

Abstract
Background
Perturbations to the microscopic level balance between synaptic excitation and inhibition and neuron organization in the cerebral cortex are suggested to underlie autism spectrum disorder (ASD) traits. The mechanism linking these perturbations to cognitive behaviors in ASD is unknown. This study strives to bridge this gap by generating clinically testable diagnostic and pharmacological predictions based on the effect of synaptic imbalance and neuron distribution on a computational local circuit model of the cerebral cortex.
Methods
We use a computational microscopic model of the cerebral cortex that incorporates N-methyl-D-aspartate and gamma-aminobutyric acid synaptic kinetics. We employ the model circuit during model tasks similar to visually guided and gap oculomotor saccade tasks and interpret qualitative model predictions of saccade hypometria and dysmetria. We consider the effects of varying the excitatory to inhibitory synaptic balance, neuron density, and neuron clustering in this model.
Results
An increase of synaptic excitation over synaptic inhibition results in increased hypometria and dysmetria. Similar effects by either reduced inhibition or increased excitation suggest that a variety of pharmacological compounds can be used for both screening and medical management. On the other hand, any change to the microscopic neuron anatomy that increases the effective maximum distance between excitatory neurons decreases hypometria but has no affect on dysmetria.
Conclusions
Perturbations to a computational model of a local cerebral cortical circuit can account for saccade hypometria and dysmetria reported in ASD studies. This approach may provide a direct link between cerebral cortical function and ASD behaviors.
Keywords: Cognitive shifting, cortical circuit, fragile X syndrome, minicolumn, saccade, synaptic balance
Microscopic level cerebral cortical perturbations of synaptic balance and minicolumn structure have been postulated as etiologies for autism spectrum disorder (ASD) attributes (1-3). Synaptic imbalance refers to a perturbation of the balance between synaptic excitation and synaptic inhibition. The mechanism linking these neurophysiological parameters to ASD clinical traits is unknown. Here, we use computational modeling to make this connection. We refer to excitation-dominant synaptic imbalance as a surfeit of cortical synaptic excitation or a deficit of cortical synaptic inhibition. Clinically, excitation-dominant synaptic imbalance is suggested by the presence of ASD “hyperexcitable” comorbidities including epileptic seizure disorder and catatonia (4,5). Neurophysiologically, a decrease in the gamma-aminobutyric acid (GABA) receptor subunits α1 to α3 and β3 has been found in the parietal cortex (Brodmann area [BA] 40), α1 in the frontal cortex (BA 9), and α1 and β3 in the cerebellum of ASD postmortem tissue (6). Genetic studies of subjects with ASD show abnormalities among gene loci containing synaptic imbalance candidates, including the GABA β3 receptor subunit and genes encoding the neurorexin, neuroligin, and Shank proteins (7,8). Excitation-dominant synaptic imbalance is also suspected to play a role in fragile X syndrome, which exhibits significant ASD and hyperexcitable clinical traits as well as synaptic imbalance neurophysiology (4,9,10).
Anatomically, there is an observed increase in minicolumn density and decrease in the neuropil space between minicolumns in ASD postmortem cerebral cortical tissue, which has led to the hypothesis that increased minicolumnar density may be an etiology for ASD attributes. It has also been suggested that a loss of neuropil space decreases lateral inhibition by decreasing the number of inhibitory interneurons, thus providing an anatomical hypothesis for excitation-dominant synaptic imbalance (3,11). However, as we will demonstrate, it is also possible that a perturbation of the neuron distribution without a decrease in inhibitory neurons produces qualitative effects that are similar to but independent of excitation-dominant synaptic imbalance.
Here, we illustrate how cortical synaptic imbalance and neuron distribution may affect saccade performance. Oculomotor saccades are ballistic eye movements used in visual scanning. They are finely regulated movements controlled by the central nervous system (CNS) and are not under voluntary control once a saccade process has been initiated. As such, saccade tasks are useful in the clinical assessment of CNS dysfunction. In particular, they are useful for assessing abnormalities in rudimentary cognitive functions and anatomical regions. Previous work has shown significant saccade pathology in subjects with ASD. Behaviors described include reduced motor amplitude or gain (hypometria), increased performance variance (dysmetria), and abnormal latency to saccade initiation (12-15). Hypometria is also noted during smooth pursuit tasks (14), which use similar saccade components for acquiring sensory information, determining salience, and executing an oculomotor response.
It is known that physical and electrophysiological perturbations of the parietal lobe in typically developed adults affect saccade accuracy and latency (16,17). The lateral intraparietal area (LIP) is a fundamental component of this mechanism and is involved in the integration of sensory maps and intentions, regardless of execution. Features of the LIP include: 1) single-neuron stimulus-tuned activation, 2) persistent patterned neural activity in response to stimuli (see Methods and Materials), 3) functional columnar organization, and 4) functional on-center off-surround organization (18-23). Single-neuron stimulus tuning refers to each neuron having a specific feature for which it optimally responds and a range of similar features for which it suboptimally responds. Functional columnar organization refers to the subpopulations of neurons arranged in clusters with similar electrophysiological signal response properties. Functional on-center off-surround organization refers to a system response characteristic in which areas with increased activity are surrounded by areas with decreased activity (i.e., lateral inhibition).
We use a well-known computational model of a local microscopic cerebral cortical circuit (24-28) that satisfies these features and uses biologically plausible single-neuron and synaptic properties to model the dynamics of a saccade target encoding module similar to the LIP. Although we relate our findings to the LIP, the model we use represents a canonical cortical circuit (29) and the dynamics we describe are applicable to numerous other local circuits such as those in the prefrontal cortex (PFC). The interdependence of the PFC and LIP (20) may even imply that perturbations in LIP dynamics may be a result of PFC abnormalities. Gamma-aminobutyric acid modifying compounds injected directly into the PFC demonstrate the dependence of both visually guided and memory-guided saccade tasks on this region (30).
We show that synaptic imbalance results in increased saccade hypometria and dysmetria. We also show that dysmetria is a robust effect of synaptic imbalance, but hypometria may be decoupled from dysmetria by altering the neuron distribution or task design. We discuss the implications of these biophysical mechanisms for screening synaptic imbalance disorders and the use of pharmacological interventions for screening and therapy.
Methods and Materials
Computational Model
We consider a neurophysiologically based cerebral cortical circuit model similar to that developed by Compte et al. (25). In the absence of external input, the neurons in the circuit will remain in a quiescent state. If an input is provided, the neurons will transition to an activated state where a subpopulation of neurons around the site of the stimulus will fire persistently in the absence of any input. This response pattern is sometimes called a “bump,” which refers to the concave down, bump-like distribution of spike-firing rates over the population of neurons in the computational model. Schematics of the model circuit are shown in Figure 1.
Figure 1.

(A) Schematic of the model. Model neurons are recurrently connected (double arrows) within and between the E neuron (circles) and I neuron (square) layers. Synaptic weights between neurons are governed by the “short” (jshort) and “long” (jlong) distances between neurons on the ring structure. During the saccade tasks, an external cue current is fed into the E neuron layer as depicted. (B) Example of the effective current distribution from E neuron xi to neurons within the E layer that results from activating neighboring E and I neurons. (C) Example of neuron spatial distributions during clustering simulations. Black circles represent E neurons as in (A). The dashed line represents the maximum nearest-neighbor distance (dfar) between neurons. As clustering is increased, dfar is increased while the minimum nearest-neighbor distance (dnear) is decreased. E, excitatory; I, inhibitory.
The model consists of both excitatory and inhibitory populations. The individual neurons incorporate biophysically plausible membrane kinetics and synaptic N-methyl-D-aspartate (NMDA) and GABA channel kinetics. Our base model has 800 pyramidal (excitatory [E]) and 200 interneuron (inhibitory [I]) neurons. The neurons are arranged on a one-dimensional ring. The synaptic weights between neurons fall off with distance according to Gaussian synaptic weight functions. These are formulated such that the number of contiguous neurons activated by a stimulus in the base model is approximately one quarter of the total E neuron population length. Each E neuron is assigned a preferred stimulus (i.e., orientation angle) for which it receives a maximum current input. Parameters for the model and equations are in Supplement 1. Simulations are implemented on MATLAB (The MathWorks Inc., Natick Massachusetts) using Euler’s method with a time step of .05 msec.
Model Task
The purpose of the modeled saccade task is to evaluate how well a new piece of information is acquired, given that some other information was encoded recently. Our tasks involve two nonoverlapping consecutive cues at different gaze locations. These cues are either transient or constant. The constant cue case is similar to a visually guided saccade task, where the first cue represents the central fixation target and the second cue represents the saccade target. As there is a penalty for drifting from the central fixation target before the presentation of the saccade target in actual tasks, we assume that this first cue is perceived by the subject throughout the actual central fixation period and this is treated as a constant cue during the modeled initial period. The transient cue case is similar to a gap saccade task, where the fixation stimulus is presented briefly and then followed by the target stimulus after a temporal gap. For both constant and transient cue tasks, we model a horizontal saccade task where possible orientation angles range from 90 degrees in the left visual field to 90 degrees in the right visual field. Further details of task designs are illustrated in Figure 2.
Figure 2.

Schematic of saccade tasks. Cue current location and duration are indicated by black boxes. These were 500 pA Gaussian cues centered at the indicated marks. Arrows represent the time interval at which saccade measurements are taken. (A) Constant cue task: cues are presented for 1.5 seconds. Hypometria and dysmetria are calculated from the average single-neuron spike rate over the 2.9- to 3.0-second interval. (B) Transient cue task: 200-millisecond transient cues are presented with intervals ranging from .5 to 6.5 seconds (6.5 seconds is the default condition). Hypometria and dysmetria are calculated from the average single-neuron spike rate over the 14.9- to 15.0-second interval regardless of the cue interval.
We measured task performance over several test parameters including: 1) GABA maximum conductance (gGABA), 2) NMDA emaximum conductance (gNMDA), 3) the GABA decay time constant (τGABA), and 4) neuron distribution. The test variables for these tasks were hypometria and dysmetria. Hypometria is defined as the angle between the final gaze direction and the saccade target divided by the total angle to the target (i.e., hypometria = 1 corresponds to no saccade and hypometria = 0 corresponds to a complete saccade to the new orientation). We assume that the maximally firing E neuron codes the intended gaze direction. Given that the modeled activity bump is generally unimodal and symmetrical, we used the center of mass of the bump as an estimate for the maximally firing E neuron. This proved to be a much more robust measure than the maximum. We measured dysmetria as the width of the bump. The rationale for this choice is that a wider bump is also flatter near the maximum, which would make a subject’s estimate of the maximum (and hence the perceived saccade target) less reliable. The width of the bump is reported as the number of neurons active within a 100-msec window divided by the total number of neurons in the E neuron layer. We defined a synaptic balance measure to be (gNMDA * τNMDA)/(gGABA * τGABA). This measure captures the ratio of the integrated synaptic contributions from NMDA and GABA.
Results
The model results show several significant effects. The maximum GABA conductance gGABA had a significant effect on hypometria and dysmetria during the constant and transient cue tasks (all p values < 10−3). Table 1 lists the model predicted hypometria and dysmetria values for both tasks. The transient cue task showed the greatest sensitivity to model perturbations, so we focused on that task. Figure 3A shows hypometria increasing linearly as a function of synaptic balance. Dysmetria behaved almost identically, as seen by the strong correlation with hypometria in Figure 3B. We note that this almost exact linear dependence was not predicted a priori. We verified that increased hypometria due to excitation-dominant synaptic imbalance is robust to added external noise in the model (data not shown). As the synaptic balance measure may be altered by numerous parameters that affect excitatory and inhibitory currents, we evaluated the effect on hypometria caused by varying gGABA, gNMDA, or τGABA. As shown in Figure 3A, changes in gGABA and τGABA lead to near identical behavior, while changes in gNMDA lead to a steeper slope. The strong correlation between dysmetria and hypometria is maintained under all of these parameter changes (data not shown).
Table 1.
Saccade Performance Measures of Hypometria and Dysmetria for Constant Cue and Transient Cue Tasks
| gGABA (%) | Hypometria | Dysmetria | |
|---|---|---|---|
| Constant Cue Saccade Task | |||
| 40 | .44 | .52 | |
| 50 | .39 | .48 | |
| 60 | .34 | .45 | |
| 70 | .31 | .43 | |
| 80 | .27 | .41 | |
| 90 | .24 | .39 | |
| 100 | .23 | .37 | |
| Transient Cue Saccade Task | |||
| 40 | .83 | .48 | |
| 50 | .74 | .40 | |
| 60 | .66 | .35 | |
| 70 | .61 | .32 | |
| 80 | .57 | .29 | |
| 90 | .55 | .27 | |
| 100 | .54 | .25 |
GABA, gamma-aminobutyric acid.
Figure 3.

Synaptic balance and saccade performance measures. (A) Hypometria versus synaptic balance for changes in gGABA (circle), τGABA (triangle), and gNMDA (square). Linear regressions are plotted for gGABA (solid line, r2 = .9974), τGABA (dashed line, r2 = .9993), and gNMDA (solid line, r2 = .998). (B) Correlation between hypometria and dysmetria for changes in gGABA (r2 = .9965). GABA, gamma-aminobutyric acid; NMDA, N-methyl-D-aspartate.
We note that increasing synaptic excitation increased the transition time of the bump in response to perturbations (data not shown). We hypothesized that giving a saccade cue before the transition time would reduce hypometria. This should preferentially affect systems with greater excitation. The simulations confirm that hypometria increases as a function of the interval between cues and higher excitation (Figure 4). Changing the interval between cues had no effect on dysmetria (data not shown).
Figure 4.
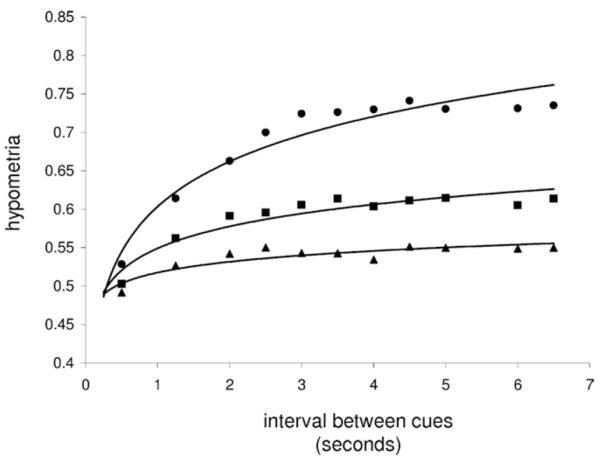
Hypometria versus cue interval with log-regressions are shown for low gGABA (50% gGABA, circle), medium gGABA (70% gGABA, square), and high gGABA (90% gGABA, triangle) simulations. GABA, gamma-aminobutyric acid.
Anatomical studies indicate that there is a perturbation of the typical neuron distribution in ASD cerebral cortical regions. Specifically, there is a reduction in the distance between clustered neurons in subjects with ASD (3,11). In this model, we implemented neuron clustering by fixing the preferred orientation angle for each E neuron but changed the spatial distribution between neurons as shown in Figure 1C. Increased clustering increases the distance between clusters but decreases the distance between neurons within a cluster (see Supplement 1 for equations). We tested the effects of neuron distribution by altering the overall neuron density or by altering the clustering of neurons. Figure 5A shows that increasing the nearest-neighbor distance by decreasing neuron density reduces hypometria and also negates the effect of synaptic imbalance on hypometria. There is an almost exact linear relationship between hypometria and the nearest-neighbor distance for both high and medium gGABA synaptic balance systems. This relationship was not expected a priori. Some fluctuations from the linear fit are likely due to finite size effects on the bump activity caused by small neuron population sizes used in the simulations. We found similar linear relationships between hypometria and maximum nearest-neighbor distance between clusters (Figure 5B). Dysmetria is not affected by density or clustering (data not shown).
Figure 5.

Hypometria versus E neuron distribution for three gGABA titrations as in Figure 4. (A) Hypometria versus nearest-neighbor distance for homogeneously distributed neurons. Linear regressions shown for low gGABA (r2 = .9946), medium gGABA (r2 = .9019), and high gGABA (r2 = .5378). Distance is set by adjusting the total number of neurons. (B) Hypometria versus maximum nearest-neighbor distance for clustered neuron systems. Linear regressions shown for low gGABA (r2 = .9959), medium gGABA (r2 = .9555), and high gGABA (r2 = .4839). Distances are set as in Figure 1C and the clustering equation in Supplement 1. E, excitatory; GABA, gamma-aminobutyric acid.
The effect of excitation-dominant synaptic imbalance on hypometria and dysmetria can be understood from how it affects the neural activity in the computational model. An increase in excitation leads to an increase in the net recurrent excitatory feedback (Figure 1B). Recurrent excitation can be thought of as self-excitation, which makes the neural activity more persistent and less responsive to external stimuli. This decreases the rate by which the system shifts between targets and hence increases hypometria. At the same time, the increase in excitation also makes the bump state representing a gaze direction wider, which increases dysmetria. This is because the bump width is a function of the recurrent on-center excitation and off-surround or lateral recurrent inhibition. By decreasing the potential recurrent inhibition (gGABA or τGABA) or increasing the potential recurrent excitation (gNMDA or τNMDA), previously quiescent neurons near the edge of the bump will receive more input that can put them above the threshold for firing. In the case of excitation-dominant synaptic imbalance, hypometria and dysmetria are correlated, since increased excitation increases the net recurrent excitation and makes a bump larger.
By contrast, changes to the microscopic neuron anatomy that increase the effective maximum distance between excitatory neurons decrease hypometria but have no affect on dysmetria. As shown previously (24,25,31), a decrease in neuron density can increase the ability of persistent activity states to move or “wander” about in the cortical column (even in the absence of added external noise), thereby making them more responsive to stimuli. Our results confirm this but also find that the maximum spacing between neuron clusters is the critical quantity that affects responsiveness of neural activity. Hence, even if the total number of neurons in a circuit is increased, thereby increasing the neuron density, the responsiveness of the bump state to external stimuli could still increase if the neurons are organized into tight clusters.
Discussion
This work highlights potential clinical applications of computational modeling such as: 1) elucidating measures of saccade performance that are directly linked to the underlying neuropathology, 2) making predictions of the diagnostic effects of saccade task designs and pharmacological interventions, 3) providing mechanisms for ameliorative pharmacotherapies, and 4) providing bottom-up clues for analyzing data from diagnostic platforms such as functional magnetic resonance imaging (fMRI). Our results demonstrate that saccade performance may be a means to discriminate between control and ASD populations and between ASD subpopulations. For example, the presence of hypometria without dysmetria may indicate a perturbation in neuron organization or density. Dysmetria without hypometria may indicate excitation-dominant synaptic imbalance together with a concomitant increase in the spacing between neurons. The presence of both may indicate excitation-dominant synaptic imbalance or a combination of excitation-dominant synaptic imbalance with decreased neuron spacing.
Our computational results agree with reported ASD saccade abnormalities in human subjects (12-15), although reports are inconsistent (32,33). However, an ASD study showing no saccade abnormalities did find differences in functional imaging that might indicate compensatory mechanisms that helped to equalize saccade performance (33). Also, previous data suggest that baseline saccade gain of both ASD and control groups are close to peak performance, which reduces the ability to resolve differences between groups. Our model suggests changes to task design and pharmacological challenges that may improve diagnostic resolution (Figure 6). We predict that transient cue tasks should provide a sharper test than constant cue tasks. This is predicted by the model dynamics, which show that decreasing the interval between cues normalizes hypometria between synaptic balance systems but has no affect on dysmetria. Also, according to this mechanism, excitation-dominant synaptic imbalance decreases the rate of shifting between cues. By extension, increasing the saccade cue duration will also normalize hypometria. These phenomena have not been explicitly tested in clinical studies. A possible complication of our hypotheses is the facilitation effect reported during saccade tasks with predictable cue intervals. However, a recent study has shown a decrease in facilitation by infant siblings of subjects with ASD (34). In addition, we predict that facilitation may be abrogated by introducing an unpredictable interval duration. Data from one ASD study also indirectly support the effect of saccade cue duration. This study involved a saccade followed immediately by smooth pursuit of a moving target and showed that increased saccade hypometria in both the control and ASD groups appeared to be correlated with increased velocity of the target, which can be interpreted as a decrease in the saccade cue duration. Subjects with ASD also appeared to exhibit more saccade hypometria than control subjects at higher target velocities (14), suggesting that briefer saccade cues provide greater hypometric discrimination between control and ASD groups.
Figure 6.
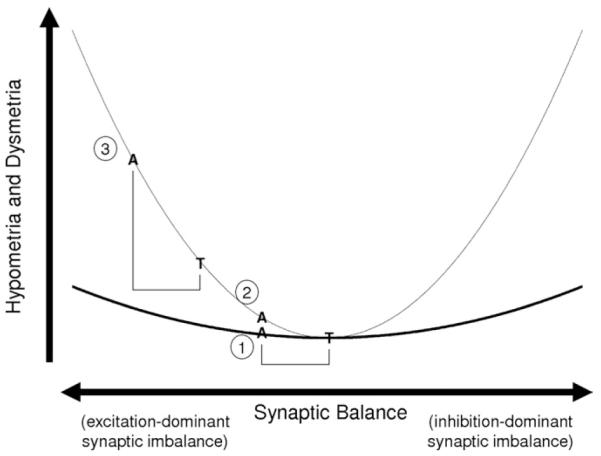
Saccade pathology (e.g., hypometria and dysmetria) is predicted to be a U-shaped function of synaptic balance with typically developed adults (T) at the bottom. Subjects with ASD (A) are hypothesized to be on the left branch. The black curve represents a task with poor resolution (e.g., a visually guided task). The gray curve is from a task with better resolution (e.g., a random gap, transient cue task). Case 1 has the weakest discrimination, which is improved by altering the task design, i.e., shifting to the gray curve (case 2). Further improved discrimination is achieved by using a GABA antagonist or NMDA agonist (case 3). A, subjects with ASD; ASD, autism spectrum disorder; GABA, gamma-aminobutyric acid; NMDA, N-methyl-D-aspartate; T, typically developed adults.
This neurophysiologically based model provides a means for predicting diagnostic and ameliorative effects of pharmacological compounds. The model predicts that synaptic balance-modifying compounds (e.g., GABA and NMDA channel compounds) can shift the performance of subjects with ASD and those without ASD to regions of the performance curve with better discrimination. Conversely, pharmacotherapies can be used to improve saccade performance in subjects with ASD that would otherwise reduce performance in subjects without synaptic imbalance (35,36). We predict that too little localized excitation may result in a decrease in performance because of a loss of signal strength and a failure to suppress background noise activity in the PFC or LIP (37). This would imply that hypometria and dysmetria are related to synaptic balance by a U-shaped curve, where too much or too little localized excitation will increase hypometria and dysmetria (Figure 6). Our simulations are restricted to the left side of this curve (excitation-dominant side). We have not analyzed the right side of this curve (inhibition-dominant synaptic imbalance) because it is likely to involve other neural areas, as the bump state is not stable in this regime without added external inputs.
The neurophysiologically based model also suggests that subphenotypes for ASD such as saccade hypometria and dysmetria (i.e., more basic traits that underlie complex clinical phenotypes) can be used to provide a mechanistic link between microscopic perturbations and complex traits. The model provides a context for investigating ameliorative rather than symptomatic pharmacotherapies for ASD with respect to subphenotypes as well as ASD phenotypes. We predict that improvements may be most dramatic in children because of possible pathological sequelae and compensatory changes in other neural mechanisms that occur over time. There is some clinical anecdotal evidence for the ameliorative affects of low-dose ketamine or dextromethorphan (which affect synaptic balance) in subjects with ASD (A.W. Zimmerman, M.D., oral communication, November 2008) and recent work has shown the effectiveness of GABA agonists such as benzodiazepines and NMDA antagonists such as memantine for catatonia (38,39), which is a major comorbidity of ASD and possibly the result of excitation-dominant synaptic imbalance (5,40).
While our model suggests that clinically important consequences can arise from single-neuron interactions, inaccessibility of such data in patients is an obstacle in making definitive diagnoses. Computational modeling provides clues as to what qualitative features are directly related to these dynamics on multiple scales. This insight is useful for interpreting data from psychophysics studies (e.g., oculomotor tasks) and data from noninvasive platforms such as fMRI. For instance, excitation-dominant synaptic imbalance increases persistent activity, which may correlate well with failures to deactivate neural networks as previously reported (41,42). Combining results from fMRI measurements and psychophysical measurements may also help to resolve underlying neuropathology. For example, the failure to deactivate cerebral cortical regions and saccade hypometria and dysmetria may help to resolve excitation-dominant synaptic imbalance from inhibition-dominant synaptic imbalance.
Although this model provides novel insights into the possible underlying biophysical processes in ASD, it will be useful to explore additional biological factors that are thought to be relevant to ASD neuropathology. This includes evaluating the robustness of these effects to biophysical properties we omitted. For example, it will be useful to see if our results are robust to spike-frequency adaptation and other synaptic plasticity dynamics. We assumed that local PFC and LIP perturbations permeate well through the network, as indicated by lesion studies. However, it is possible that local perturbations have important nonintuitive effects on network behavior. Further modeling will be useful in understanding what additional local circuit biological parameters and biophysical properties are critical for oculomotor effects, how local circuit perturbations affect overall network dynamics, and how perturbations of other saccade network components such as the cerebellum or basal ganglia affect overall saccade network dynamics. Combining microscopic and macroscopic data from ASD studies and basic sciences will be important in reducing the large pool of potential models to a few with the most likely clinical significance.
Prior studies have suggested that cerebellar dysfunction may account for hypometria and dysmetria in ASD (13). However, it has also been argued that patterns of oculomotor findings, including saccade abnormalities, are not entirely consistent with cerebellar lesions (e.g., task-dependent presence of saccade abnormalities and task-dependent regions of the affected visual field) (14,43,44). It is also suggested that cerebellar findings may be acquired rather than neurodevelopmental (45). This article provides alternative, cerebral cortical explanations for saccade pathology. In addition, since the model is a canonical cerebral cortical model, we hypothesize that these properties extend to other cortical regions and cognitive shifting tasks in which dysmetria is the variability of behavioral results and hypometria is a correlate of perseverative behavior. Further work should be directed at elucidating these alternative explanations for ASD phenotypes and subphenotypes. Appropriate clinical management depends on understanding these mechanisms and being able to differentiate subjects with ASD phenotypes due to different underlying mechanisms.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases.
We acknowledge Dr. Andrew W. Zimmerman, Director of Medical Research at the Center for Autism and Related Disorders, Kennedy Krieger Institute, for his clinical input on this article.
Footnotes
The authors reported no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online.
References
- 1.Rubenstein JL, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hussman JP. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord. 2001;31:247–248. doi: 10.1023/a:1010715619091. [DOI] [PubMed] [Google Scholar]
- 3.Casanova MF, Buxhoeveden DP, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: Implications for autism. Neuroscientist. 2003;9:496–507. doi: 10.1177/1073858403253552. [DOI] [PubMed] [Google Scholar]
- 4.Accardo PJ. Capute and Accardo’s Neurodevelopmental Disablities in Infancy and Childhood. Paul Brooks Publishing; Baltimore, MD: 2008. [Google Scholar]
- 5.Wing L, Shah A. Catatonia in autistic spectrum disorders. Br J Psychiatry. 2000;176:357–362. doi: 10.1192/bjp.176.4.357. [DOI] [PubMed] [Google Scholar]
- 6.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009;39:223–230. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abrahams BS, Geschwind DH. Advances in autism genetics: On the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourgeron T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb Symp Quant Biol. 2007;72:645–654. doi: 10.1101/sqb.2007.72.020. [DOI] [PubMed] [Google Scholar]
- 9.Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered up states reflect network hyperexcitability in the mouse model of fragile x syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curia G, Papouin T, Seguela P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile x syndrome. Cereb Cortex. 2009;19:1515–1520. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casanova MF, van Kooten IA, Switala AE, van Engeland H, Heinsen H, Steinbusch HW, et al. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287–303. doi: 10.1007/s00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- 12.Rosenhall U, Johansson E, Gillberg C. Oculomotor findings in autistic children. J Laryngol Otol. 1988;102:435–439. doi: 10.1017/s0022215100105286. [DOI] [PubMed] [Google Scholar]
- 13.Takarae Y, Minshew NJ, Luna B, Sweeney JA. Oculomotor abnormalities parallel cerebellar histopathology in autism. J Neurol Neurosurg Psychiatry. 2004;75:1359–1361. doi: 10.1136/jnnp.2003.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takarae Y, Minshew NJ, Luna B, Krisky CM, Sweeney JA. Pursuit eye movement deficits in autism. Brain. 2004;127:2584–2594. doi: 10.1093/brain/awh307. [DOI] [PubMed] [Google Scholar]
- 15.Luna B, Doll SK, Hegedus SJ, Minshew NJ, Sweeney JA. Maturation of executive function in autism. Biol Psychiatry. 2007;61:474–481. doi: 10.1016/j.biopsych.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 16.Duhamel JR, Goldberg ME, Fitzgibbon EJ, Sirigu A, Grafman J. Saccadic dysmetria in a patient with a right frontoparietal lesion. The importance of corollary discharge for accurate spatial behaviour. Brain. 1992;115:1387–1402. doi: 10.1093/brain/115.5.1387. [DOI] [PubMed] [Google Scholar]
- 17.Kapoula Z, Yang Q, Coubard O, Daunys G, Orssaud C. Role of the posterior parietal cortex in the initiation of saccades and vergence: Right/left functional asymmetry. Ann N Y Acad Sci. 2005;1039:184–197. doi: 10.1196/annals.1325.018. [DOI] [PubMed] [Google Scholar]
- 18.Barash S, Bracewell RM, Fogassi L, Gnadt JW, Andersen RA. Saccade related activity in the lateral intraparietal area. II. Spatial properties. J Neurophysiol. 1991;66:1109–1124. doi: 10.1152/jn.1991.66.3.1109. [DOI] [PubMed] [Google Scholar]
- 19.Bracewell RM, Mazzoni P, Barash S, Andersen RA. Motor intention activity in the macaque’s lateral intraparietal area. II. Changes of motor plan. J Neurophysiol. 1996;76:1457–1464. doi: 10.1152/jn.1996.76.3.1457. [DOI] [PubMed] [Google Scholar]
- 20.Chafee MV, Goldman-Rakic PS. Inactivation of parietal and prefrontal cortex reveals interdependence of neural activity during memory-guided saccades. J Neurophysiol. 2000;83:1550–1566. doi: 10.1152/jn.2000.83.3.1550. [DOI] [PubMed] [Google Scholar]
- 21.Hamed S Ben, Duhamel JR. Ocular fixation and visual activity in the monkey lateral intraparietal area. Exp Brain Res. 2002;142:512–528. doi: 10.1007/s00221-001-0954-z. [DOI] [PubMed] [Google Scholar]
- 22.Melcher D, Colby CL. Trans-saccadic perception. Trends Cogn Sci. 2008;12:466–473. doi: 10.1016/j.tics.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Fanini A, Assad JA. Direction selectivity of neurons in the macaque lateral intraparietal area. J Neurophysiol. 2009;101:289–305. doi: 10.1152/jn.00400.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laing CR, Chow CC. Stationary bumps in networks of spiking neurons. Neural Comput. 2001;13:1473–1494. doi: 10.1162/089976601750264974. [DOI] [PubMed] [Google Scholar]
- 25.Compte A, Brunel N, Goldman-Rakic PS, Wang XJ. Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical networked model. Cereb Cortex. 2000;10:913–923. doi: 10.1093/cercor/10.9.910. [DOI] [PubMed] [Google Scholar]
- 26.Wang XJ. Toward a prefrontal microcircuit model of cognitive deficits in schizophrenia. Pharmacopsychiatry. 2006;39:S80–S87. doi: 10.1055/s-2006-931501. [DOI] [PubMed] [Google Scholar]
- 27.Carter E, Wang XJ. Cannabinoid-mediated disinhibition and working memory: Dynamical interplay of multiple feedback mechanisms in a continuous attractor model of prefrontal cortex. Cereb Cortex. 2007;17:i16–i26. doi: 10.1093/cercor/bhm103. [DOI] [PubMed] [Google Scholar]
- 28.Edin F, Klingberg T, Johansson P, McNab F, Tegner J, Compte A. Mechanism for top-down control of working memory capacity. Proc Natl Acad SciUSA. 2009;106:6802–6807. doi: 10.1073/pnas.0901894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douglas RJ, Martin KA. Mapping the matrix: The ways of neocortex. Neuron. 2007;56:226–238. doi: 10.1016/j.neuron.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 30.Dias EC, Kiesau M, Segraves MA. Acute activation and inactivation of macaque frontal eye field with GABA-related drugs. J Neurophysiol. 1995;74:2744–2748. doi: 10.1152/jn.1995.74.6.2744. [DOI] [PubMed] [Google Scholar]
- 31.Chow CC, Coombes S. Existence and wandering of bumps in a spiking neural network model. SIAM Journal on Applied Dynamical Systems. 2006;5:552–574. [Google Scholar]
- 32.D’Cruz AM, Mosconi MW, Steele S, Rubin LH, Luna B, Minshew N, et al. Lateralized response timing deficits in autism. Biol Psychiatry. 2009;66:393–397. doi: 10.1016/j.biopsych.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takarae Y, Minshew NJ, Luna B, Sweeney JA. Atypical involvement of frontostriatal systems during sensorimotor control in autism. Psychiatry Res. 2007;156:117–127. doi: 10.1016/j.pscychresns.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elsabbagh M, Volein A, Holmboe K, Tucker L, Csibra G, Baron-Cohen S, et al. Visual orienting in the early broader autism phenotype: Dis-engagement and facilitation. J Child Psychol Psychiatry. 2009;50:637–642. doi: 10.1111/j.1469-7610.2008.02051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ball DM, Glue P, Wilson S, Nutt DJ. Pharmacology of saccadic eye movements in man: 1. Effects of the benzodiazepine receptor ligands midalozam and flumanzenil. Psychopharmacology (Berl) 1991;105:361–367. doi: 10.1007/BF02244431. [DOI] [PubMed] [Google Scholar]
- 36.Reilly JL, Lencer R, Bishop JR, Keedy S, Sweeney JA. Pharmacological treatment effects on eye movement control. Brain Cogn. 2008;68:415–435. doi: 10.1016/j.bandc.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunel N, Wang XJ. Effects of neuromodulation in a cortical network model of object working memory dominated by recurrent inhibition. J Comput Neurosci. 2000;11:63–85. doi: 10.1023/a:1011204814320. [DOI] [PubMed] [Google Scholar]
- 38.Carroll BT, Goforth HW, Thomas C, Ahuja N, McDaniel WW, Kraus MF, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19:406–412. doi: 10.1176/jnp.2007.19.4.406. [DOI] [PubMed] [Google Scholar]
- 39.Fink M, Taylor MA, Ghaziuddin N. Catatonia in autistic spectrum disorders: A medical treatment algorithm. Int Rev Neurobiol. 2006;72:233–244. doi: 10.1016/S0074-7742(05)72014-6. [DOI] [PubMed] [Google Scholar]
- 40.Northoff G, Steinke R, Czcervenka C, Krause R, Ulrich S, Danos P, et al. Decreased density of GABA-A receptors in the left sensorimotor cortex in akinetic catatonia: Investigation of in vivo benzodiazepine receptor binding. J Neurol Neurosurg Psychiatry. 1999;67:445–450. doi: 10.1136/jnnp.67.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller RA, Pierce K, Ambrose JB, Allen G, Courchesne E. Atypical patterns of cerebral motor activation in autism: A functional magnetic resonance study. Biol Psychiatry. 2001;49:665–676. doi: 10.1016/s0006-3223(00)01004-0. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy DP, Redcay E, Courchesne E. Failing to deactivate: Resting functional abnormalities in autism. Proc Natl Acad Sci U S A. 2006;103:8275–8280. doi: 10.1073/pnas.0600674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minshew NJ, Luna B, Sweeney JA. Oculomotor evidence for neocortical systems but not cerebellar dysfunction in autism. Neurology. 1999;52:917–922. doi: 10.1212/wnl.52.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowinski CV, Minshew NJ, Luna B, Takarae Y, Sweeney JA. Oculomotor studies of cerebellar function in autism. Psychiatry Res. 2005;137:11–19. doi: 10.1016/j.psychres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Casanova MF. The neuropathology of autism. Brain Pathol. 2007;17:422–433. doi: 10.1111/j.1750-3639.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.