

Abstract
Chronic alcohol causes hepatic steatosis and liver hypoxia. Hypoxia-regulated Hypoxia-inducible factor 1-α, (HIF1α) may regulate liporegulatory genes but the relationship of HIF1 to steatosis remains unknown. We investigated HIF1α in alcohol-induced hepatic lipid accumulation. Alcohol administration resulted in steatosis, increased liver triglyceride levels and serum ALT suggesting liver injury in WT mice. There was increased hepatic HIF1α mRNA, protein and DNA-binding activity in alcohol-fed mice compared to controls. Mice engineered with hepatocyte-specific HIF1 activation (HIF1dPA) had increased HIF1α mRNA, protein, and DNA-binding activity, and alcohol feeding in HIF1dPA mice increased hepatomegaly and hepatic triglyceride compared to WT. In contrast, hepatocyte-specific deletion of HIF1α (HIF-1α(Hep-/-), protected mice from alcohol- and LPS-induced liver damage, serum ALT elevation, hepatomegaly and lipid accumulation. HIF-1α(Hep-/-), WT, and HIF1dPA mice had equally suppressed levels of PPARα mRNA after chronic ethanol, while the HIF target, ADRP, was upregulated in WT, but not in HIF-1α(Hep-/-) ethanol fed/LPS challenged mice. The chemokine, MCP-1, was cooperatively induced by alcohol feeding and LPS in WT but not in HIF-1α(Hep-/-) mice. Using Huh7 hepatoma cells in vitro, we found that MCP-1 treatment induced lipid accumulation and increased HIF1α protein expression as well as DNA-binding activity. SiRNA inhibition of HIF1α prevented MCP-1-induced lipid accumulation suggesting a mechanistic role for HIF1α in hepatocyte lipid accumulation.
Conclusions
Alcohol feeding results in lipid accumulation in hepatocytes involving HIF1α activation. The alcohol-induced chemokine, MCP-1, triggers lipid accumulation in hepatocytes via HIF1α activation, suggesting a mechanistic link between inflammation and hepatic steatosis in alcoholic liver disease.
Keywords: Hypoxia-Inducible Factor-1 alpha, Monocyte-Chemoattractant Protein-1, Steatosis, Alcoholic Liver Disease
Introduction
Alcoholic liver disease (ALD) is a spectrum of disorders ranging from mild and reversible steatosis to life-threatening and irreversible cirrhosis. The cellular and molecular mechanisms that contribute to ALD continue to be elucidated, and over the past decades numerous paradigms have been proposed, including the pivotal inflammatory role of tumor necrosis factor alpha (TNFα) signaling downstream of Toll-like-Receptor-4 (TLR4) stimulation by gut-derived endotoxin.[1] However, thus far no unifying mechanism for hepatic lipid accumulation has emerged, with various lines of evidence suggesting roles for nuclear regulatory factors such as the family of peroxisome-proliferator activated receptors, sterol-regulatory element binding proteins, metabolic enzymes such as cytochrome P4502E1, or hormonal factors such as adiponectin.[2-7] Increasing evidence suggests that inflammation and hepatic lipid accumulation are linked processes, as knockout of several genes involved in the inflammatory response, such as those of the TLR4 pathway or NFκB pathway, also prevent lipid accumulation in response to chronic alcohol feeding.[1] A recent report demonstrated that treatment of the hepatoma cell line, Huh7, with the chemokine MCP-1 directly induced lipid accumulation in vitro, raising the possibility that a relationship between pro-inflammatory mediators and hepatic lipid accumulation may exist.[8]
The Hypoxia Inducible factors (HIFs) are a family of heterodimeric transcription factors that promote a homeostatic transcriptional response to low oxygen tension. Mature HIF is composed of one of three isoforms of an alpha- subunit (HIF1α, HIF2α, or HIF3α) and a β subunit, the major isoform of which is termed HIF1β or the Aryl-Hydrocarbon Receptor Nuclear Translocator (ARNT). Under conditions of normal oxygen tension, the alpha subunits of HIF are rapidly scaffolded on the Von-Hippel Lindau tumor suppressor protein, where they are hydroxylated and subsequently ubiquitinated and degraded. Under conditions of low oxygen tension, HIF alpha subunits escape hydroxylation and dimerize with HIF1β/ARNT, translocate to the nucleus and activate hypoxia response elements (HRE) in the genome.[9] HIFs are named by their alpha subunit, with HIF1 and HIF2 having a wide, overlapping but non-identical set of transcriptional targets.[10] In a recently described model, HIF1dPA, a mutant of HIF1α construct in which the proline that is normally targeted for hydroxylation is mutated to alanine enables tissue-specific constitutive activation of HIF.[10] In the HIF-1α(Hep-/-) model, floxed exons of the native HIF1α gene enable tissue specific ablation of HIF activity.[11] Recent investigation with the HIF1dPA model demonstrated that while activation of HIF1 alone resulted in minimal lipid accumulation, and activation of HIF2 alone resulted in gross vascular changes without any appreciable increase in hepatic lipid, simultaneous activation of HIF1 and HIF2 results in a phenotype of hepatomegaly with macrovesicular lipid accumulation.[10] However, the relationship of this phenotype to human diseases characterized by steatosis, e.g., alcoholic steatosis or non-alcoholic fatty liver disease, remains to be elucidated.
The role of hypoxia-inducible factors in ALD is yet to be fully explored. Liver hypoxia has been documented in rats on a continuous ethanol diet, and some investigators suggest that a process analogous to ischemia-reperfusion injury may be implicated.[12-15] Others have postulated an increase in HIF1α mRNA as a mechanism of ethanol-induced liver injury.[16] However, the direct contribution of HIF1 to alcoholic liver injury is unknown.
We hypothesized that HIF1α protein, mRNA, and downstream gene activation would be upregulated in the livers of mice after chronic ethanol feeding, and that modifying HIF expression in hepatocytes might affect the progression of alcoholic liver disease. In order to dissect the contribution of HIF1α to alcoholic liver disease, we utilized cre-lox mouse models of hepatocyte-specific HIF1α activation (HIF1dPA) as well as hepatocyte-specific HIF1α deletion (HIF-1α(Hep-/-)).
Materials and Methods
Animal Studies
All animals received care in compliance with protocols approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School. Mice were gradually habituated to a Lieber-DeCarli liquid diet with 5% ethanol (volume/volume) over a period of two weeks, then maintained on the 5% diet for four weeks. Consumption was recorded daily throughout and isocaloric amounts of a non-alcohol containing diet (in which dextran-maltose replaced calories from ethanol) were dispensed to pair-fed animals. Weights were recorded weekly. Wild-type mice (C57/Bl6), alb-Cre, and HIF1flox/flox mice were purchased from Jackson Laboratories (Bar Harbor, Maine). LSL-HIF1dPA mice were the kind gift of Dr. William Kim (University of North Carolina-Chapel Hill). The HIF1dPA allele was engineered by Kim et al and has been described previously.[10] Briefly, a stop codon is flanked by loxP sites upstream of a HIF1α transgene in which a proline-to-alanine substitution enables the transgene to escape recognition by proline hydroxylases and subsequent proteasomal degradation. Coexpression of the albumin-cre transgene excises the stop codon, and subsequently enables expression of the transgene in hepatocytes. LSL-HIF1dPA and HIF1flox/flox mice were bred against Cre mice as previously described,[10, 11] tagged by ear notching, and housed in separate cages.[10,11] Prior to the conclusion of the study, some mice were randomly assigned to receive lipopolysaccharide (LPS, Sigma) injection (500ug/kg) or saline injection. Mice were sacrificed 18 hours after LPS injection. At the conclusion of the feeding, mice were weighed and euthanized. Livers were excised and weighed, and portions were snap frozen in liquid nitrogen for protein and biochemical assays, preserved in 10% neutral-buffered formalin for histopathological analysis, or soaked in RNALater (Qiagen GmbH, Hilden, Germany) for RNA extraction. Blood was collected and serum separated for biochemical analysis. Tail snips were collected for genotyping. Nuclear extracts were prepared via sucrose gradient centrifugation and two-step purification as previously described.[17]
Biochemical Analysis
Serum alanine aminotransferase (ALT) was determined using a commercially available reagent (Advanced Diagnostics Inc, Plainfield, NJ) as previously described.[17] Liver triglycerides were quantified as previously described using a commercially available kit (Wako Chemicals USA Inc., VA).[17]
Histopathological Analysis
Sections of formalin-fixed livers were stained with hematoxylin/eosin and analyzed by microscopy. Frozen sections were prepared from liver tissue frozen in OCT media, and stained with Oil-Red O. Photomicrographs were collected as TIFF files and subsequently analyzed with Metamorph software.
Multiplex Cytokine Bead Array
Multiplex cytokine bead array was performed using the BioRad Precision Pro multiplex cytokine bead array kit (BioRad) according to the instructions of the manufacturer. Briefly, serum aliquots stored at -80 degrees were diluted at a 1:4 ratio using dilution buffer provided in the kit. Serum was allowed to mix with beads coated with antibodies to one of 8 different cytokines and subsequently incubated with a second antibody that detects conjugated bead-cytokine pairs.
Cell Culture Studies
All studies were performed using the human hepatocellular carcinoma cell line Huh7. Cells were maintained in complete growth medium (Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2% Penicillin/Streptomycin, and 1% 100X amino-acid supplement mixture.) For in vitro assays, cells were plated on 10cm plates, and then transfected with plasmid DNA or siRNA and/or treated with recombinant monocyte chemoattractant protein-1 (MCP-1, R&D Systems).
Plasmid transfections
The HIF1dPA and HIF2dPA plasmids were the kind gift of Dr. William Kim (UNC). Plasmid DNA was transfected into Huh7 cells using Fugene transfection reagent according to the manufacturers instructions. Briefly, Huh7 cells on 10cm plates at 50-60% confluence were transfected with 5ug plasmid DNA (HIF1dPA or HIF2dPA transgenes encoded into pcDNA3.1) with 15μl Fugene 6 and 160μl serum free media. For verification of plasmid transfection, pcDNA3.1 encoding GFP was used and cells imaged 24 hours post-transfection with a fluorescent microscope.
siRNA transfections
HIF1α siRNA, HIF2α siRNA, or scrambled (scr) siRNA were purchased from Santa Cruz Biotechnology. Transfection was achieved using siPORT Amine transfection agent (Applied Biosystems) according to the protocol of the manufacturer. Briefly, for a 10cm-plate, 17μl siPORT Amine reagent at room temperature was added to 333μl Opti-MEM serum-free medium. 7.5μl of 10um HIF1α, HIF2α, or scr siRNA was diluted in 142μl Opti-MEM. Subsequently, both the transfection reagent and the siRNA mixture were mixed and transfection complexes formed at RT. The mixture (500μl) was dispersed on a 10cm plate, and overlaid with 7×105 cells in a final volume of 7ml. After 24 hours, medium was aspirated and replaced with 10ml complete culture medium for subsequent treatment.
Real Time PCR
RNA was purified using the RNeasy Mini kit (Qiagen Sciences, Maryland, USA) with on-column DNA digestion (Qiagen). cDNA was prepared using random hexamer primers and the Reverse Transcription System kit (Promega Corp., Madison WI). Real-time quantitative polymerase chain reaction was performed using an iCycler (Bio-Rad laboratories Inc., Hercules, CA), using specific primers. Primer sequences available on request. Fold-change in gene expression was determined by normalizing to 18S mRNA.
Oil Red O Staining
Cultured and treated cells were washed with 2ml PBS. Plates were incubated in 2ml 10% formalin for 10 minutes, formalin solution was replaced and plates incubated overnight, then washed twice with ddH2O and once with 60% isopropanol. Dried plates were incubated for 10 minutes in 1ml of Oil Red O working solution. Plates were immediately destained with 4 washes of ddH2O and photographed.
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared as previously described.[18] a consensus double-stranded Hypoxia Response Element (HRE) (Santa Cruz Biotech, CA) oligonucleotide was used for EMSA. End-labeling, oligonucleotide purification, and EMSA assay were performed as previously described. [19]
Western Blotting
30-50ug of nuclear extract was resolved on 10% polyacrylamide gels and transferred overnight to nitrocellulose. Membranes were blocked overnight with blocking buffer (5% bovine serum albumin in Tris-Borate-SDS with 0.01% Tween 20) with refrigeration, and subsequently probed overnight with anti-HIF1α (R&D Biosciences) mouse monoclonal antibodies. Detection was performed using anti-mouse horseradish-peroxidase conjugated secondary antibody and chemiluminescent substrates.
Quantification and statistics
Band density was quantified using Labworks 4.0 image analysis. Statistical analysis was performed with Microsoft Excel using two-tailed student's T-Test and p<0.05 was considered significant.
Results
Chronic alcohol administration induces liver steatosis and HIF1α activation in mice
As has been reported elsewhere, ethanol feeding increased liver weight to body weight ratio, liver triglyceride, and serum ALT values and resulted in liver steatosis in wild-type mice compared to isocaloric diet-fed controls (Figure 1, A-E). To test our hypothesis that alcohol may increase the expression and activity of hypoxia-inducible factor-1, nuclear extracts from liver tissue were evaluated for HIF-1 expression. We found that HIF1α mRNA was upregulated by ethanol feeding in wild-type mice (Figure 1D). HIF1α protein was also more abundant in alcohol-fed than in pair-fed livers (Figure 2, A, B). HIFs are primarily degraded by post-translational hydroxylation and subsequent degradation of the alpha subunits by the ubiquitin/proteasomal system. To confirm that HIF1α was transcriptionally active, we performed an electrophoretic mobility shift assay (EMSA) using a commercially available HRE oligonucleotide. Our results showed a significant upregulation of HIF DNA-binding activity in ethanol-fed animals versus pair-fed animals suggesting HIF-1 activation (Figure 2 C,D).
Figure 1.
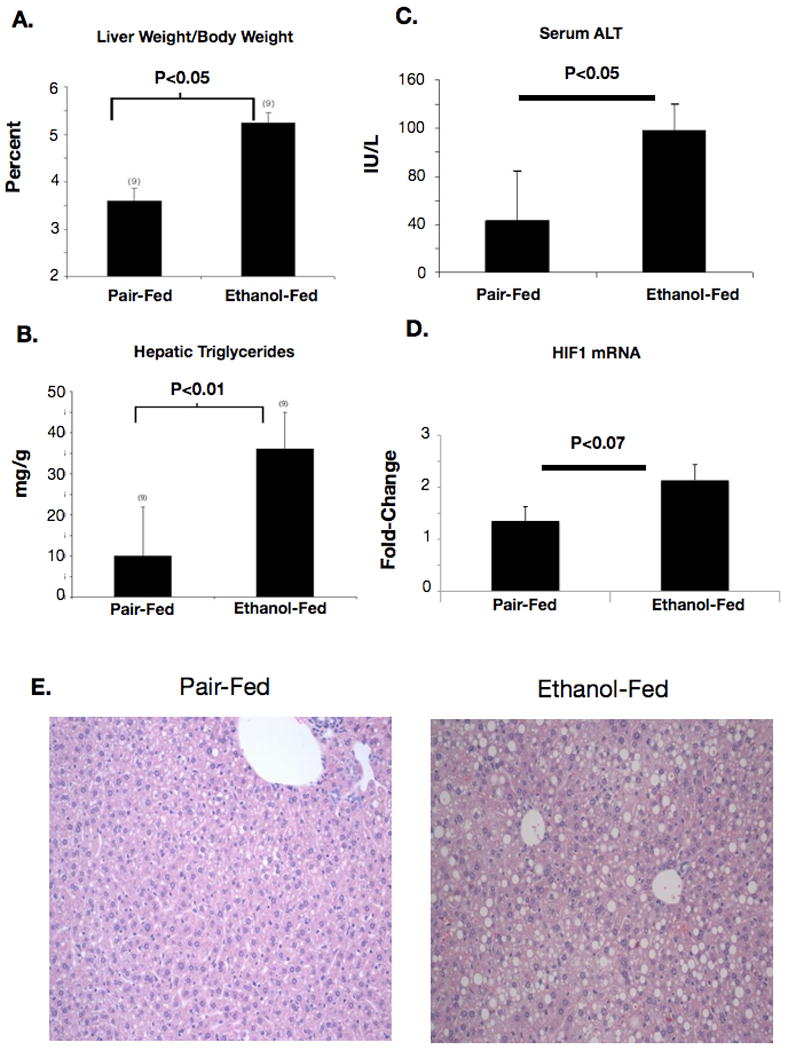
Changes in liver histology with chronic ethanol feeding. (A) Wild-type ethanol-fed mice developed hepatomegaly as measured by liver-weight to body weight ratio after 4 weeks of ethanol feeding. (B) Liver triglyceride was quantified using a biochemical assay and normalized to protein content of liver samples. (C) Serum ALT was quantified as an index of liver injury in sera from ethanol-fed or pair-fed mice. (D) HIF1α mRNA was amplified using real-time PCR from RNA extracts from livers of ethanol-fed or pair-fed mice. (E) Photomicrographs prepared from H&E stained liver sections from ethanol-fed or pair-fed mice.
Figure 2.
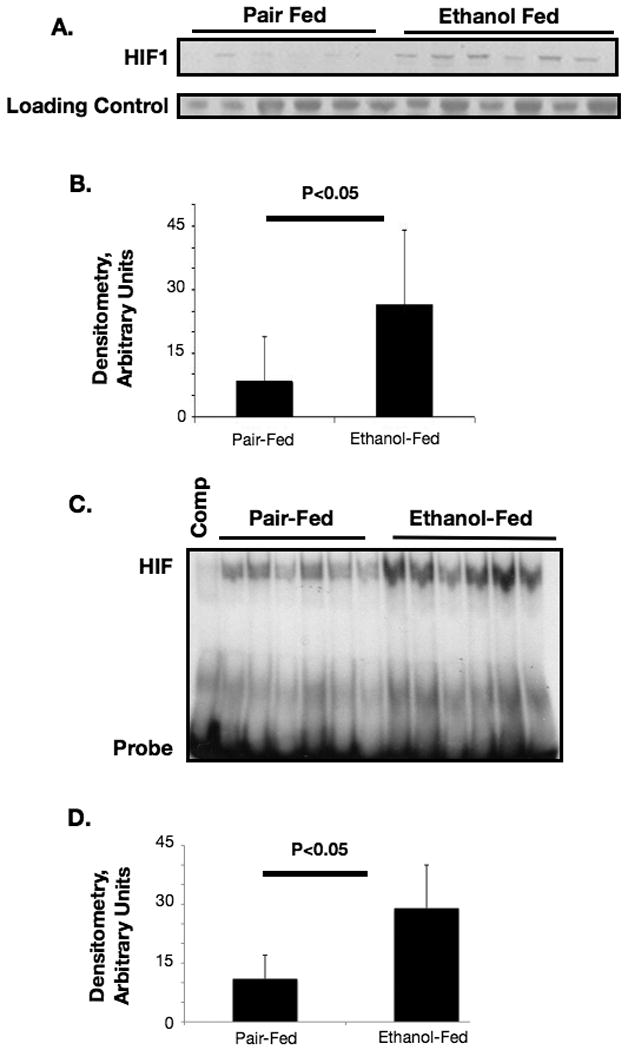
(A) Western blot for HIF1α protein prepared from nuclear extracts from livers of ethanol-fed or pair-fed mice, band quantification in (B). (C) HIF1α DNA-binding activity was measured using the electrophoretic mobility shift assay (EMSA) and a HIF-specific probe, (D) EMSA band densitometry.
Increased liver steatosis in alcohol-fed HIF1dPA mice
In order to determine the contribution of HIF1α to ethanol-induced liver pathology, we utilized a mouse model of hepatocyte-specific HIF1α activation(HIF-1dPA) described by Kim et al.[9] Due to a mixed genetic background, Alb-Cre littermates that did not harbor the HIF1dPA transgene were selected as controls. To confirm the activation of HIF1α in HIF1dPA mice, HIF1α DNA-binding activity was examined in liver nuclear extracts from HIF1dPA and Alb-Cre control mice, and a significant upregulation of HIF1α DNA-binding activity was observed (P<0.01; HIF1dPA pair-fed vs. Alb-Cre pair-fed, Supplementary Figure S1.) We found increased liver weight/body weight (LW/BW) ratios in HIF1dPA mice versus Alb-Cre controls even without alcohol feeding (Fig 3A). Specifically, both HIF1dPA-pair-fed mice and control ethanol-fed mice had a statistically significant upregulation of LW/BW ratio versus pair-fed controls after 4 weeks of ethanol feeding (Figure 3A). Ethanol-fed HIF1dPA mice had the highest LW/BW ratios (p<0.05 vs. HIF1dPA pair-fed mice).
Figure 3.
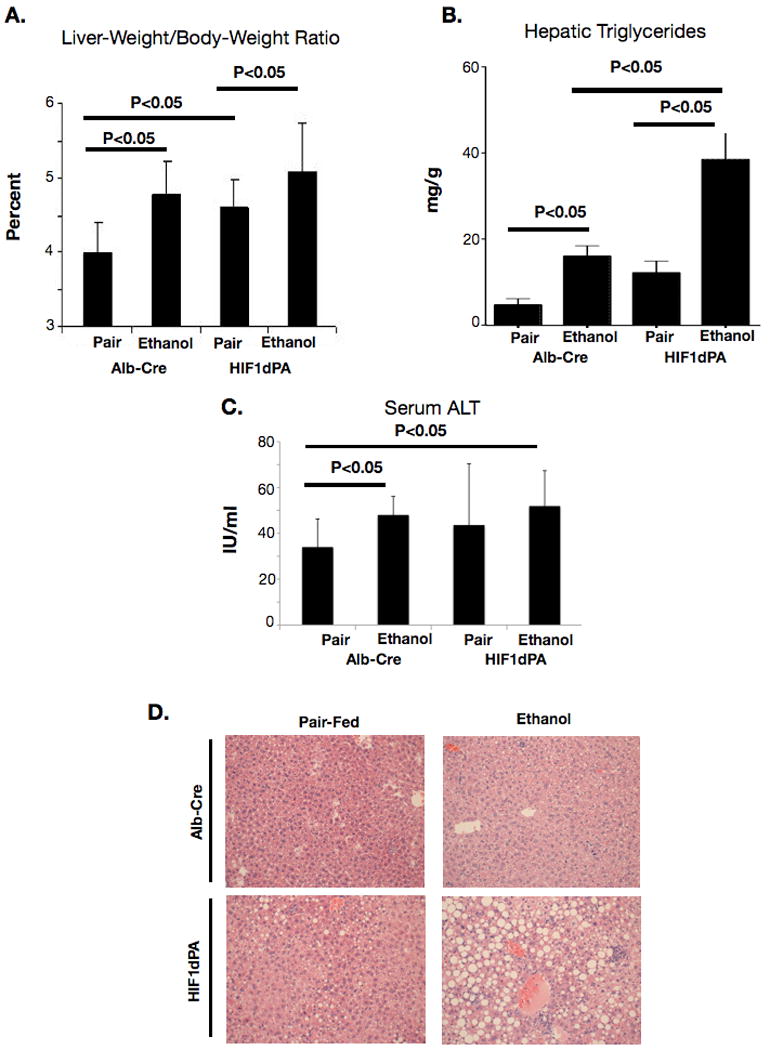
Alcohol feeding and changes in the liver of mice with hepatocyte-specific, constitutively active HIF1α. (A) Liver-weight to body weight ratio from ethanol or pair-fed HIF1dPA or Alb-Cre control mice. (B) Triglycerides were quantified from whole livers of ethanol-fed or pair-fed Alb-Cre or HIF1dPA mice. (C) Serum ALT in ethanol and pair-fed Alb-Cre mice and HIF1dPA mice. (D) Microscopy of HIF1dPA and Alb-Cre ethanol- and pair-fed mice.
Examination of liver triglycerides in whole-liver extracts revealed that alcohol caused an upregulation of triglyceride in hepatic extracts in control mice at 4 weeks (Figure 3B). Triglyceride levels were higher in pair-fed HIF1dPA mice versus pair-fed control mice, (p<0.05, HIF1dPA pair-fed vs. alb-cre pair-fed) indicating an effect of constitutive HIF1α on lipid accumulation in the absence of any other stimulus. Alcohol-fed HIF1dPA mice had the highest average hepatic triglyceride content (p<0.05 versus all other groups). The presence of HIF1dPA transgene also led to serum ALT levels comparable to Alb-Cre ethanol-fed mice (Figure 3C). Histopathology analysis also confirmed that ethanol-fed HIF1dPA mice had more lipid vacuolization than ethanol-fed Alb-Cre mice (Figure 3D). These results suggested that constitutive HIF-1 activation in hepatocytes (HIF1dPA mice) results in liver abnormalities reminiscent of alcoholic liver disease and that alcohol feeding and constitutive HIF-1 activation cooperatively upregulated hepatic steatosis.
Deletion of HIF1 in hepatocytes protects from alcohol-induced liver damage
Because our findings suggested an effect of hepatocyte-specific HIF1α expression on lipid accumulation, we sought to test whether elimination of HIF1α activity in hepatocytes could ameliorate the pathology associated with chronic ethanol feeding. We utilized a mouse engineered by Johnson and coworkers [10] where native HIF1α is flanked by LoxP sites, and co-expression of Cre recombinase results in tissue-specific deletion of HIF1α. Analysis of mice with hepatocyte-specific deletion of HIF1α and controls maintained on the ethanol diet revealed increased liver-weight/body-weight ratios in WT ethanol-fed mice versus control mice at 4 weeks. In contrast, HIF-1α(Hep-/-) mice showed no significant difference in liver-weight/body weight ratio between pair-fed and ethanol fed groups (Figure 4A). Consistent with the role of HIF1α in hepatocyte steatosis, HIF-1α(Hep-/-) mice were protected from the increase in liver triglyceride content observed in wild-type mice after alcohol feeding (Figure 4B). Wild-type mice showed a robust cooperative upregulation of serum ALT with chronic ethanol and lipopolysaccharide challenge (p<0.02, WT ETOH/LPS versus WT pair-fed). In contrast, HIF-1a(Hep-/-) mice were protected against serum ALT increase, even in the presence of chronic ethanol and LPS (Figure 4C). Next, we performed immunoblotting on nuclear extracts from wild-type and HIF-1α(Hep-/-) mice. Ethanol feeding resulted in a significant increase in HIF1α expression in nuclear extracts prepared from WT mice (Figure 4D). In contrast, nuclear extracts from HIF-1α(Hep-/-) mice had very low levels of HIF1α expression, and no further upregulation with ethanol feeding was observed, confirming suppression of HIF1α signaling in our mouse model (Fig 4 D, E). On histology examination, livers from WT mice exhibited significant steatosis after chronic alcohol feeding, but no significant difference was observed between livers from pair-fed and ethanol-fed HIF-1α(Hep-/-) mice. (Figure 5).
Figure 4.
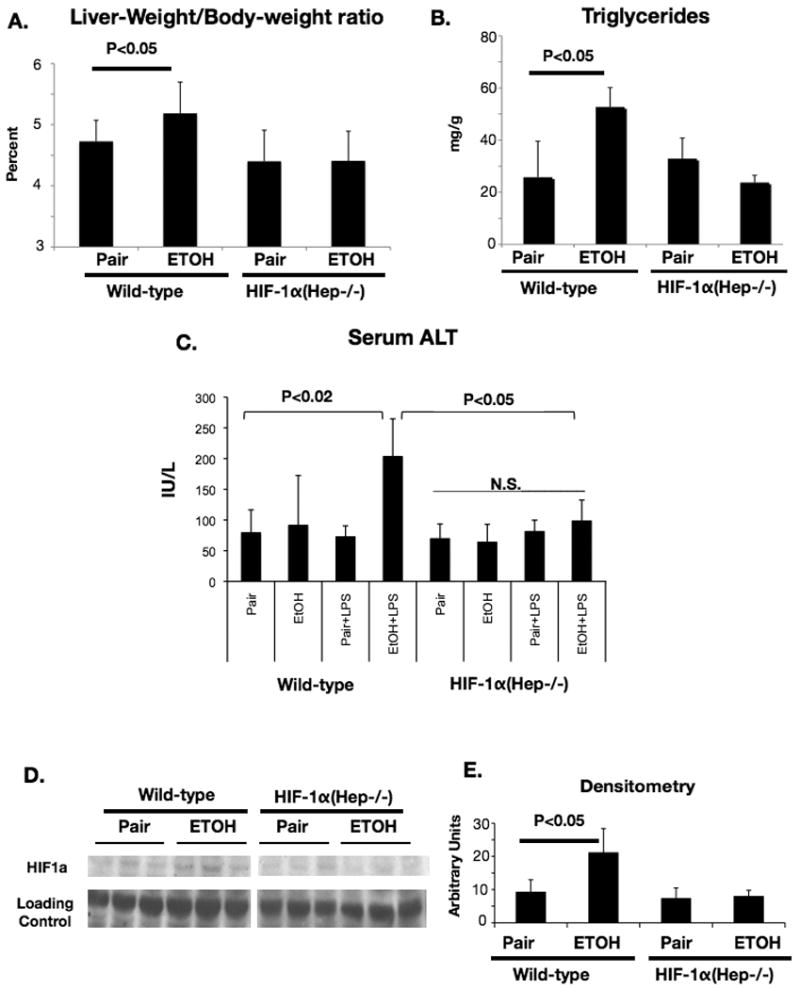
Protection from liver injury in HIF-1α(Hep-/-) mice after chronic ethanol challenge.
(A) Liver-weight to body-weight ratio in HIF-1α(Hep-/-) or control mice with chronic ethanol. (B) Triglycerides were quantified from whole livers of ethanol-fed or pair-fed HIF-1α(Hep-/-) or control mice. (C) Serum ALT data from HIF-1α(Hep-/-) and wild-type mice after chronic ethanol and LPS challenge. (D) Western blot showing HIF1α protein expression with chronic ethanol in control and HIF-1α(Hep-/-) mice, Densitometry in (E).
Figure 5.
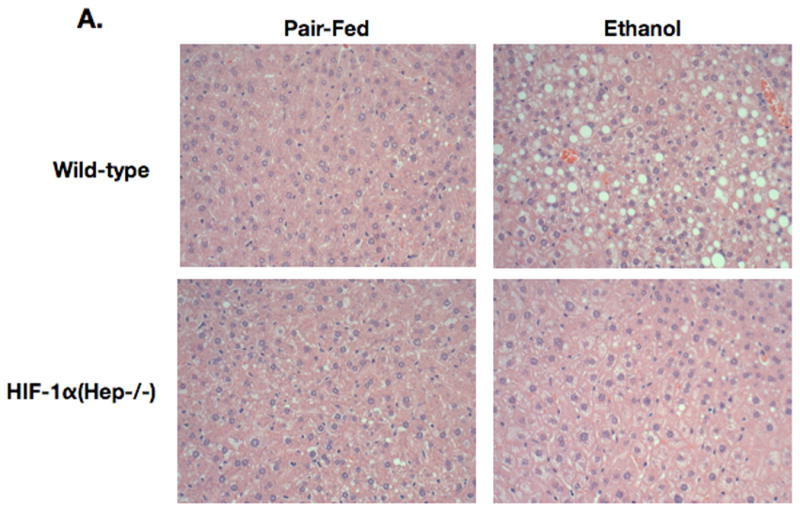
Microscopy of HIF-1α(Hep-/-) and control mice with chronic ethanol feeding or control feeding.
Because the peroxisome-proliferator associated receptor-alpha (PPARα) is associated with lipid accumulation, we examined PPARα mRNA levels in ethanol- and pair-fed control and HIF-1α(Hep-/-) mice. To amplify the effect of ethanol feeding, we also applied an LPS challenge. LPS has been identified in the portal circulation after chronic alcohol intake in mice and men and it contributes to the development of alcoholic liver disease.[20] To our surprise, we found that PPARα was similarly suppressed by ethanol feeding in each of these experimental groups, indicating that the HIF1α effect on lipid accumulation was independent of PPARα (Figure 6A). Next, we examined adipocyte-differentiation-related protein (ADRP), which has been associated with HIF1α expression[21]. We found that ADRP mRNA was significantly upregulated with ethanol feeding alone (p<0.05, Figure 6B). Although no cooperative effect of LPS injection and chronic ethanol was observed in ADRP mRNA expression 2 hours after LPS injection (Figure 6B), by 18 hours there was a robust cooperative upregulation of ADRP mRNA with chronic ethanol and LPS injection (p<0.05, Supplementary figure S2). HIF-1α(Hep-/-) mice were protected from any upregulation of ADRP with chronic ethanol alone or with chronic ethanol and LPS challenge. (Figure 6B; Supplementary figure S2). These results indicated that ADRP may be implicated in the differential effect of HIF1α on lipid accumulation. Thus, we examined the effect of constitutive HIF activation on the expression of ADRP in HIF1dPA and in control, Alb-cre mice. (Figure 6C) We found a significant increased in ADRP expression with ETOH feeding in Alb-Cre mice, similar to that observed in wild-type mice (p<0.02). Furthermore, we found that the presence of the HIF1dPA transgene upregulated hepatic ADRP protein expression to a similar extent as ETOH feeding (p<0.01). (Figure 6D, E)
Figure 6.
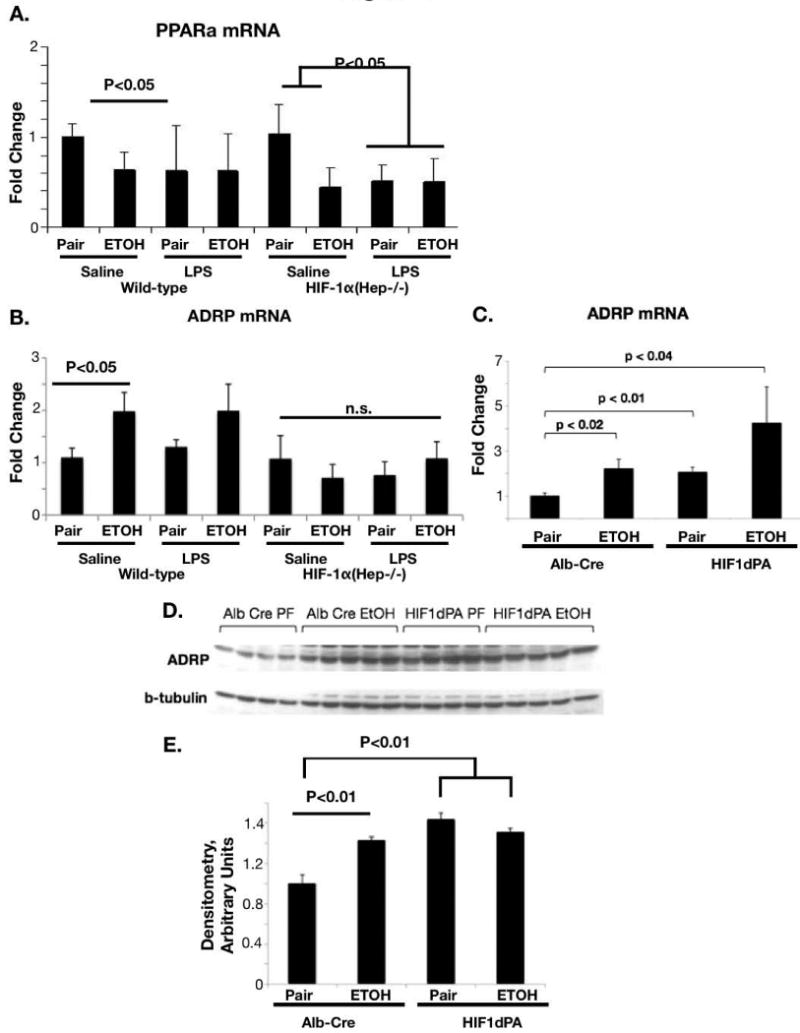
Altered patterns of genes involved in lipid homeostasis in HIF-1α(Hep-/-) or control mice. (A) PPARα mRNA in ethanol-fed or pair-fed control and HIF-1α(Hep-/-) mice was quantified by real-time PCR. (B) ADRP mRNA with ethanol and LPS challenge in wild-type and HIF-1α(Hep-/-) mice, quantified by real-time PCR. (C) ADRP mRNA expression in control (Alb-Cre) and HIF1dPA mice with ethanol or pair-feeding. (D) ADRP protein expression in Alb-Cre and HIF1dPA mice with ethanol or pair feeding, Densitometry in (E).
Monocyte-chemoattractant protein-1 (MCP-1) induces steatosis via HIF1α expression in hepatocytes
In order to further dissect the mechanism of HIF1α regulation in hepatic lipid accumulation, we supplemented our in vivo work with an in vitro model of hepatic lipid accumulation. The chemokine MCP-1 has recently been demonstrated to result in lipid accumulation in the hepatocyte cell line Huh7[8]. First, we examined MCP-1 expression levels in ethanol-fed control and HIF-1α(Hep-/-) mice. We found that alcohol feeding alone resulted in a small, but significant upregulation in MCP-1 serum levels. (Figure 7A) This corresponded to increased MCP-1 hepatic mRNA with chronic ethanol (Figure 7B). LPS stimulation and ethanol cooperatively upregulated MCP-1 in WT mice (Figure 7C). LPS induced MCP-1 in HIF-1α(Hep-/-) mice to an extent comparable to wild-type, but there was no further increase in HIF-1 α(Hep-/-) with alcohol feeding (Figure 7C). To evaluate mechanistic events, we next treated Huh7 cells with recombinant MCP-1 or with a plasmid containing the degradation-resistant HIF1dPA mutant. MCP-1 treatment resulted in increased HIF1α protein in nuclear extracts from Huh7 cells. (Figure 8A). We also determined that HIF1dPA over-expression resulted in increased HIF1α mRNA (Figure 8B) and this was associated with increased triglyceride levels compared to control cells (Figure 8C). Furthermore, we found that either MCP-1 treatment or HIF1dPA plasmid treatment resulted in increased lipid accumulation in Huh7 cells (Figure 8E).
Figure 7.
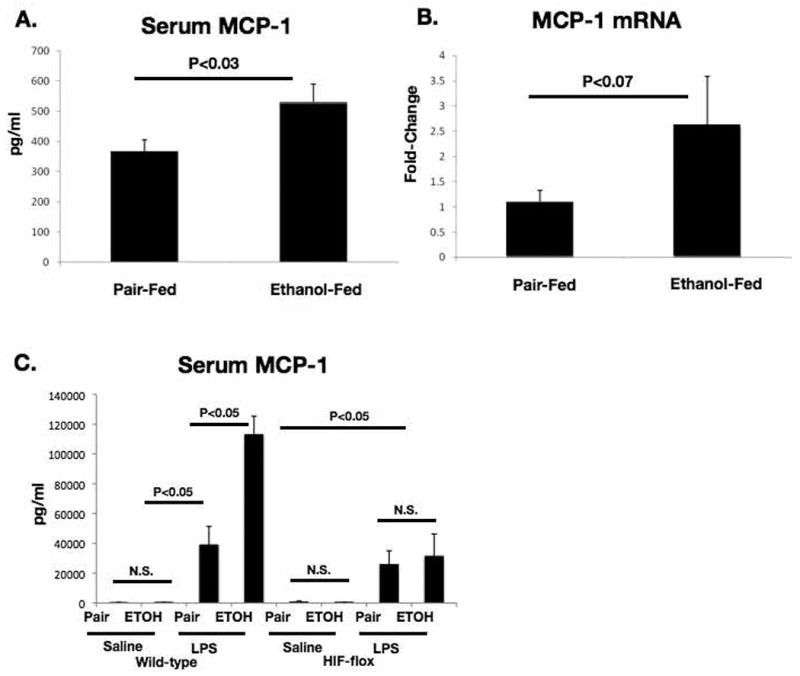
Changes in MCP-1 with chronic ethanol and/or LPS challenge.
(A) Serum MCP-1 quantified by low-threshold cytokine assay in ethanol or pair-fed mice. (B) Hepatic MCP-1 mRNA in ethanol or pair-fed mice. (C) Serum MCP-1 quantified using high threshold cytokine assay in control or ethanol fed HIF-1α(Hep-/-) or wild-type mice, with or without LPS injection.
Figure 8.
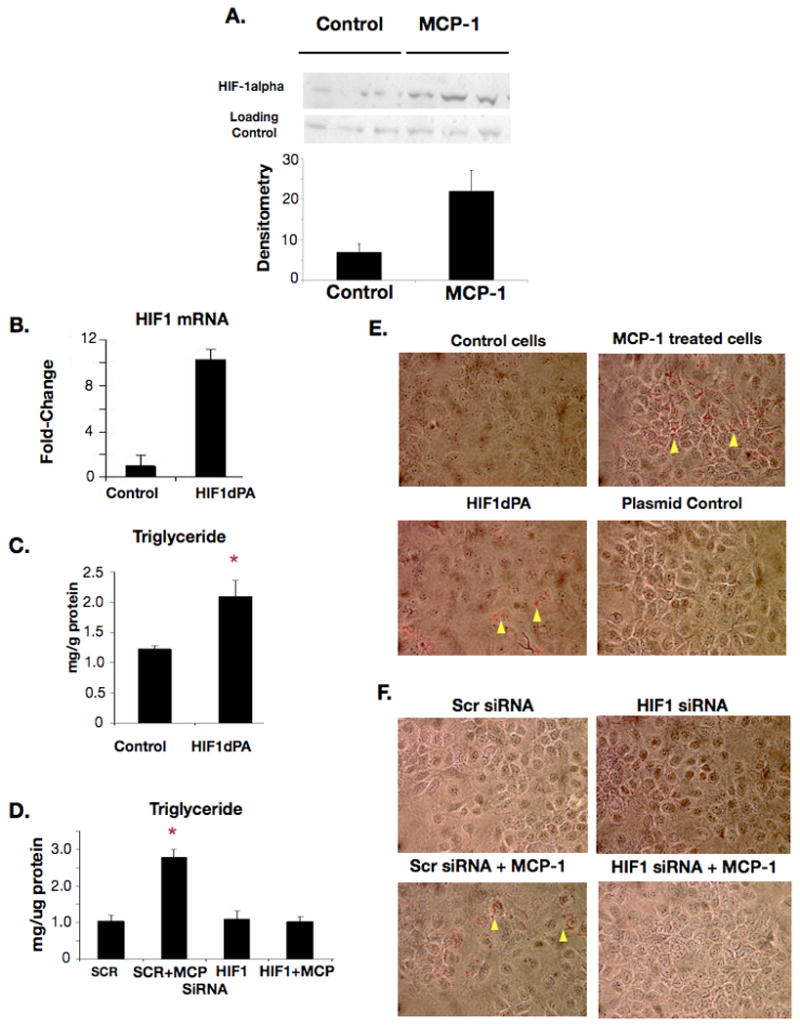
In vitro studies on relationship of MCP-1 and HIF1α
(A) HIF1α protein measured by western blot of nuclear extracts from Huh7 cells treated with MCP-1; Densitometry appears below. (B) Real-time PCR for HIF1α mRNA after HIF1dPA plasmid treatment. (C) Triglyceride content in HIF1dPA treated cells or control cells. (D) Triglyceride in scrambled siRNA but not HIF1α siRNA pretreated cells treated with MCP-1. (E) Lipid accumulation in MCP-1 treated cells and HIF1dPA plasmid treated cells, yellow arrowheads show refractile lipid droplets by Oil-red O stain. (F) Lipid accumulation in HIF1α siRNA pretreated cells treated with MCP-1, yellow arrowheads show refractile lipid droplets by Oil-red O stain.
To establish a further mechanistic insight into the role of HIF-1 in hepatocyte lipid accumulation, we sought to determine whether we could block lipid accumulation in MCP-1 treated cells by silencing HIF1α. When Huh7 cells were treated with a HIF1α siRNA we found that expression of HIF1α mRNA was significantly suppressed at 24 and 36 hours (Supplementary Figure S3). Next, Huh7 cells that had been pretreated with HIF1α siRNA were challenged with MCP-1 stimulation. We found increased triglyceride in scrambled siRNA control but not in HIF1α-siRNA pretreated cells after the MCP-1 challenge (Figure 8E). Using Oil-red-O staining we also confirmed that HIF1α siRNA pretreatment could prevent MCP-1 treatment –induced lipid accumulation (Figure 8F). These results suggest a link between alcohol-induced increases in HIF-1, MCP-1 and lipid accumulation in hepatocytes.
Discussion
In this study, we provide evidence for an effect of HIF1α on hepatic lipid accumulation in alcoholic liver disease. While the relationship between alcohol and hypoxia in the liver has been described, our novel observations ascribe a specific pathophysiological role to the dysregulation of a hypoxia-responsive transcription factor in alcoholic liver disease. We found that chronic alcohol feeding results in increased HIF1α levels and activation in the liver. We further demonstrated that constitutive activation of HIF1α in hepatocytes accelerates lipid accumulation with chronic ethanol feeding, and report that HIF1dPA mice have higher steatosis on histology evaluation and increased hepatic triglyceride levels compared to control mice. We report for the first time that alcohol-induced lipid accumulation can be prevented in mice with hepatocyte-specific deletion of HIF1α. Using an in vitro system, we found that inhibition of HIF1α prevents lipid accumulation. We also demonstrated that the protective effect of HIF1α deletion may be independent of PPARα, and may depend upon regulation of other genes involved in lipid homeostasis, including the adipocyte differentiation related protein. Our data further suggested that the upregulation of MCP-1 observed in LPS-injected, ethanol-fed mice may be an upstream mediator of HIF1α expression, as MCP-1 treatment resulted in increased HIF1α expression in vitro. Finally we present data to show that inhibition of HIF1α prevents lipid accumulation in vitro in response to MCP-1 treatment.
Our novel observations link alcohol-induced induction of HIF1α and alcohol-induced steatosis in a mechanistic way. Previous studies suggested a role for hypoxia-inducible factors in hepatic lipid accumulation and our findings offer a novel perspective on the role of hypoxia-inducible factors in alcohol-induced steatosis. We found induction of HIF1α after alcohol feeding and demonstrated that hepatocyte-specific inhibition of HIF-1 prevented the alcohol-induced steatosis suggesting that HIF1α alone can mediate alcohol-induced steatosis. This observation is somewhat different from results by Rankin et al. who recently described a dominant role for the HIF2α isoform in hepatic lipid regulation using a scheme of cre-lox mediated activation of HIF1α or HIF2α in hepatocytes; in that model, disruption of either HIF isoform in combination with pVHL knockout resulted in activation of the remaining isoform.[22] Their findings, however, were in sharp contrast to work by Scortegagna et al. that demonstrated that adult HIF2 knockout mice developed severe hepatic steatosis that could be reversed by treatment with a superoxide dismutase inhibitor.[23] Kim et al, as well, found no significant contribution to hepatic lipid accumulation with a constitutively active mutant of HIF2, despite finding a robust effect on angiogenesis. On the other hand, they demonstrated a mild HIF1α dependent effect on lipid accumulation.[10] The different genetic techniques used to create specific gene expression or knockout in each of these studies may offer some explanation of the different results each describes.
Many of the genes involved in lipid homeostasis are regulated by hypoxia inducible factors.[24, 25]. However, it is yet to be dissected whether significant differences exist in the contribution of HIF1α and HIF2α in a given cell type and/or cell-specific effects. Our data suggest that in hepatocytes both in vivo and in vitro (in mice as well as in human cells), HIF1α activation alone is sufficient to induce lipid accumulation. We explored the contribution of ADRP, a lipid droplet-associated surface protein that is regulated by HIF[21]. ADRP has been shown to be upregulated in human steatosis as well as in mice developing steatosis after high fat diet[26, 27]. Here we report the novel observation that ADRP is upregulated with chronic ethanol alone. We found further cooperative upregulation of ADRP in wild-type mice after alcohol feeding and LPS injection that correlated with HIF1α induction. ADRP was upregulated with constitutive HIF1a expression but conversely, ADRP upregulation with chronic ethanol and/or LPS injection was prevented in mice with hepatocyte specific HIF1α deletion. This suggested a mechanistic role for HIF1a in ADRP induction and liver steatosis.
Increasing evidence suggests that lipid accumulation is affected by pro-inflammatory stimuli. In support of this notion, the chemokine, MCP-1 was recently shown to cause lipid accumulation in human hepatoma cells[8]. We found a synergistic upregulation of MCP-1 in the serum of chronic alcohol-fed, LPS challenged mice suggesting that increased gut-derived LPS could amplify MCP-1 induction in ALD. Consistent with the role of HIF1α in induction of MCP-1 by LPS, we found lower increase in MCP-1 serum levels in mice with hepatocyte-specific HIF-1 deletion compared to controls after an in vivo LPS challenge. These data imply a role of HIF1α in MCP-1 induction in alcoholic liver disease. We speculate that gut-derived LPS could be an inducer of HIF1α in vivo as serum LPS levels are increased after chronic alcohol feeding in mice and reportedly in humans as well [28]. More important, we found that MCP-1 can induce HIF1α mRNA, and protein in hepatoma cells in vitro and this was associated with induction of lipid accumulation in hepatocytes. Together, these results suggest a cross-regulation between HIF1α activation and MCP-1 in promoting hepatocyte lipid accumulation where MCP-1 can induce HIF1α activation and in turn, HIF1α can contribute to MCP-1 induction.
The clinically relevant question is the implication of these findings for the development of clinical strategies for the treatment of alcoholic fatty liver disease. Whereas cessation of alcohol use tends to result in a rapid reversal of alcoholic fatty liver, the increasingly recognized epidemic of the related entity, non-alcoholic fatty liver disease, suggests that therapies that modify hepatic lipid accumulation are likely to find clinical use.
Supplementary Material
Supplementary Figure S1.
Confirmation of active HIF1α in livers of HIF1dPA mice by gel-shift assay.
Liver nuclear extracts from pair-fed and ethanol-fed HIF1dPA and Alb-Cre control mice were prepared. HIF1α-DNA-binding activity was assayed using the electrophoretic mobility shift assay with a hypoxia-response element oligonucleotide (S1A.) There was greater HIF1α DNA-binding activity in liver nuclear extracts from HIF1dPA mice than Alb-Cre control mice (P<0.01, pair-fed HIF1dPA versus pair-fed Alb-Cre). Bands were quantified by densitometry (S1B).
Supplementary Figure S2
Hepatic ADRP mRNA quantified by real-time PCR 18 hours after LPS or sham injection in wild-type and HIF-1α(Hep-/-) mice.
Supplementary Figure S3
Real-time PCR quantification of HIF1α mRNA after treatment of cells with siRNA targeting HIF1α, scrambled control siRNA, or reagent control at 24 and 36 hours.
Acknowledgments
This work was supported by grants R21 AA017544(GS) and F30 AA017030(BN) and the core resources supported by the Diabetes Endocrinology Research Center grant DK32520 were also used (GS is a member of the UMass DERC).
List of abbreviations
- ALD
Alcoholic Liver Disease
- HIF1α
Hypoxia Inducible Factor 1-alpha
- ADRP
Adipocyte Differentiation Related Protein
- MCP-1
Monocyte Chemoattractant Protein-1
- HRE
Hypoxia Response Element
- TLR
toll-like receptor
- LPS
lipopolysaccharide
- ALT
alanine aminotransferase
- TNFα
tumor necrosis factor
- PPARα
Peroxisome-Proliferator Activated Receptor alpha TRIF, TIR domain-containing adaptor inducing IFN-beta
- NFκB
Nuclear factor kB
- WT
Wild-Type
Footnotes
Disclosures: nothing to disclose
Contributor Information
Bharath Nath, Email: Bharath.Nath@umassmed.edu.
Ivan Levin, Email: Ivan.Levin@umassmed.edu.
Timea Csak, Email: Timea.Csak@umassmed.edu.
Jan Petrasek, Email: Jan.Petrasek@umassmed.edu.
Christian Mueller, Email: Christian.Mueller@umassmed.edu.
Karen Kodys, Email: Karen.Kodys@umassmed.edu.
Donna Catalano, Email: Donna.Catalano@umassmed.edu.
Pranoti Mandrekar, Email: Pranoti.mandrekar@umassmed.edu.
Bibliography
- 1.Nath B, Szabo G. Alcohol-induced modulation of signaling pathways in liver parenchymal and nonparenchymal cells: implications for immunity. Semin Liver Dis. 2009;29(2):166–77. doi: 10.1055/s-0029-1214372. [DOI] [PubMed] [Google Scholar]
- 2.Tomita K, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology. 2004;126(3):873–85. doi: 10.1053/j.gastro.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y, et al. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology (Baltimore, Md) 2008;47(5):1483. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 4.Lieber CS, et al. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun. 2008;370(1):44–8. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 5.You M, et al. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277(32):29342–7. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 6.Wan YJ, et al. Expression of the peroxisome proliferator-activated receptor gene is decreased in experimental alcoholic liver disease. Life Sci. 1995;56(5):307–17. doi: 10.1016/0024-3205(94)00953-8. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Sebastian BM, Nagy LE. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am J Physiol Endocrinol Metab. 2007;292(2):E621–8. doi: 10.1152/ajpendo.00387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clement S, et al. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology. 2008;48(3):799–807. doi: 10.1002/hep.22404. [DOI] [PubMed] [Google Scholar]
- 9.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 10.Kim W, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. The EMBO journal. 2006;25(19):4650. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peyssonnaux C, et al. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178(12):7516–9. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 12.Arteel GE, et al. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25(4):920–6. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- 13.Arteel GE, et al. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: role of Kupffer cells. Am J Physiol. 1996;271(3 Pt 1):G494–500. doi: 10.1152/ajpgi.1996.271.3.G494. [DOI] [PubMed] [Google Scholar]
- 14.Bardag-Gorce F, et al. The importance of cycling of blood alcohol levels in the pathogenesis of experimental alcoholic liver disease in rats. Gastroenterology. 2002;123(1):325–35. doi: 10.1053/gast.2002.34177. [DOI] [PubMed] [Google Scholar]
- 15.French SW. The role of hypoxia in the pathogenesis of alcoholic liver disease. Hepatol Res. 2004;29(2):69–74. doi: 10.1016/j.hepres.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Li L, et al. Is the hypoxia-inducible factor-1 alpha mRNA expression activated by ethanol-induced injury, the mechanism underlying alcoholic liver disease? Hepatobiliary Pancreat Dis Int. 2006;5(4):560–3. [PubMed] [Google Scholar]
- 17.Hritz I, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology (Baltimore, Md) 2008 doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolganiuc A, et al. Additive inhibition of dendritic cell allostimulatory capacity by alcohol and hepatitis C is not restored by DC maturation and involves abnormal IL-10 and IL-2 induction. Alcohol Clin Exp Res. 2003;27(6):1023–31. doi: 10.1097/01.ALC.0000071745.63433.32. [DOI] [PubMed] [Google Scholar]
- 19.Romics L, et al. Increased lipopolysaccharide sensitivity in alcoholic fatty livers is independent of leptin deficiency and toll-like receptor 4 (TLR4) or TLR2 mRNA expression. Alcoholism, clinical and experimental research. 2005;29(6):1018. doi: 10.1097/01.alc.0000167744.60838.4a. [DOI] [PubMed] [Google Scholar]
- 20.Thurman RG, et al. The role of gut-derived bacterial toxins and free radicals in alcohol-induced liver injury. J Gastroenterol Hepatol. 1998;13(Suppl):S39–50. [PubMed] [Google Scholar]
- 21.Bostrom P, et al. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26(8):1871–6. doi: 10.1161/01.ATV.0000229665.78997.0b. [DOI] [PubMed] [Google Scholar]
- 22.Rankin EB, et al. HIF-2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009 doi: 10.1128/MCB.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scortegagna M, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35(4):331–40. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 24.Belanger A, et al. Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor alpha/retinoid X receptor. Biochemical and biophysical research communications. 2007;364(3):567–72. doi: 10.1016/j.bbrc.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 25.Furuta E, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–11. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- 26.Chang BH, et al. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26(3):1063–76. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motomura W, et al. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun. 2006;340(4):1111–8. doi: 10.1016/j.bbrc.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 28.Enomoto N, et al. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcoholism, clinical and experimental research. 2001;25(6 Suppl):51. doi: 10.1097/00000374-200106001-00012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.
Confirmation of active HIF1α in livers of HIF1dPA mice by gel-shift assay.
Liver nuclear extracts from pair-fed and ethanol-fed HIF1dPA and Alb-Cre control mice were prepared. HIF1α-DNA-binding activity was assayed using the electrophoretic mobility shift assay with a hypoxia-response element oligonucleotide (S1A.) There was greater HIF1α DNA-binding activity in liver nuclear extracts from HIF1dPA mice than Alb-Cre control mice (P<0.01, pair-fed HIF1dPA versus pair-fed Alb-Cre). Bands were quantified by densitometry (S1B).
Supplementary Figure S2
Hepatic ADRP mRNA quantified by real-time PCR 18 hours after LPS or sham injection in wild-type and HIF-1α(Hep-/-) mice.
Supplementary Figure S3
Real-time PCR quantification of HIF1α mRNA after treatment of cells with siRNA targeting HIF1α, scrambled control siRNA, or reagent control at 24 and 36 hours.