

Abstract
Alzheimer's disease (AD) is an age-related neurodegenerative disease and the most common form of dementia. AD is pathologically characterized by the deposition of pathogenic Aβ peptides that are derived from larger integral membrane proteins, termed β-amyloid precursor proteins (APPs). In an attempt to understand the function of APP, in vitro studies have focused on the identification of interacting proteins. To investigate the APP in vivo interactome in an unbiased manner, we generated mice that harbor a mouse prion protein promoter-driven cDNA encoding human APP-695 fused to a C-terminal affinity tag. Using this tag, we prepared mild detergent lysates from transgenic mouse brain cortical membrane preparations and isolated a number of previously identified APP-interacting proteins. In addition to these factors, mass spectrometric analysis revealed the presence of NEEP21 as a novel interacting protein. We now report that NEEP21 profoundly affects the processing of APP and Aβ production. Thus, this study demonstrates that using proteomic methods on our transgenic model can uncover important in vivo APP-interacting proteins that will provide insights into the biology of APP.
Introduction
The principal pathological hallmark of AD is the presence of extracellular deposits of ∼4 kDa Aβ peptides in senile plaques. Aβ is liberated from type I integral membrane proteins, termed β-amyloid precursor proteins (APPs), by the concerted action of β-secretase (BACE1) and γ-secretase (for review, see Selkoe, 2002). The function of APP and protein family members APLP1 and APLP2 remains to be established, but studies have revealed that mice with genetic deletions of APLP2 together with APP or APLP1 display defects in cortical organization due to alterations in neuronal cell migration (von Koch et al., 1997; Heber et al., 2000; Herms et al., 2004). Moreover, mice lacking APLP2 and APP die immediately after birth and exhibit deficits in synaptic transmission at neuromuscular junctions (Wang et al., 2009).
From studies of APP trafficking and metabolism, the following pathways have emerged: in the first, a fraction of APP molecules residing on the cell surface are processed by ADAM/TACE “sheddases” N-terminal to the ectodomain-transmembrane domain to generate an 83 aa membrane-tethered stub, termed α-CTF. α-CTF is subsequently processed within the lipid bilayer by γ-secretase to generate ∼23–25 aa “p3” peptides; alternatively, APP molecules that are subject to endocytosis are proteolyzed by BACE1 at a site further N-terminal to the membrane domain to generate a 99 aa membrane-tethered stub, termed β-CTF. β-CTF is subsequently processed by γ-secretase to generate ∼34–42 aa Aβ peptides. The accumulation of Aβ within the brain is hypothesized to be the causative agent in Alzheimer's disease (Walsh and Selkoe, 2004).
APP has been reported to interact with a myriad of proteins (Perreau et al., 2010), but with very few exceptions, these interactions have not been confirmed in vivo. To assess the native APP interactome in brain, we generated mice expressing human APP-695 fused to a C-terminal affinity tag (APP-AT). The addition of an affinity tag allowed us to avoid the use of antibodies targeted toward the intracellular C-terminal tail of APP, a region to which a number of reported interacting proteins bind (King and Turner, 2004). Purification of APP-AT protein complexes and identification of the constituents by mass spectrometry revealed several previously reported APP-interacting proteins as well as proteins involved in synaptic maintenance. Additionally, we report the identification of novel APP binding protein, NEEP21, a neuronally expressed single-pass transmembrane protein resident in endocytic vesicles and the Golgi (Sabéran-Djoneidi et al., 1998). In cultured neurons, NEEP21 is mainly localized to the somatodendritic compartment and can modulate targeting of L1/Ng-CAM to the axonal compartment (Yap et al., 2008) NEEP21 has been reported to affect glutamate receptor recycling (Alberi et al., 2005) through an interaction with GRIP1 (Steiner et al., 2005).
Here, we show that APP can coimmunoprecipitate NEEP21 from nontransgenic mouse brain and from mammalian cells stably coexpressing both proteins. Coexpression of NEEP21 with APP profoundly affected the generation of β-CTF with a concomitant decrease in Aβ levels. Thus, this study demonstrates that our transgenic model can uncover important in vivo APP-interacting proteins that will contribute to our understanding of APP processing in in vivo settings.
Materials and Methods
Antibodies.
Rabbit polyclonal antiserum Ctm1 was raised against a synthetic peptide corresponding to the C-terminal 15 aa of APP followed by the c-Myc epitope (MEQKLISEEDLN). BACE1 monoclonal antibody 3D5 was a kind gift from Robert Vassar (Northwestern University, Chicago, IL). APP antibody 369, raised against the entire intracellular C terminus of APP (Buxbaum et al., 1990), was a kind gift from Sam Gandy (Mount Sinal School of Medicine, New York, NY). Monoclonal antibody P2-1 recognizes a disulfide-dependent tertiary epitope in the N-terminal region of APP (Van Nostrand et al., 1989). Fe65 antibody was a kind gift from Qubai Hu (University of Washington, Seattle, WA) (Hu et al., 2005). Monoclonal antibody 26d6 was raised against the first 12 aa of Aβ (Kang et al., 2000); Nsg1 (NEEP21) and anti-His antibodies were purchased from GenScript.
Plasmids and cell culture.
Generation of myc-tagged APP and APPswe has been described (Lo et al., 1994). Plasmid pAPPswe (Lo et al., 1994) encodes Myc epitope-tagged human APP695 that harbors the Swedish FAD-specific amino acid substitutions (K595N and M596L). To generate affinity tagged NEEP21-his, the open reading frame of NEEP21 was subcloned from the pSport6 plasmid (American Type Culture Collection) containing the full mRNA sequence for Nsg1 and inserted into the pAG3-myc-his vector. pSuper vector for expression of shRNA was a gift from Reuvan Agami (Netherlands Cancer Institute, Amsterdam, The Netherlands). Human embryonic kidney (HEK293) cells and mouse neuroblastoma cells (N2a) were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen) and kept in a humidified chamber at 5% CO2.
Generation of affinity-tagged APP cDNA.
Original tandem affinity purification (TAP) vector was kindly supplied by Bertrand Seraphin (Institute of Genetics and Molecular and Cellular Biology, Illkrich, France). To generate APP-AT, TAP sequence was amplified from the vector by PCR using primers containing appropriate restriction enzyme sites and ligated into an open pAG3 vector containing APP without a stop codon. Next, site-directed mutagenesis was used to generate a silent mutation in the APP sequence to remove an XhoI site which was needed in subsequent cloning. To generate moPrP.XhoI, APP-AT, cDNAs were amplified by PCR using Pfu polymerase (Stratagene) and primers containing flanking XhoI sites. Vector was linearized with XhoI and purified, and APP PCR products were digested with XhoI, purified and ligated to open moPrP.XhoI vector. Bacterial clones containing moPrP.XhoI with APP inserts were selected using 32P-labeled probes generated from APP by hybridization and exposure to film. All constructs were confirmed by sequencing.
Transfections.
Mouse N2a neuroblastoma cells were transiently transfected using Lipofectamine2000 reagent (Invitrogen). To generate cell lines that stably express NEEP21 and APPswe, N2a cells were transfected with pCB6-APPswe-myc and pAG3 NEEP21-his which contain resistance markers for G418 and Zeocin, respectively. Stable transfectants were selected in medium containing 0.8 mg/ml G418 (Geneticin; Invitrogen) and 0.4 mg/ml Zeocin (Invitrogen) and maintained as stable “pools” in 0.4 mg/ml G418 and 0.2 mg/ml Zeocin. HEK293 cells stably expressing human APP or APP-AT were generated by transfection with pAG3 containing the appropriate open reading frame followed by selection using 0.8 mg/ml G418. Clones were selected that expressed each construct at varying levels and maintained in 0.4 mg/ml G418.
Coimmunoprecipitation.
Cells were lysed in coimmunoprecipitatin buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% NP-40, Protease Inhibitor Cocktail) and insoluble material was removed by centrifugation. Lysates were precleared with protein G agarose and incubated with epitope-specific antibodies for 16 h at 4°C after which protein G agarose beads were added for 1 h. Beads were collected by centrifugation and washed three times with coimmunoprecipitation (co-IP) buffer, and transferred to a new tube for a final wash before analysis by Western blot.
Metabolic labeling.
For metabolic labeling, N2a cells were starved for 60 min in methionine-free DMEM (Invitrogen) and then labeled with 250 μCi/ml [35S]methionine (DuPont NEN) in methionine-free DMEM supplemented with 1% dialyzed fetal bovine serum (Invitrogen) for 10 min or 4 h. At the end of the labeling period, cells were washed in PBS and then lysed in immunoprecipitation (IP) buffer containing detergents and protease inhibitors (Sisodia et al., 1990). To immunoprecipitate full-length APP, we used antibody Ctm1. A mAb, 26d6, that recognizes residues 1–17 of human Aβ, was used to immunoprecipitate Aβ from culture medium as well as β-CTF and full-length APP from cell lysates. Quantification of immunoprecipitated polypeptides was performed by phosphorimaging using Molecular Dynamics software.
Statistical analysis.
Data are expressed as mean values ± SEM (m ± SEM). Student's t test or ANOVA was performed for all comparisons of quantitative data. Values of p < 0.05 were used as the criterion for statistical significance.
Cell lysis and Western blotting.
Monolayers were washed twice with ice-cold PBS followed by incubation in lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% NP-40, 0.5% Triton X-100) with Protease Inhibitor Cocktail (P1860, from Sigma) on ice for 5 min. Cells were scraped into microcentrifuge tubes and spun briefly to remove nuclei and cell debris. Postnuclear supernatants were analyzed for protein concentration using the BCA method (Pierce). For Western blotting, lysates were run on tris-glycine or tris-tricine SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad). Membranes were blocked in PBST (10 mm phosphate, 150 mm NaCl, 0.2% Tween20) with 5% milk for 1 h at room temperature (RT) with rocking. Membranes were then transferred to PBST/1% milk containing the appropriate primary antibody for 3 h at RT or overnight at 4°C with rocking followed by washing three times in PBST and incubation in the appropriate HRP-conjugated secondary antibody for 1 h at RT. After subsequent washing, immunoreactivity was visualized using Western Lightning ECL substrate (GE Healthcare).
Generation and screening of transgenic mice.
All animal handling was conducted in accordance with institutional and National Institutes of Health guidelines. moPrP.Xho vector has been previously described (Borchelt et al., 1996). APP-AT cDNA was inserted in XhoI-digested moPrP.Xho and transformed into XL10-gold competent cells (Stratagene). Amplified vector was purified by alkali lysis and double CsCl purification (Sambrook et al., 1989). The moPrP.XhoAPP-AT vector was linearized and the pBluescript backbone excised by digestion with NotI. Fertilized eggs for injection were generated using superovulated C57BL/6J mice. Pronuclear injection of the purified APP-AT (4–5 ng/μl) was performed and the injected eggs were transferred into pseudopregnant females to develop. Tail samples were taken from the pups 2 weeks after birth and genotyped. DNA was extracted by incubation in 500 μl of TNES extraction buffer (50 mm Tris, pH 8.0, 100 mm EDTA, 400 mm NaCl, 0.5% SDS) with 320 μg of proteinase K at 65°C for 3 h followed by addition of 150 μl of 6 m NaCl, pelleting of debris and precipitation with ethanol. Primers annealing to ORFs of APP, PrP, and an upstream region of PrP were used in a 3-primer PCR to amplify PrP (all mice) and PrP-APP (Tg mice) products.
Purification of APP-AT protein complexes from brain.
Mice were deeply anesthetized with a mixture of ketamine and xylazine, and perfused transcardially with ice-cold PBS for 90 s after which the brains were quickly removed, the cortex separated from the rest of the brain and both samples were placed in prechilled tubes and snap-frozen in liquid nitrogen until use. All subsequent steps were performed on ice or at 4°C. Each brain was treated separately before being combined for final gel electrophoresis to avoid the possibility of cross contamination of incorrectly genotyped mice. To isolate membrane preparations, cortices were weighed and 9 volumes of homogenization buffer [0.32 m sucrose, 10 mm HEPES, pH 7.4, 1 mm EDTA, 1 mm NaFl, 2 mm Na3VO4, Protease Inhibitor Cocktail (4-(2-aminoethyl)benzenesulfonyl fluoride, pepstatin A, E-64, bestain, leupeptin, and aprotinin; Sigma) and 100 mm phenylmethylsulfonyl fluoride (PMSF) (Sigma)] were added followed by homogenization using a Teflon-glass homogenizer for 40 strokes on ice. The crude homogenate was spun at 800 × g for 10 min. The supernatant was centrifuged at 14,000 × g for 20 min and the pellet (P2) was resuspended in purification buffer (PBS with 0.5% Igepal, 1 mm NaFl, 2 mm Na3VO4 and Protease Inhibitor Cocktail). Samples were solubilized for 1 h and insoluble material was removed by centrifugation at 10,000 × g for 10 min. The detergent-soluble membrane fraction was used for subsequent purification immediately. To control for variations in sample purification from day to day, 3 non-Tg and 3 APP-AT mice were processed in parallel at each purification.
Purification of APP-AT.
Soluble membrane preparations were incubated with rabbit IgG-coated magnetic beads (Dynabeads M-270, Invitrogen) for 2 h, which preliminary experiments had shown was necessary and sufficient for complete capture of APP-AT complexes. Beads were collected and washed five times for 5 min each with purification buffer and complexes were eluted using 0.1 m citrate, pH 3.1. Purifications of non-Tg and APP-AT samples were always performed in parallel. A total of 10% of each preparation was assayed by silver stain to assure purification before combining 10 preparations each for subsequent proteomic analysis.
Proteomic analysis of purified protein.
Purified proteins were run on 12% acrylamide tris-glycine SDS-PAGE with control (non-Tg) and experimental (APP-AT) samples run on separate gels to avoid cross-contamination. Protein bands were visualized by staining with Coomassie. Each lane was cut into 10 slices. Excised gel plugs were cut into ∼1 mm × 1 mm × 1 mm cubes, in-gel digested with trypsin, and extracted from gel matrix with acetonitrile and 5% formic acid (Shevchenko et al., 2006). Recovered peptides were subjected to LC-MS/MS analysis on an LTQ ion trap mass spectrometer (ThermoFisher Scientific) under conditions described by Waridel et al. (2007), and acquired MS/MS spectra were searched against the latest version of the Human IPI database (Kersey et al., 2004) by MASCOT v. 2.2 software (Matrix Science Ltd.) installed on a local server. Protein hits were considered confident if at least two MS/MS matched the corresponding database sequences with peptide ion scores exceeding the confidence threshold suggested by MASCOT (p < 0.05). For each identified protein, the number of matched peptides and of MS/MS spectra were exported from MASCOT output to Excel spreadsheets using a script developed in-house, which further created a nonredundant list of protein hits detected in all analyzed gel slices of the same IP experiment. If the same protein was sequenced in several bands, only the analysis that produced the highest number of matched peptides and spectra was reported. From the list of protein IDs retrieved from APP-AT IP, we manually subtracted proteins identified from control nontransgenic IP run in parallel.
RNAi knockdown.
siRNA duplex ON-TARGETplus smart-pools were obtained from Dharmacon Inc. (ThermoFisher Scientific) targeting four different regions of mouse Nsg1 open reading frame. Target sequences were AUAUGAACCUGAUCGCAAA, GGAAUAAUUUCGCAGAGAA, CAGAUAAGGUCGUGGUGAA, GAAGUCAGCUUAGCGAGCA, and were mixed in equimolar ratios for siRNA treatment. Control siRNA consisted of ON-TARGETplus siRNA control oligo or AllStars control siRNA from Qiagen. Both control siRNAs are proprietary sequences. shRNA expression was performed using pSuper vector (Brummelkamp et al., 2002) with target sequences inserted downstream of the H1 promoter. Two target sequences were cotransfected with a hygromycin resistance vector. Stable pools were selected using hygromycin at 400 μg/ml and maintained at 200 μg/ml. Forward target sequences were GATCCCCAGTGTACAAGTATGACCGCTTCAAGAGAGCGGTCATACTTGTACACTTTTTTA and GATCCCCCACCCAGTGCATCCCAGAATTCAAGAGATTCTGGGATGCACTGGGTGTTTTTA.
ELISA.
ELISA analysis was performed on cell culture medium according to the manufacturer's instructions using a human Aβ1-40 colorimetric immunoassay kit from Invitrogen. Culture medium was diluted sufficiently to ensure that values were within the linear range of Aβ standards.
Results
Generation and expression of APP-AT
To facilitate the purification of APP-containing protein complexes from transgenic mice, we cloned sequences encoding an affinity tag containing a double protein A sequence as a portion of a tandem affinity purification tag downstream of the C-terminal domain of the human APP695 sequence (see Materials and Methods). To determine whether APP-AT was processed appropriately, cDNA encoding APP-AT was stably expressed in HEK293 cells, and Western blot analysis of cell lysates revealed full-length and C-terminal fragments of APP-AT that were immunoreactive to both anti-APP antibodies as well as rabbit IgG, that binds to the protein A sequence present in the affinity tag (Fig. 1 A). In parallel, we observed soluble APP derivatives in the culture medium from these cells (Fig. 1 B). To test whether APP-AT is a substrate for γ-secretase, HEK293 cells stably expressing APP or APP-AT were treated with the γ-secretase inhibitor L-685,458 (Shearman et al., 2000). In both wild-type and APP-AT-expressing cells, γ-secretase inhibitor treatment led to an accumulation of CTF levels (Fig. 1 C). Additionally, Aβ peptides could be precipitated from cell culture medium from cells expressing APP-AT, although at very low levels consistent with the low efficiency of wild-type APP as a substrate for BACE1 (Fig. 1 D) (Sisodia et al., 1990; Cai et al., 1993).
Figure 1.

Expression of APP-AT in cell culture. A, HEK293 cells stably expressing low (L), medium (M), or high (H) levels of APP-AT were generated and assayed for production of C-terminal fragments. The parental cell line (0) was used as a control. Lysates were probed with either C-terminal APP antibody 369 or rabbit IgG, which binds to the double protein-A tag on APP-AT. B, Soluble APP derivatives were also detected in cells using antibody 22C11 and P2-1, which binds to a conformation-specific disulfide-dependent epitope in the N-terminal region of APP. C, γ-Secretase inhibition leads to accumulation of CTFs in cells expressing wild-type APP or APP-AT. The APP-AT intracellular domain (AICD) is not detectable after inhibition. D, Culture medium from cells expressing wild-type APP or APP-AT was probed for Aβ using antibody 26d6.
To generate transgenic mice expressing APP-AT, we cloned cDNA encoding APP-AT into the MoPrP.Xho vector to allow widespread expression of the transgene-encoded polypeptide in the nervous system (Borchelt et al., 1996). Three founder lines were examined and we observed no differences in body weight, behavior or health between non-Tg and APP-AT mice (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Western blot analysis revealed expression of APP-AT in brain and heart with very low levels in lung (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). We chose to examine one line for the purification studies based on expression of APP-AT at near endogenous APP levels within the brain (Fig. 2 A; supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Importantly, the pattern of CTFs determined by Western blot analysis of brains from non-Tg and APP-AT mice indicated that APP-AT was processed in a manner similar to endogenous APP (Fig. 2 B).
Figure 2.

Expression of APP-AT in transgenic mice. A, Western blot of brain homogenate from non-Tg and transgenic APP-AT mice. APP-AT is expressed at approximately endogenous levels. B, Brain homogenates were probed with antibody 369 for the presence of CTF and CTF-AT, containing the affinity tag.
Purification and analysis of APP in vivo interactome
To purify APP-AT protein complexes, membrane preparations were generated from the cortex of adult mice between 90 and 120 d of age. Membrane preparations allowed for reduced sample complexity during purification and mass spectrometry. Detergent solubilized membrane proteins from nontransgenic or APP-AT cortex were incubated with magnetic beads to which rabbit IgG was immobilized. After 2 h, beads were collected and washed to remove nonspecifically bound proteins and the APP-AT protein complexes were eluted in citric acid, pH 3.1, leaving IgG chains bound to the beads. Analysis of affinity-purified proteins revealed that nearly 100% of APP-AT was bound to the beads while endogenous APP from non-Tg mice was found only in the unbound fraction (Fig. 3 A). For mass spectrometric analysis, 10 individual purifications each from non-Tg and APP-AT were performed in parallel, representing 5 males and 5 females mice from each group. Combined preparations were submitted to SDS-PAGE and Coomassie staining. Non-Tg and APP-AT purifications were run on separate gels to avoid any possibility of cross-contamination of the two samples (Fig. 3 B). Coomassie stained gels were cut into pieces, proteins digested with trypsin in situ, and the digested peptides analyzed by liquid chromatography/MS2.
Figure 3.

Purification of APP-AT from brain. A, Detergent-soluble cortical membrane fractions from non-Tg or APP-AT brains were incubated with magnetic beads bound with rabbit IgG. After 2 h, beads were washed and protein complexes eluted with citrate, pH 3.1. APP-AT was undetectable in the unbound fractions, indicating nearly 100% capture of APP-AT complexes. Input, Unbound, and Wash lanes represent 1% of the total respective volumes. Eluate represents 50% of the eluate volume. B, Purifications from individual brains of non-Tg and APP-AT mice were performed in parallel and pooled for subsequent analysis by SDS-PAGE and Coomassie stain. Each lane represents 10 individual purifications. C, Protein identification by MS/MS of tryptic digests of gel-extracted proteins yielded 101 total proteins exceeding MASCOT confidence threshold.
Comparison of the identified proteins from each sample revealed 99 total proteins in the APP-AT sample while 57 identifications were made in the non-Tg sample. The two samples had 55 proteins in common indicating nonspecific binders to the affinity matrix while 44 proteins were-specific to APP-AT and only 2 proteins were detected exclusively in the non-Tg sample (Fig. 3 C). Of the APP-AT-specific proteins, several have been previously characterized as APP-interacting proteins (Table 1), including Fe65, thus validating the approach of using our mice to identify in vivo interactors with APP. All protein identifications specific to the APP-AT sample are shown in Table 2. The in vivo APP interactome displays a preference for synaptic proteins, consistent with reported localization and processing of APP at presynaptic and postsynaptic sites (Shigematsu et al., 1992; Buxbaum et al., 1998; Wei et al., 2010) and, in adult mice, with a role for APP in synaptic maintenance and genesis (Lazarov et al., 2002; Wang et al., 2009; Lee et al., 2010). These data highlight the importance of proteomic exploration in an in vivo setting in which cellular contact, differentiation and polarity are intact.
Table 1.
Previously characterized APP-interacting proteins
| Protein interaction | Gene | Reference |
|---|---|---|
| APP | App | Soba et al. (2005) |
| APLP2 | Aplp2 | Soba et al. (2005) |
| APLP1 | Aplp1 | Soba et al. (2005) |
| Fe65 | Apbb1 | Zambrano et al. (1997) |
| Fe65L1 | Apbb2 | Guénette et al. (1996) |
| 78 kDa glucose-regulated protein | Hspa5 | Yang et al. (1998) |
| BRI2 | Itm2b | Fotinopoulou et al. (2005) |
| BRI3 | Itm2c | Matsuda et al. (2009) |
| Flotillin-2 | Flot2 | Schneider et al. (2008) |
Table 2.
Protein identifications specific to the APP-AT sample
| APP-interacting proteins | Gene |
|---|---|
| APP family | |
| APP | App |
| APLP2 | Aplp2 |
| APLP1 | APlp1 |
| Cell adhesion and signal transduction | |
| Contactin-associated protein 1 | Cntnap1 |
| Syndecan-3 | Sdc3 |
| Fe65 | Apbb1 |
| Fe65L1 | Apbb2 |
| Calcium/calmodulin-dependent protein kinase type II β chain | Camk2b |
| 14-3-3 protein ϵ | Ywhae |
| Synaptic regulation | |
| Synapsin-2 | Syn2 |
| Syp protein | Syp |
| Synaptic vesicle glycoprotein 2A | Sv2a |
| Neuroplastin | Nptn |
| Protein bassoon | Bsn |
| Synaptotagmin XI | Syt11 |
| Neurexin-1-α | Nrxn1 |
| Neurexin 2 | Nrxn2 |
| Membrane traffic and organization | |
| Flotillin-2 | Flot2 |
| NEEP21—Neuron-specific protein family member 1 | Nsg1 |
| Ras-related protein Rab-3B | Rab3b |
| Axon growth | |
| Lipid phosphate phosphatase-related protein type 3 | BC005764 |
| Neuromodulin | Gap43 |
| Cytoskeleton | |
| α-Actinin-4 | Actn4 |
| α-Internexin | Ina |
| Catenin α2 | Ctnna2 |
| Protein folding/binding | |
| 78 kDa glucose-regulated protein | Hspa5 |
| Clusterin | Clu |
| Transporters/receptors | |
| Plasma membrane calcium-transporting ATPase 2 | Atp2b2 |
| Sodium/potassium-transporting ATPase subunit α1 | Atp1a1 |
| V-type proton ATPase 116 kDa subunit a isoform 1 | Atp6v0a1 |
| V-type proton ATPase subunit C 1 | Atp6v1c1 |
| GABA type B receptor subunit 2 | Gabbr2 |
| Mitochondrial glutamate carrier 1 | Slc25a22 |
| ADP/ATP translocase 2 | Slc25a5 |
| ADP/ATP translocase 1 | Slc25a4 |
| Excitatory amino acid transporter 1 | Slc1a3 |
| Other | |
| Mannan-binding lectin serine protease 1 | Masp1 |
| Isoform 1 of elongation factor Tu, mitochondrial | Tufm |
| S-phase kinase-associated protein 1 | Skp1a |
| Trifunctional enzyme subunit α, mitochondrial | Hadha |
| Phosphoglycerate mutase family member 5 | Pgam5 |
| BRI2 | Itm2b |
| BRI3 | Itm2c |
| BTB/POZ domain-containing protein KCTD12 | Kctd12 |
As reported by others, APP was found to interact with the homologues APLP1 and APLP2 (Bai et al., 2008), probably forming dimers in cis- and/or trans-interaction structures (Soba et al., 2005; Wang et al., 2009). Moreover, the use of human APP sequence in the APP-AT mice allowed for the detection of endogenous mouse APP and identified it as a specifically copurified protein with APP-AT. Although human and mouse APP are 98% homologous, divergent sequence within the tryptic peptide (K)THTHIVIPYR allowed for the detection of endogenous APP as a coprecipitating protein. Implications for the functional relevance of identified proteins are discussed below.
Identification of novel APP-interacting protein: NEEP21
To further explore the functional significance of the newly identified proteins identified in the proteomic dataset, we chose to focus our studies on NEEP21 (gene name: Nsg1), a protein previously reported to reside in Golgi and endosomal compartments within neurons (Sabéran-Djoneidi et al., 1998) and which is involved in the recycling and sorting of multiple cargoes including glutamate receptors and neuron-glia cell adhesion molecule (L1/NgCAM) (Steiner et al., 2002, 2005; Debaigt et al., 2004; Alberi et al., 2005; Yap et al., 2008). APP is also present within the secretory pathway and recycling endosomes (Koo and Squazzo, 1994), undergoes sorting to the axonal compartment of cultured neurons (Caporaso et al., 1994) and is transported to nerve terminals via the fast component of anterograde transport both in peripheral (Koo et al., 1990; Sisodia et al., 1993) and central neurons (Buxbaum et al., 1998; Lazarov et al., 2002). Thus, NEEP21 represented a promising candidate for characterization of a novel APP-interacting protein with possible functional significance in amyloidogenic processing pathways. To confirm the interaction of APP and NEEP21, coimmunoprecipitation experiments were performed on membrane preparations from nontransgenic mouse brain. The results show that pull-down with anti-APP antibodies, but not control IgG, resulted in NEEP21 coprecipitation (Fig. 4 A). To confirm these results in a heterologous expression system, neuroblastoma (N2a) cells stably expressing APP and NEEP21 bearing a myc-his tag were submitted to coimmunoprecipitation with a human APP-specific N-terminal antibody, P2-1 (Van Nostrand et al., 1989) or Ab369, raised against the entire cytoplasmic domain of APP. NEEP21 was specifically coprecipitated using either antibody (Fig. 4 A, middle and bottom) suggesting binding to full-length APP and possibly to CTFs, as well.
Figure 4.

A, Coimmunoprecipitation of APP and NEEP21 was performed on brain homogenate from nontransgenic mice. IP with a C-terminal antibody was able to copurify NEEP21. In cell lysates from N2a cells stably expressing APP and NEEP21-myc-his, copurification was observed using both an N-terminal (P2-1) and C-terminal (369) antibody. B, Coexpression of Fe65 with APP and NEEP21 did not increase the efficiency of co-IP relative to coexpression of GFP with APP and NEEP21. C, Using N2a cells transiently expressing NEEP21-myc-his and Fe65, we also failed to observe co-IP, although an APP-Fe65 complex was detectable. Text to the right of each image indicates primary antibody used for Western blot.
NEEP21 contains a proline-rich N-terminal region, and the human form of a related protein, calcyon, is reported to bind group II WW domains through its proline rich domain through the canonical PPLP binding motif (Muthusamy et al., 2009). Because the known APP-binding protein Fe65 contains a WW domain, we tested whether coexpression of Fe65 with APP and NEEP21 would increase the efficiency of APP-NEEP21 coimmunoprecipitation. N2a cells were transiently transfected with cDNA encoding APP, NEEP21 and either green fluorescent protein (GFP) or Fe65 and detergent lysates subjected to coimmunoprecipitation analysis. Expression of Fe65 did not increase the amount of NEEP21 that precipitated with APP suggesting that the NEEP21/APP interaction is independent of Fe65 (Fig. 4 B). To determine whether NEEP21 can bind to Fe65 directly, N2a cells were transiently transfected with cDNA encoding a his-tagged version of NEEP21 together with Fe65. Lysates immunoprecipitated with anti-his to pull down NEEP21 were probed with anti-Fe65, but no signal was detectable (Fig. 4 C, top) under conditions in which Fe65 was able to coimmunoprecipitate APP (Fig. 4 C, bottom). We interpret these data to suggest that APP binds to NEEP21 in a Fe65-independent manner.
Having established that APP and NEEP21 interact in brain and in cells that coexpress both proteins, we next sought to determine the functional consequence of this interaction on APP processing. N2a cells were transiently cotransfected with cDNA encoding APP along with either empty vector, a control plasmid encoding an unrelated membrane protein, influenza hemagglutinin (HA), or NEEP21. When wild-type APP was coexpressed with NEEP21, we observed a small decrease in levels of soluble APPα (Fig. 5 A). Because wild-type APP (wtAPP) is not processed very efficiently by BACE1 (Sisodia et al., 1990; Cai et al., 1993), thus making analysis of β-CTF and Aβ difficult to quantify, we chose to coexpress NEEP21 and APP harboring the familial Alzheimer's disease (FAD)-linked “Swedish” double mutation (APPswe) that is efficiently processed at the β-secretase site by BACE1 (Citron et al., 1992; Cai et al., 1993). In this case, coexpression of NEEP21 with APPswe led to a striking reduction in β-CTF in cell lysates, as well as soluble sAPPβ in the culture medium, while BACE1 levels were unchanged (Fig. 5 B). Quantitation of the ratio of β-CTF:α-CTF showed a reduction to 31.3 ± 6.2% compared with the β-CTF:α-CTF ratio in cells expressing either the vector or HA controls (Fig. 5 C). As steady-state analysis of APP-CTFs by Western blot analysis can be misleading because these polypeptides have differential half-lives in recycling and degradative compartments, we chose to examine the levels of newly generated β-CTF. Stable cell pools expressing NEEP21 and APPswe were metabolically labeled for 4 h with 35S-methionine. Detergent lysates and conditioned medium were immunoprecipitated with antibody 26D6 raised against the first 12 aa of Aβ, and hence, recognizes both Aβ and sAPPα in culture medium as well as full-length APP and β-CTF in cell lysates. As shown in Figure 5 D, lysates from the 4 h labeling experiments showed markedly reduced levels of β-CTF in cells that coexpress APPswe and NEEP21. Parallel analysis of immunoprecipitated Aβ peptides revealed a lowering of Aβ in cells coexpressing APPswe and NEEP21 to 60.5 ± 4.3% compared with cells expressing APPswe alone (Fig. 5 E,F). To confirm the specificity of the effects of NEEP21 on APP-CTF levels, we coexpressed APPswe and P19, a polypeptide that shares ∼64% homology to NEEP21. These studies failed to reveal any effect on reducing β-CTF levels, indicating that the observed effects of NEEP21 on APP metabolism are specific (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 5.

Effect of NEEP21 expression on APP processing. A, Wild-type APP was coexpressed with NEEP21 in N2a cells and probed for changes in processing to C-terminal fragments. A slight, but consistent decrease in sAPPα levels was observed. B, When NEEP21 was coexpressed with APPswe, a profound decrease in β-CTF was apparent with a slight decrease in α-CTF. The decrease in β-CTF was accompanied by a decrease in sAPPβ, while BACE1 levels were unchanged. C, The ratio of β-CTF:α-CTF was measured by densitometry. The decrease in β-CTF levels led to a decrease in the β-CTF:α-CTF ratio compared with controls. D, Immunoprecipitation of 4 h continuously labeled cell lysates using antibody 26D6, which recognizes the N-terminal region of β-CTF. E, Immunoprecipitation of Aβ from media of labeled cells indicated a concomitant reduction in Aβ production in NEEP21-expressing cells. F, Quantitation of Aβ IP phosphor imaging indicated a reduction of 39.5% in cells expressing NEEP21.
To test whether reduction of NEEP21 levels would affect APP CTF levels, N2a cells stably expressing shRNA targeted against NEEP21 were established. These cells were transiently transfected with wild-type APP or APPswe and CTF levels assessed by Western blot analysis. As shown in Figure 6 A, α-CTF levels were reduced in stable NEEP21 shRNA cells that transiently express either wild-type APP or APPswe. In contrast to α-CTF levels, β-CTF levels remained unchanged in cells expressing APP or APPswe. Because APP constructs are tagged with a C-terminal myc tag, the endogenous α-CTF was detected and showed a similar reduction to the α-CTF derived from the chimeric APP-myc protein. Quantitation of NEEP21 levels indicated that levels were reduced to 62% that of naive N2a cells. Thus, in the shRNA knockdown experiments, we observed effects on CTFs that were opposite to that seen in the overexpression studies. Whereas NEEP21 overexpression decreased the β-CTF:α-CTF ratio relative to control cells, reduction of NEEP21 increased this ratio by 43.5 ± 10.4% (Fig. 6 B). Interestingly, while overexpression exerted its effect mainly on β-CTF, reduction of NEEP21 affected primarily α-CTF. The differential effects of NEEP21 overexpression or silencing on APP-CTFs may be a reflection of alterations in sorting of APP or CTFs, and further studies will be necessary to clarify this issue. To examine whether acute reduction of NEEP21 levels would lead to changes on APP-CTFs, we treated cells stably expressing APPswe with NEEP21 siRNA. As shown earlier in the shRNA knockdown studies, we observed a reduction in levels of α-CTF without a change in β-CTF (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). To assess the effect of these changes on Aβ production, ELISA assays were performed on cell culture medium from “pools” of cells that stably overexpress NEEP21 and APPswe or cells treated with NEEP21 siRNAs. Coexpression of APPswe and NEEP21 led to a 56% reduction in Aβ levels relative to cells that coexpress the pAG3 vector or HA (Fig. 7 A), a result consistent with the observed reduction in β-CTF. In NEEP21 siRNA and shRNA knockdown cells, Aβ levels were also slightly reduced (Fig. 7 B), although this change did not reach statistical significance (n = 3, p = 0.052). To assess this effect in another cell type, human H4 neuroglioma cells stably expressing APP751 were treated with siRNA against NEEP21. The reduction in NEEP21 levels was modest in H4-APP751 cells (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material), but surprisingly, while we did not observe changes in APP-CTFs, Aβ levels were significantly reduced (supplemental Fig. S5C,D, available at www.jneurosci.org as supplemental material). These studies would suggest that the effects of NEEP21 knockdown on APP-CTF and Aβ levels are cell-type specific. Future studies will be required to fully understand the mechanism of NEEP21-dependent alterations in APP processing and metabolism, especially in polarized neuronal cell types and in vivo.
Figure 6.
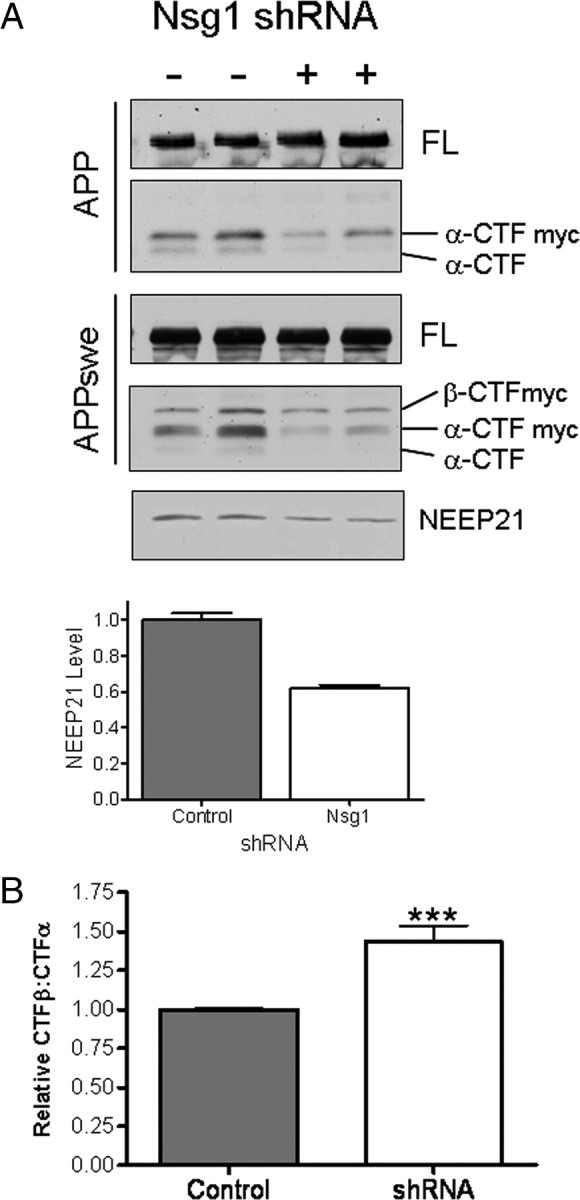
A, N2a cells stably expressing shRNA directed against NEEP21 were transfected with APP or APPswe. α-CTF levels decreased in NEEP21 knockdown cells relative to naive N2a cells. FL, Full-length APP. B, The reduction in α-CTF led to a reversal of β-CTF:α-CTF ratio compared with NEEP21 overexpression.
Figure 7.
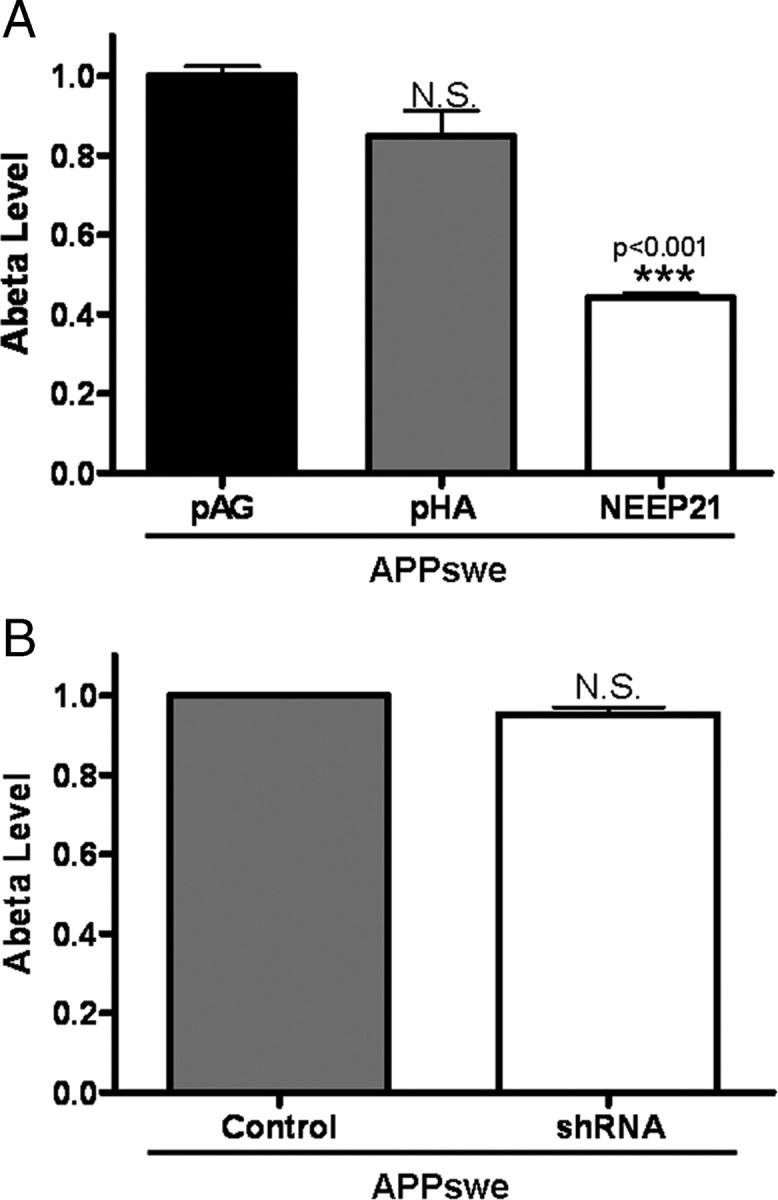
Aβ levels and NEEP21 expression. A, ELISA analysis revealed reduced Aβ levels in N2a culture medium from cells expressing APPswe and NEEP21 compared with HA or vector alone. B, N2a cells stably expressing control vector or vector containing NEEP21 shRNA were transiently transfected with APPswe and assessed for Aβ levels. No significant difference was observed by ELISA. For both A and B, n = 3.
Discussion
In an attempt to clarify the normal physiological function of APP, a number of experimental approaches have been used to identify interacting proteins and to determine their role(s) in modulating APP trafficking and processing (King and Turner, 2004). To these latter efforts, we have examined the native APP interactome in brain using a new transgenic mouse line that expresses human APP-695 fused to a C-terminal affinity tag. Notably, our chimeric APP leaves relatively unperturbed the cytoplasmic domain of APP to interact with its native binding partners. In this regard, earlier attempts to isolate APP interactors from nontransgenic mice failed to isolate Fe65, a finding the authors attribute to saturation of shared binding sites by Fe65 and C-terminal antibodies (Bai et al., 2008). In the present study, we offer several insights pertaining to the mouse brain interactome of APP and the effects of a novel interacting protein, termed NEEP21 on APP processing.
First, our purification strategy revealed several proteins that have been previously characterized as APP-interacting proteins. For example, we detected mouse APLP1 and APLP2, consistent with earlier in vitro studies and cross-linking experiments in mouse brain showing the presence of APP and APLP homo- and heterodimers (Soba et al., 2005; Bai et al., 2008; Dahms et al., 2010). Unclear at this point is the relative stoichiometry and spatial distribution of monomers and dimers, questions that remain an important question for future research. In addition to APP/APLP, we identified Fe65 and its homolog, Fe65L1 (McLoughlin and Miller, 2008). Interestingly, Fe65/Fe65L1 double knock-out mice exhibit a cortical dysplasia phenotype similar to that of APP/APLP1/APLP2 knock-out mice (Guénette et al., 2006). In addition to these well studied interactions, we also identified interactions with the Bri2 and Bri3 proteins as well as flotillin-2. Bri2 and Bri3 were reported to directly bind and modulate amyloidogenic processing of APP (Fotinopoulou et al., 2005; Matsuda et al., 2008), while flotillin-2 has been shown to mediate APP clustering in a cholesterol-dependent fashion (Schneider et al., 2008). To these interactors, we identified numerous proteins involved in regulation of synaptic function, consistent with reported anterograde trafficking of APP (Koo et al., 1990; Sisodia et al., 1993) to nerve terminals in vivo (Lazarov et al., 2002) and with reports that APP may function in the context of trans-synaptic adhesion (Wang et al., 2009). The presence of neurexin-1 and neurexin-2 are interesting and we would have predicted that the known neurexin-binding partner, mint/X11-would be copurified with APP. While the latter was not identified in the mass spectra, it is possible that Mint1 may act as a scaffold between APP and neurexins as Mint1 has been shown to bind to APP via a phosphotyrosine binding domain (Mueller et al., 2000) and to neurexins via PDZ domains (Biederer and Südhof, 2000). Further studies are certainly warranted to explore these important issues.
Second, we chose to focus our efforts on NEEP21, a protein previously shown to be present within Golgi and endosomal compartments of neurons (Sabéran-Djoneidi et al., 1998), and synaptic membranes (Utvik et al., 2009), very similar to the normal distribution of APP (Koo et al., 1990). NEEP21 has also been shown to modulate axonal trafficking and polarization of L1/NgCAM (Yap et al., 2008), is localized to endosomes in differentiated PC12 cells where it can modulate transferrin recycling as well as GluR2 recycling in neuronal cell culture (Steiner et al., 2002, 2005), and it may also play a role in AMPA receptor cycling during synaptic plasticity (Alberi et al., 2005). We now show that NEEP21 can be coimmunoprecipitated with APP from mouse brain and stably transfected mammalian cells. Studies to identify the APP domains that are responsible for binding to NEEP21 are ongoing. Paradoxically, both overexpression of NEEP21, or siRNA-mediated downregulation of NEEP21 levels in mammalian cells lead to a reduction, albeit modest in the case of NEEP21 knockdown, in secreted levels of Aβ peptides. While there is no satisfactory explanation for this apparent discrepancy, it is conceivable that overexpressing NEEP21 may lead to a dominant-negative scenario that perturbs the stoichiometry of complexes that contain this molecule. In this regard, gel filtration studies indicate that NEEP21 exists in high molecular weight complexes (Steiner et al., 2005) and these may be disrupted by titration effects of overexpressed NEEP21. It is thus intriguing that the levels of β-CTF, the penultimate precursor of Aβ were decreased in cells that overexpress NEEP21, but the levels of this C-terminal derivative was unchanged in cells in which NEEP21 levels were reduced. A firm understanding of the differential effects of NEEP21 overexpression or silencing on APP-CTFs will require additional studies.
In summary, we have used a novel transgenic mouse model in conjunction with proteomics to investigate the in vivo interactome of APP and have identified a novel interacting protein, NEEP21, that has profound effects on amyloidogenic processing of the precursor. The present studies serve as a foundation for future investigations focused on the role of NEEP21 in APP trafficking and metabolism in cultured neurons and in vivo, studies that we anticipate will offer important insights into potential therapeutic opportunities for AD.
Footnotes
This work was funded in part by the American Health Assistance Foundation (E.M.N.) and the Chicago Biomedical Consortium (S.S.S.). We thank Anna Shevchenko (Max Planck Institute, Dresden, Germany) for assistance on mass spectrometry.
References
- Alberi S, Boda B, Steiner P, Nikonenko I, Hirling H, Muller D. The endosomal protein NEEP21 regulates AMPA receptor-mediated synaptic transmission and plasticity in the hippocampus. Mol Cell Neurosci. 2005;29:313–319. doi: 10.1016/j.mcn.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Bai Y, Markham K, Chen F, Weerasekera R, Watts J, Horne P, Wakutani Y, Bagshaw R, Mathews PM, Fraser PE, Westaway D, St George-Hyslop P, Schmitt-Ulms G. The in vivo brain interactome of the amyloid precursor protein. Mol Cell Proteomics. 2008;7:15–34. doi: 10.1074/mcp.M700077-MCP200. [DOI] [PubMed] [Google Scholar]
- Biederer T, Südhof TC. Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–3138. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Dahms SO, Hoefgen S, Roeser D, Schlott B, Gührs KH, Than ME. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2010;107:5381–5386. doi: 10.1073/pnas.0911326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debaigt C, Hirling H, Steiner P, Vincent JP, Mazella J. Crucial role of neuron-enriched endosomal protein of 21 kDa in sorting between degradation and recycling of internalized G-protein-coupled receptors. J Biol Chem. 2004;279:35687–35691. doi: 10.1074/jbc.M402751200. [DOI] [PubMed] [Google Scholar]
- Fotinopoulou A, Tsachaki M, Vlavaki M, Poulopoulos A, Rostagno A, Frangione B, Ghiso J, Efthimiopoulos S. BRI2 interacts with amyloid precursor protein (APP) and regulates amyloid beta (Abeta) production. J Biol Chem. 2005;280:30768–30772. doi: 10.1074/jbc.C500231200. [DOI] [PubMed] [Google Scholar]
- Guénette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénette SY, Chen J, Jondro PD, Tanzi RE. Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 1996;93:10832–10837. doi: 10.1073/pnas.93.20.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Müller U. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Müller U. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Wang L, Yang Z, Cool BH, Zitnik G, Martin GM. Endoproteolytic cleavage of FE65 converts the adaptor protein to a potent suppressor of the sAPPalpha pathway in primates. J Biol Chem. 2005;280:12548–12558. doi: 10.1074/jbc.M411855200. [DOI] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- King GD, Turner RS. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Moussa CE, Lee Y, Sung Y, Howell BW, Turner RS, Pak DT, Hoe HS. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience. 2010;169:344–356. doi: 10.1016/j.neuroscience.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AC, Haass C, Wagner SL, Teplow DB, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in Madin-Darby canine kidney cells. J Biol Chem. 1994;269:30966–30973. [PubMed] [Google Scholar]
- Matsuda S, Giliberto L, Matsuda Y, McGowan EM, D'Adamio L. BRI2 inhibits amyloid beta-peptide precursor protein processing by interfering with the docking of secretases to the substrate. J Neurosci. 2008;28:8668–8676. doi: 10.1523/JNEUROSCI.2094-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Matsuda Y, D'Adamio L. BRI3 inhibits amyloid precursor protein processing in a mechanistically distinct manner from its homologue dementia gene BRI2. J Biol Chem. 2009;284:15815–15825. doi: 10.1074/jbc.M109.006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin DM, Miller CC. The FE65 proteins and Alzheimer's disease. J Neurosci Res. 2008;86:744–754. doi: 10.1002/jnr.21532. [DOI] [PubMed] [Google Scholar]
- Mueller HT, Borg JP, Margolis B, Turner RS. Modulation of amyloid precursor protein metabolism by X11alpha /Mint-1. A deletion analysis of protein-protein interaction domains. J Biol Chem. 2000;275:39302–39306. doi: 10.1074/jbc.M008453200. [DOI] [PubMed] [Google Scholar]
- Muthusamy N, Ahmed SA, Rana BK, Navarre S, Kozlowski DJ, Liberles DA, Bergson C. Phylogenetic analysis of the NEEP21/calcyon/P19 family of endocytic proteins: evidence for functional evolution in the vertebrate CNS. J Mol Evol. 2009;69:319–332. doi: 10.1007/s00239-009-9273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreau VM, Orchard S, Adlard PA, Bellingham SA, Cappai R, Ciccotosto GD, Cowie TF, Crouch PJ, Duce JA, Evin G, Faux NG, Hill AF, Hung YH, James SA, Li QX, Mok SS, Tew DJ, White AR, Bush AI, Hermjakob H, Masters CL. A domain level interaction network of amyloid precursor protein and Abeta of Alzheimer's disease. Proteomics. 2010;10:2377–2395. doi: 10.1002/pmic.200900773. [DOI] [PubMed] [Google Scholar]
- Sabéran-Djoneidi D, Picart R, Escalier D, Gelman M, Barret A, Tougard C, Glowinski J, Lévi-Strauss M. A 21-kDa polypeptide belonging to a new family of proteins is expressed in the Golgi apparatus of neural and germ cells. J Biol Chem. 1998;273:3909–3914. doi: 10.1074/jbc.273.7.3909. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch E, Maniatis T. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. Molecular cloning, a laboratory manual. [Google Scholar]
- Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest. 2002;110:1375–1381. doi: 10.1172/JCI16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry. 2000;39:8698–8704. doi: 10.1021/bi0005456. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Shigematsu K, McGeer PL, McGeer EG. Localization of amyloid precursor protein in selective postsynaptic densities of rat cortical neurons. Brain Res. 1992;592:353–357. doi: 10.1016/0006-8993(92)91697-d. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that beta-amyloid protein in Alzheimer's disease is not derived by normal processing. Science. 1990;248:492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Sarria JC, Glauser L, Magnin S, Catsicas S, Hirling H. Modulation of receptor cycling by neuron-enriched endosomal protein of 21 kD. J Cell Biol. 2002;157:1197–1209. doi: 10.1083/jcb.200202022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Alberi S, Kulangara K, Yersin A, Sarria JC, Regulier E, Kasas S, Dietler G, Muller D, Catsicas S, Hirling H. Interactions between NEEP21, GRIP1 and GluR2 regulate sorting and recycling of the glutamate receptor subunit GluR2. EMBO J. 2005;24:2873–2884. doi: 10.1038/sj.emboj.7600755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utvik JK, Haglerød C, Mylonakou MN, Holen T, Kropf M, Hirling H, Skare O, Laake P, Ottersen OP, Haug FM, Davanger S. Neuronal enriched endosomal protein of 21 kDa colocalizes with glutamate receptor subunit GLUR2/3 at the postsynaptic membrane. Neuroscience. 2009;158:96–104. doi: 10.1016/j.neuroscience.2008.11.030. [DOI] [PubMed] [Google Scholar]
- Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid beta-protein precursor. Nature. 1989;341:546–549. doi: 10.1038/341546a0. [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang B, Yang L, Guo Q, Aithmitti N, Songyang Z, Zheng H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci. 2009;29:10788–10801. doi: 10.1523/JNEUROSCI.2132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waridel P, Frank A, Thomas H, Surendranath V, Sunyaev S, Pevzner P, Shevchenko A. Sequence similarity-driven proteomics in organisms with unknown genomes by LC-MS/MS and automated de novo sequencing. Proteomics. 2007;7:2318–2329. doi: 10.1002/pmic.200700003. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273:25552–25555. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]
- Yap CC, Wisco D, Kujala P, Lasiecka ZM, Cannon JT, Chang MC, Hirling H, Klumperman J, Winckler B. The somatodendritic endosomal regulator NEEP21 facilitates axonal targeting of L1/NgCAM. J Cell Biol. 2008;180:827–842. doi: 10.1083/jcb.200707143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano N, Buxbaum JD, Minopoli G, Fiore F, De Candia P, De Renzis S, Faraonio R, Sabo S, Cheetham J, Sudol M, Russo T. Interaction of the phosphotyrosine interaction/phosphotyrosine binding-related domains of Fe65 with wild-type and mutant Alzheimer's beta-amyloid precursor proteins. J Biol Chem. 1997;272:6399–6405. doi: 10.1074/jbc.272.10.6399. [DOI] [PubMed] [Google Scholar]