/normal cell. Such a definition, however, may result in over-ranking enzymes with small control in the diseased phenotype as putative targets. An enzyme A with 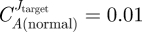 and
and  , for instance, would be considered preferable to an enzyme B with
, for instance, would be considered preferable to an enzyme B with  and
and  , as the ratio
, as the ratio  would be around three times the ratio
would be around three times the ratio  . However, using an appropriate dosage of a drug that inhibits enzyme B, one could reduce the effect of the enzyme inhibition in the normal cell to a negligible extent while maintaining in the diseased cell a strong effect, in fact stronger than would be feasible by inhibition of enzyme A.
. However, using an appropriate dosage of a drug that inhibits enzyme B, one could reduce the effect of the enzyme inhibition in the normal cell to a negligible extent while maintaining in the diseased cell a strong effect, in fact stronger than would be feasible by inhibition of enzyme A.
The criterion of maximal safety (or minimal toxicity) was introduced to assure that the normal phenotype was robust to a perturbation of the drug target. The predicted toxic effect of inhibiting an enzyme i was quantified through the toxicity coefficient defined in equation (2.8). We defined this coefficient as the average of the absolute values of the flux control coefficients of enzyme i with respect to all the exchange fluxes. Our definition reflects a black-box perspective, where the behaviour of the system is assessed solely through its input and output fluxes. However, this definition of toxicity is far from unique and its suitability may depend on the specific situation. For example, an alternative choice would be to define the toxicity as the largest of all of the target enzyme's flux control coefficients. Also, there may be reactions or pathways for which a change in the flux does not induce any major effect on the vital functions of the cell, while even small alterations in other fluxes might entail significant stress in the normal cellular physiology. In this case, the criterion of minimal toxicity can be augmented by assigning different weights to the respective control coefficients. Another possibility to define the toxicity coefficient would be to take into account the concentration of key metabolites that are known to have toxic effects. In this case, the toxicity is assessed in terms of the concentration control coefficients of the target enzymes with respect to these metabolites.
Finally, the criterion of maximal reliability was introduced in order to prefer enzymes whereby the computed average selectivity was affected by the least possible uncertainty.
To evaluate the suitability of an enzyme as a drug target, different weights can be assigned to these criteria, depending on the relevance they have for the researcher. In the present work, we chose to prioritize the maximal selectivity over the minimal toxicity, and the latter over the requirement of minimal uncertainty. By doing so, PGK was identified as the best guess for a suitable drug target. Although in this case PGK was both the most selective and most reliable enzyme target, besides being hardly toxic, we note that in general the outcome of our analysis can sensibly depend on the priority ascribed to the three criteria mentioned above. From a clinical perspective, performing our analysis with different weight factors may then provide a palette of best drug targets referring to different thresholds among the required properties of selectivity, safety and reliability.
The robustness of our results was tested against different choices of the sampling conditions. The fact that PGK always emerged as the best guess as putative target represents an important result, as it suggests that the sampling approach proposed in this paper can provide a relevant insight about the control profile of a metabolic system that is reasonably robust with respect to alterations in the numerical methods. If the top entries of the scoring list were to change completely based on the sampling condition, the method would have proved to be of less utility. We note, however, that in general different choices of the sampling distribution may lead to different enzymes as best putative drug target. The use of different sampling distributions may then be thought and adopted as a way to provide a more restrictive assessment of how the emergence of a specific enzyme as best target is due to topological constraints rather than specific parameter values (or range of values).
The conclusions achieved through our probabilistic approach, leading to the identification of PGK as the best putative target, were compared with the outcome of a mechanistic approach by decreasing the activity (Vmax) of PGK in the dynamic model introduced in §2 and described in the electronic supplementary material. The response of the system to this perturbation showed that the normal phenotype was indeed less sensitive to an inhibition of PGK than the diseased phenotype. This lower sensitivity has been observed not only in respect to the glucose uptake flux, but in the general behaviour of the system. The exchange fluxes and the main metabolic processes were in fact less affected in the normal phenotype, showing good agreement with our statistical result. Regarding specifically the production of lactate, we registered a significant decrease in the flux through LDH in both normal and diseased phenotype (−61% and −82%, respectively). Since this strong decrease occurred on a branch which is virtually unused in normal cells (except for muscle cells under anaerobic conditions and erythrocytes), this result does not invalidate PGK as putative drug target candidate.
In conclusion, the statistical approach proposed in this paper provides us with a useful strategy for assessing how the control profile is differently distributed in two distinct metabolic phenotypes. It also highlights which enzyme can best represent a putative target with respect to requirements such as high effectiveness and low toxicity. The significance of the results obtained through this kind of analysis, however, may be improved at different levels, not necessarily only related to the probabilistic nature of our approach. For example, a higher degree of detail in the representation of the metabolic map would reduce the approximation introduced by considering lumped reactions representing more complex biochemical pathways or processes. In the example provided in this paper, two of such reactions were used to represent the TCA cycle and oxidative phosphorylation. Expanding a lumped reaction into the entire set of enzymatic steps it represents, could lead to new and important insights about how the control properties are distributed in the system. This is mostly the case when metabolic intermediates that are only involved in a lumped pathway have regulatory effects on enzymatic steps outside that pathway. A more detailed description of the lumped pathway would then result not only in a ‘higher resolution’ of how the control properties are locally distributed among the different steps which were lumped together, but also in a different overall control profile. Just as in the case of conventional kinetic modelling, the level of detail in which the metabolic map is represented can determine the level of accuracy of the regulatory map, and consequently have a non-negligible effect on the results.
A related aspect is the choice of rate equations of the enzymatic steps. While generic rate equations are commonly used to capture generic aspects of metabolic networks, the actual, experimentally determined, rate equations may result in slightly modified control properties. In particular, the experimental determination of reaction functions may also identify further unknown regulatory interactions, as well as possible cooperativity between metabolic compounds. Regarding the use of the generic rate equation, we also note that different choices are possible. The specific instance of generalized Michaelis–Menten equation proposed by Liebermeister & Klipp [35], and used in this paper, takes into account generic characteristics of enzyme catalysed reactions—such as reversibility, product inhibition, saturation, reaction direction that only depends upon the mass–action ratio relative to the equilibrium constant. However, alternative choices have been proposed by Rohwer et al. [64] that also account for competition between substrates and products and take possible cooperativity into account. We note though, in contrast to explicit kinetic models, we are only interested in the derivatives of the rate equations, such that minor differences in the precise functional form often have no major effect. In most applications, we also expect that at least partial information on some kinetic parameters is available. Such partial information allows us to further constraint the sampling intervals and to obtain results that are specific for the system under study. In this sense, our approach can be straightforwardly incorporated into an iterative scheme that allows us to quantify uncertainty in the control profile, and hence allows us to pinpoint further experiments to increase the specificity and reliability of the results.
Acknowledgements
We acknowledge the support of the Doctoral Training Center ISBML by EPSRC and BBSRC (EP/F500 009/1), of the Manchester Center of Integrative Systems Biology by BBSRC, EPSRC (BB/C0082191) and additional BBSRC support (BBD0190791), of SysMO (BBF003535281, 35361, 35521) and of the EC Network of Excellence BioSim.
References
- 1.Majumder H. K. 2008. Drug targets in kinetoplastid parasites. Advances in experimental medicine and biology, vol. 625 Berlin, Germany: Springer; [PubMed] [Google Scholar]
- 2.Groen A. K., Wanders R. J., Westerhoff H. V., van der Meer R., Tager J. M. 1982. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 257, 2754–2757 [PubMed] [Google Scholar]
- 3.Bakker B. M., Assmus H. E., Bruggeman F., Haanstra J. R., Klipp E., Westerhoff H. 2002. Network-based selectivity of antiparasitic inhibitors. Mol. Biol. Rep. 29, 1–5 10.1023/A:1020397513646 (doi:10.1023/A:1020397513646) [DOI] [PubMed] [Google Scholar]
- 4.Gerber S., Assmus H., Bakker B., Klipp E. 2008. Drug-efficacy depends on the inhibitor type and the target position in a metabolic network—a systematic study. J. Theoret. Biol. 252, 442–455 10.1016/j.jtbi.2007.09.027 (doi:10.1016/j.jtbi.2007.09.027) [DOI] [PubMed] [Google Scholar]
- 5.Westerhoff H. V. 2007. Systems biology: new paradigms for cell biology and drug design, systems biology, pp. 45–67 Berlin, Germany: Springer; [DOI] [PubMed] [Google Scholar]
- 6.Guimera R., Sales-Pardo M., Amaral L. A. N. 2007. A network-based method for target selection in metabolic networks. Bioinformatics 23, 1616–1622 10.1093/bioinformatics/btm150 (doi:10.1093/bioinformatics/btm150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitano H. 2004. Cancer as a robust system: implications for anticancer therapy. Nat. Rev. Cancer 4, 227–235 10.1038/nrc1300 (doi:10.1038/nrc1300) [DOI] [PubMed] [Google Scholar]
- 8.Kitano H. 2007. The theory of biological robustness and its implication in cancer, in systems biology: applications and perspectives, pp. 69–88 Berlin, Germany: Springer; [DOI] [PubMed] [Google Scholar]
- 9.Hornberg J. J., Bruggeman F., Bakker B., Westerhoff H. 2007. Metabolic control analysis to identify optimal drug targets, in systems biological approaches in infectious diseases, pp. 171–189 Basel, Switzerland: Birkhäuser; [DOI] [PubMed] [Google Scholar]
- 10.Westerhoff H. V. 2006. Emerging principles: a theory for robustness, in ICSB2006. Yokohama, Japan: Yokohama Publisher [Google Scholar]
- 11.Cascante M., et al. 2002. Metabolic control analysis in drug discovery and disease. Nat. Biotechnol. 20, 243–249 10.1038/nbt0302-243 (doi:10.1038/nbt0302-243) [DOI] [PubMed] [Google Scholar]
- 12.Schellenberger J., Palsson B. Ø. 2008. The use of randomized sampling for analysis of metabolic networks. J. Biol. Chem. 284, 5457–5461 10.1074/jbc.R800048200 (doi:10.1074/jbc.R800048200) [DOI] [PubMed] [Google Scholar]
- 13.Luan D., Zai M., Varner J. D. 2007. Computationally derived points of fragility of a human cascade are consistent with therapeutic strategies. Comput. Biol. 3, 1347–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L., Hatzimanikatis V. 2006. Metabolic engineering under uncertainty. I: framework development. Metab. Eng. 8, 133–141 10.1016/j.ymben.2005.11.003 (doi:10.1016/j.ymben.2005.11.003) [DOI] [PubMed] [Google Scholar]
- 15.Wang L., Hatzimanikatis V. 2006. Metabolic engineering under uncertainty—II: analysis of yeast metabolism. Metab. Eng. 8, 142–159 10.1016/j.ymben.2005.11.002 (doi:10.1016/j.ymben.2005.11.002) [DOI] [PubMed] [Google Scholar]
- 16.Tran L. M., Rizk M. L., Liao J. C. 2008. Ensemble modeling of metabolic networks. Biophys. J. 95, 5606–5617 10.1529/biophysj.108.135442 (doi:10.1529/biophysj.108.135442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steuer R., Gross T., Selbig J., Blasius B. 2006. Structural kinetic modeling of metabolic networks. Proc. Natl Acad. Sci. USA 103, 11 868–11 873 10.1073/pnas.0600013103 (doi:10.1073/pnas.0600013103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steuer R. 2007. Computational approaches to the topology, stability and dynamics of metabolic networks. Phytochemistry 68, 2139–2151 10.1016/j.phytochem.2007.04.041 (doi:10.1016/j.phytochem.2007.04.041) [DOI] [PubMed] [Google Scholar]
- 19.Grimbs J., Selbig S., Bulik H. G., Holzutter S., Steuer R. 2007. The stability and robustness of metabolic states: identifying stabilizing sites in metabolic networks. Mol. Syst. Biol. 3, 146. 10.1038/msb4100186 (doi:10.1038/msb4100186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reder C. 1988. Metabolic control theory: a structural approach. J. Theoret. Biol. 135, 175–201 10.1016/S0022-5193(88)80073-0 (doi:10.1016/S0022-5193(88)80073-0) [DOI] [PubMed] [Google Scholar]
- 21.Burns J. A., et al. 1985. Control analysis of metabolic systems. Trends Biochem. Sci. 10, 16. 10.1016/0968-0004(85)90008-8 (doi:10.1016/0968-0004(85)90008-8) [DOI] [PubMed] [Google Scholar]
- 22.Kacser H., Burns J. A. 1973. The control of flux. Symp. Soc. Exp. Biol. 27, 65–105 [PubMed] [Google Scholar]
- 23.Heinrich R., Rapoport T. A. 1974. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 42, 89–95 10.1111/j.1432-1033.1974.tb03318.x (doi:10.1111/j.1432-1033.1974.tb03318.x) [DOI] [PubMed] [Google Scholar]
- 24.Westerhoff H. V., VanDam K. 1987. Thermodynamics and control of biological free-energy transduction. Amsterdam, The Netherlands: Elsevier [Google Scholar]
- 25.Hoops S., et al. 2006. COPASI—a COmplex PAthway SImulator. Bioinformatics 22, 3067–3074 10.1093/bioinformatics/btl485 (doi:10.1093/bioinformatics/btl485) [DOI] [PubMed] [Google Scholar]
- 26.Snoep J. L. 2005. The silicon cell initiative: working towards a detailed kinetic description at the cellular level. Curr. Opin. Biotechnol. 16, 336–343 10.1016/j.copbio.2005.05.003 (doi:10.1016/j.copbio.2005.05.003) [DOI] [PubMed] [Google Scholar]
- 27.Fiehn O. 2001. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genom. 2, 155–168 10.1002/cfg.82 (doi:10.1002/cfg.82) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kell D. B. 2004. Metabolomics and systems biology: making sense. Curr. Opin. Microbiol. 7, 296–307 10.1016/j.mib.2004.04.012 (doi:10.1016/j.mib.2004.04.012) [DOI] [PubMed] [Google Scholar]
- 29.Goodacre R., Vaidyanathan S., Dunn W. B., Harrigan G. H., Douglas D. B. 2004. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 22, 252–254 10.1016/j.tibtech.2004.03.007 (doi:10.1016/j.tibtech.2004.03.007) [DOI] [PubMed] [Google Scholar]
- 30.Sauer U. 2006. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2, (doi:10.1038/msb4100109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zamboni N., Fendt S. M., Ruhl M., Sauer U. 2009. 13C-based metabolic flux analysis. Nat. Protocols 4, 878–892 10.1038/nprot.2009.58 (doi:10.1038/nprot.2009.58) [DOI] [PubMed] [Google Scholar]
- 32.Kruger N. J., Ratcliffe R. G. 2009. Insights into plant metabolic networks from steady-state metabolic flux analysis. Biochimie 91, 697–702 10.1016/j.biochi.2009.01.004 (doi:10.1016/j.biochi.2009.01.004) [DOI] [PubMed] [Google Scholar]
- 33.Bulik S., Grimbs S., Huthmacher C., Selbig J., Holzutter H. G. 2009. Kinetic hybrid models composed of mechanistic and simplified enzymatic rate laws—a promising method for speeding up the kinetic modelling of complex metabolic networks. FEBS J. 276, 410–424 10.1111/j.1742-4658.2008.06784.x (doi:10.1111/j.1742-4658.2008.06784.x) [DOI] [PubMed] [Google Scholar]
- 34.Dräger A., Kronfeld M., Ziller M. J., Supper J., Planatscher H., Magnus J. B., Oldiges M., Kohlbacher O., Zell A. 2009. Modeling metabolic networks in C. glutamicum: a comparison of rate laws in combination with various parameter optimization strategies. BMC Syst. Biol. 3, 5. 10.1186/1752-0509-3-5 (doi:10.1186/1752-0509-3-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liebermeister W., Klipp E. 2006. Bringing metabolic networks to life: convenience rate law and thermodynamic constraints. Theoret. Biol. Med. Modell. 3, 41. 10.1186/1742-4682-3-41 (doi:10.1186/1742-4682-3-41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pharkya P., Nikolaev E. V., Maranas C. D. 2003. Review of the BRENDA Database. Metab. Eng. 5, 71–73 10.1016/S1096-7176(03)00008-9 (doi:10.1016/S1096-7176(03)00008-9) [DOI] [PubMed] [Google Scholar]
- 37.Rojas I., Golebiewiski M., Kania R., Krebs O., Mir S., Weidemann A., Wittig U. 2007. SABIO-RK: a database for biochemical reactions and their kinetics. BMC Syst. Biol. 1(Suppl 1), S6. 10.1186/1752-0509-1-51-56 (doi:10.1186/1752-0509-1-51-56) [DOI] [Google Scholar]
- 38.Alberty R. A. 2003. The role of water in the thermodynamics of dilute aqueous solutions. Biophys. Chem. 100, 183–192 10.1016/S0301-4622(02)00280-6 (doi:10.1016/S0301-4622(02)00280-6) [DOI] [PubMed] [Google Scholar]
- 39.Goldberg R. N., Tewari Y. B., Bhat T. N. 2004. Thermodynamics of enzyme-catalyzed reactions—a database for quantitative biochemistry. Bioinformatics 2, 2874–2877 10.1093/bioinformatics/bth314 (doi:10.1093/bioinformatics/bth314) [DOI] [PubMed] [Google Scholar]
- 40.Jankowski M. D., Henry C. S., Broadbelt L. J., Hatzimanikatis V. 2008. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 95, 1487–1499 10.1529/biophysj.107.124784 (doi:10.1529/biophysj.107.124784) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henry C. S., Jankowski M. D., Broadbelt L. J., Hatzimanikatis V. 2006. Genome scale thermodynamic analysis of Escherichia coli metabolism. Biophys. J. 90, 1453–1461 10.1529/biophysj.105.071720 (doi:10.1529/biophysj.105.071720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feist A. M., Henry C. S., Reed J. L., Krummenacker M., Joyce A. R., Karp P. D., Broadbelt L. J., Hatzimanikatis V., Palsson V. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3, 121. 10.1038/msb4100155 (doi:10.1038/msb4100155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holzhütter G. 2004. The principle of flux minimization and its application to estimate stationary fluxes in metabolic networks. Eur. J. Biochem. 271, 2905–2922 10.1111/j.1432-1033.2004.04213.x (doi:10.1111/j.1432-1033.2004.04213.x) [DOI] [PubMed] [Google Scholar]
- 44.Wood S., Trayhurn P. 2003. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br. J. Nutr. 89, 3–9 10.1079/BJN2002763 (doi:10.1079/BJN2002763) [DOI] [PubMed] [Google Scholar]
- 45.Macheda M. L., Rogers S., Best J. D. 2005. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 202, 654–662 10.1002/jcp.20166 (doi:10.1002/jcp.20166) [DOI] [PubMed] [Google Scholar]
- 46.Marín-Hernández A., Rodríguez-Enráquez S., Vital-Gonzalez P. A., Florez-Rodríguez F. L., Macías-Silva M., Sosa-Gorrocho M., Moreno-Sánchez R. 2006. Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J. 273, 1975–1988 10.1111/j.1742-4658.2006.05214.x (doi:10.1111/j.1742-4658.2006.05214.x) [DOI] [PubMed] [Google Scholar]
- 47.Wilson J. E. 2003. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 206, 2049–2057 10.1242/jeb.00241 (doi:10.1242/jeb.00241) [DOI] [PubMed] [Google Scholar]
- 48.Parry D. M., Pedersen P. L. 1983. Intracellular localization and properties of particulate hexokinase in the Novikoff ascites tumor. Evidence for an outer mitochondrial membrane location. J. Biol. Chem. 258, 10 904–10 912 [PubMed] [Google Scholar]
- 49.Nakashima R. A., Paggi M. G., Scott L. J., Pedersen P. L. 1988. Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res. 48, 913–919 [PubMed] [Google Scholar]
- 50.Vora S., Halper J. P., Knowles D. M. 1985. Alterations in the activity and isozymic profile of human phosphofructokinase during malignant transformation in vivo and in vitro: transformation- and progression-linked discriminants of malignancy. Cancer Res. 45, 2993–3001 [PubMed] [Google Scholar]
- 51.Richardson A. D., Yang C., Osterman A., Smith J. W. 2008. Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res. Treat. 110, 297–307 10.1007/s10549-007-9732-3 (doi:10.1007/s10549-007-9732-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warburg O., Wind F., Negelein E. 1927. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 10.1085/jgp.8.6.519 (doi:10.1085/jgp.8.6.519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinhouse S. 1976. The Warburg hypothesis fifty years later. J. Cancer Res. Clin. Oncol. 87, 115–126 [DOI] [PubMed] [Google Scholar]
- 54.DeBerardinis R. J., Lum J. L., Hatzivassiliou G., Thompson C. B. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 10.1016/j.cmet.2007.10.002 (doi:10.1016/j.cmet.2007.10.002) [DOI] [PubMed] [Google Scholar]
- 55.Westerhoff H. V., Kell D. B. 1987. Matrix method for determining steps most rate-limiting to metabolic fluxes in biotechnologicai processes. Biotechnol. Bioeng. 30, 101–107 10.1002/bit.260300115 (doi:10.1002/bit.260300115) [DOI] [PubMed] [Google Scholar]
- 56.Bakker B. M., Westerhoff H. V., Opperdoes F. R., Michels P. A. M. 2000. Metabolic control analysis of glycolysis in trypanosomes as an approach to improve selectivity and effectiveness of drugs. Mol. Biochem. Parasitol. 106, 1–10 10.1016/S0166-6851(99)00197-8 (doi:10.1016/S0166-6851(99)00197-8) [DOI] [PubMed] [Google Scholar]
- 57.Djvorzhinski D., Thalasaila A., Thomas P. E., Nelson D., Li H., White E., Dipaola R. S. 2004. A novel proteomic coculture model of prostate cancer cell growth. Proteomics 4, 3268–3275 [DOI] [PubMed] [Google Scholar]
- 58.DiPaola R. S., et al. 2008. Therapeutic starvation and autophagy in prostate cancer:a new paradigm for targeting metabolism in cancer therapy. The Prostate 68, 1743–1752 10.1002/pros.20837 (doi:10.1002/pros.20837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hornberg J. J., Westerhoff H. V. 2006. Oncogenes are to lose control on signaling following mutation. Should we aim off target? Mol. Biotechnol. 34, 109–116 10.1385/MB:34:2:109 (doi:10.1385/MB:34:2:109) [DOI] [PubMed] [Google Scholar]
- 60.Kitano H. 2002. Systems biology: a brief overview. Science 295, 1662–1664 10.1126/science.1069492 (doi:10.1126/science.1069492) [DOI] [PubMed] [Google Scholar]
- 61.Kitano H. 2002. Computational systems biology. Nature 420, 206–210 10.1038/nature01254 (doi:10.1038/nature01254) [DOI] [PubMed] [Google Scholar]
- 62.Assmus H. E., Herwig R., Cho K. H., Wolkenhauer O. 2006. Dynamics of biological systems: role of systems biology in medical research. Expert Rev. Mol. Diagnostics 6, 891–902 10.1586/14737159.6.6.891 (doi:10.1586/14737159.6.6.891) [DOI] [PubMed] [Google Scholar]
- 63.Arnaud C. H. 2006. Systems biology's clinical future. Chem. Eng. News 84, 17–26 [Google Scholar]
- 64.Rohwer J. M., Hanekom A. J., Crous C., Snoep J. L., Hofmeyr J. H. 2006. Evaluation of a simplified generic bi-substrate rate equation for computational systems biology. Syst. Biol. (Stevenage) 153, 338–341 [DOI] [PubMed] [Google Scholar]