

Abstract
Frontotemporal lobar degeneration with TDP- 43 inclusions (FTLD-TDP) is characterized by progressive decline in behavior, executive function, and language. Progranulin (GRN) gene mutations are pathogenic for FTLD-TDP, and GRN transcript haploinsufficiency is the proposed disease mechanism. However, the evidence for this hypothesis comes mainly from blood-derived cells; we measured progranulin expression in brain. We characterized mRNA and protein levels of progranulin from four brain regions (frontal cortex, temporal cortex, occipital cortex, and cerebellum) in FTLD-TDP patients with and without GRN mutations, as well as neurologically normal individuals. Moreover, we performed immunohistochemistry to evaluate the degree of TDP-43 pathology and microglial infiltration present in these groups. In most brain regions, patients with GRN mutations showed mRNA levels comparable to normal controls and to FTLD-TDP without GRN mutations. However, GRN transcript levels in a brain region severely affected by disease (frontal cortex) were increased in mutation-bearing patients. When compared with normal individuals, GRN mutation-bearing cases had a significant reduction in the amount of progranulin protein in the cerebellum and occipital cortex, but not in the frontal and temporal cortices. In GRN mutant cases, GRN mRNA originated from the normal allele, and moderate microglial infiltration was observed. In conclusion, GRN mutation carriers have increased levels of mRNA transcript from the normal allele in brain, and proliferation of microglia likely increases progranulin levels in affected regions of the FTLD-TDP brain, and whether or not these findings underlie the accumulation of TDP-43 pathology in FTLD-TDP linked to GRN mutations remains to be determined.
Keywords: Progranulin, TDP-43, Frontotemporal dementia, Frontotemporal lobar degeneration, Microglia
Introduction
Frontotemporal dementia (FTD) is the second most common cause of dementia in individuals under age 65 [39]. Clinically, the disease manifests with progressive decline in behavior, social and executive function, and/or language [34]. Pathologically, FTD most frequently manifests as frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) [2, 13, 21, 32, 36, 41]. Genetically, mutations in the progranulin gene (GRN) have been associated with FTLD-TDP [5, 16].
GRN consists of 13 exons encoding a highly glycosylated 593 amino acid precursor protein with a predicted molecular mass of 63.5 kDa [6, 9, 45]. The progranulin protein (also called granulin) is expressed in many tissues, with a low to medium level of expression in the brain [10] and it is believed to function generally in inflammation and wound repair [26]. The progranulin protein encompasses 7.5 cysteine-rich granulin peptide domains that are separated by linker sequences and are disulfide bridged. This precursor protein is secreted and cleaved at sites in the linker sequences to generate granulin peptides [26]. Evidence suggests that progranulin and these resulting granulin peptides may have opposing effects on processes, such as cell growth, survival, and cell cycling [25, 37, 47, 50].
GRN mutations are pathogenic for FTLD-TDP, and in clinical FTD patients unselected for family history, the GRN mutation frequency is approximately 5% [22, 28]. In familial FTD, the mutation frequency rises to 12–25% [12, 22, 27, 28]. Inherited in an autosomal dominant manner, GRN mutations are believed to act through a haploinsufficiency mechanism [17]. To date, over 60 GRN mutations have been reported, and the majority of them result in premature termination of the GRN transcript (AD and FTD mutation database; http://www.molgen.ua.ac.be/FTDMutations). In cases where this has been examined, patients with GRN mutations do not appear to express the mutant transcript, which is lost by nonsense-mediated mRNA decay [5, 16, 22]. Indeed, decreases in GRN mRNA from patient blood samples can be detected by microarray and predict the presence of a GRN mutation [15]. In addition, in the original GRN mutation reports, progranulin protein levels were evaluated in lymphoblastoid cell lines derived from mutation carriers and shown to be reduced by 30–35% when compared with controls [5, 16]. More recently, decreased progranulin protein levels have been found in serum [46], plasma [20], and CSF [24] samples from GRN mutation carriers.
However, few studies have evaluated GRN mRNA transcript or protein levels from the brains of mutation carriers [29]. Because transcript levels are likely to show tissue-specific variability, and the brain is the diseased organ in FTLD-TDP, we investigated mRNA transcript and protein levels from the brains of normal individuals, FTLD-TDP patients with GRN gene mutations (GRN+ FTLD-TDP), and FTLD-TDP patients without GRN gene mutations (GRN− FTLD-TDP). To do this, we sampled four brain regions ranging from most to least histopathologically affected in FTLD-TDP.
Materials and methods
Human samples
We dissected brain regions from human postmortem samples from the University of Pennsylvania Center for Neurodegenerative Disease Research Brain Bank as previously described [14]. Neurologically, normal controls, GRN+ FTLD-TDP (Table 1), and GRN− FTLD-TDP were sampled for four regions of the brain; see Supplementary Table 1 for details on numbers sampled in each disease group and brain region. Samples comprised predominantly gray matter. Regions consisted of frontal cortex (anterior to genu of corpus callosum), temporal cortex (temporal pole), occipital cortex (occipital pole), and cerebellum (cerebellar hemisphere). A minimum of three individuals were sampled for each region and disease category (details in Supplementary Table 1). Because GRN mutants fall exclusively in FTLD-TDP histopathological subtype 3 [31, 42] (Geser et al. in press, J Neurol), GRN− FTLD-TDP samples came primarily from this subtype.
Table 1.
Progranulin (GRN) genetic variants included in this study
| Cases | cDNA | Protein | Diagnosis | Tissue | References |
|---|---|---|---|---|---|
| 1, 2 | c.1252C>T | R418X | FTLD-TDP (autopsy) | Brain | [5] |
| 3 | c.1477C>T | R493X | FTLD-TDP (autopsy) | Brain | [22] |
| 4 | c.348A>C | A89VfsX139 | FTLD-TDP (autopsy) | Brain | a |
| 5 | c.911G>A | W304X | FTLD-TDP (autopsy) | Brain | [22] |
| 6b | c.1009C>T | Q337X | FTLD-TDP (autopsy) | Brain | [48] |
| c.903G>A | S301S | ||||
| c.1070C>A | P357R | ||||
| 7 | c.1179 + 2T>C | V395YfsX29 | FTLD-TDP (autopsy) | Brain | a |
| 8 | c.26C>A | A9D | FTLD-TDP (autopsy) | Brain | [22] |
| 9 | c.1414-2A>G | A472VfsX10 | FTD/PPA (clinical) | Blood | a |
| 10 | c.1317_1318delCA | D441HfsX4 | FTLD-TDP (autopsy) | Blood | a |
| 11 | c.154delA | T52HfsX2 | FTLD-TDP (autopsy) | Blood | [22] |
All variants are believed to be pathogenic. For cases 1–8, brain tissue was analyzed. For cases 9–11, peripheral blood mRNA was used. For cases 10 and 11, autopsy information subsequently became available and is shown in the table. Case 8, with the GRN variant c.26C>A, was included in protein studies only, as others have shown that this mutation may not show mRNA transcript haploinsufficiency [35]
FTLD-TDP frontotemporal lobar degeneration with TDP-43 inclusions, FTD frontotemporal dementia, PPA primary progressive aphasia
Yu et al. (in press) Arch Neurol
Case has three progranulin variations
Peripheral blood samples were collected from four neurologically normal controls and three FTD patients with GRN gene mutations (Table 1). Six of the seven blood samples were collected in PAXgene (PreAnalytiX, Qiagen/ BD, Valencia, CA) tubes which were processed immediately or stored at −80°C until RNA isolation. In one case from a different region of the country, total RNA was isolated from a buffy coat drawn from the patient.
For brain samples, pathological characterization as normal or FTLD-TDP was made by a board-certified neuropathologist following consensus criteria [34] as described in previous publications [13]. For blood samples, clinical diagnoses of FTD were made by a specialist in cognitive neurology according to consensus criteria [34] as summarized earlier [21]. We also verified that control individuals did not possess GRN gene mutations by direct sequencing of all coding exons. Informed consent was obtained for all studies. GRN gene mutations are described in Table 1.
RNA isolation and quantitation
RNA was prepared from postmortem brain samples as previously described [14] and RNA quality was verified using an Agilent 2100 Bioanalyzer both subjectively (sharp 28s and 18s peaks) and objectively (RIN>6 for QRT-PCR, RIN>8 for microarray [43]. For blood samples in PAXgene tubes (PreAnalytiX, Qiagen/BD), total RNA was isolated according to standard manufacturer protocols.
For microarray analyses, total RNA isolated from postmortem brain samples was analyzed on Affymetrix U133A microarray platforms as previously described [14]. Statistical correction for multiple-hypothesis testing was performed using the Benjamini–Hochberg method [8].
Quantitative reverse transcription PCR (QRT-PCR) was performed on the Applied Biosystems 7500 Fast real-time PCR system as previously described [14]. Gene expression was quantified using the delta–delta CT method [30], and the geometric mean of two housekeeping genes (β-actin and cyclophilin A), previously shown to have stable expression in FTLD-TDP and normal samples [14]. All samples were normalized to a common reference sample to allow comparisons between different brain regions. ANOVA was used to compare GRN expression across groups; Tukey’s test was employed for between-group comparisons.
Detailed information on primers is given in Supplementary Table 2.
DNA sequencing and allele-specific expression analysis
GRN mutations were identified by genomic DNA sequencing as previously described [48]. Mutations in GRN were confirmed to be absent from control blood samples by bidirectional sequencing of coding exons. For brain samples from individuals with GRN mutations, cDNA obtained from reverse transcription of RNA was amplified (RTPCR) and the region around the mutation was sequenced in order to determine whether one or both GRN alleles were expressed. Primer sequences are given in Supplementary Table 2.
Sequential biochemical fractionation and immunoblotting
Sequential biochemical fractionation of postmortem human brain samples was performed. Gray matter samples were sequentially extracted in buffers of increasing strength: Tris/30% sucrose (10 mM Tris–HCl, 2 mM EDTA, pH 7.5 and 30% sucrose), RIPA [0.1% sodium dodecyl sulfate (SDS), 1% NP-40, 0.5% deoxycholic acid, 5 mM EDTA, 150 mM NaCl, 50 mM Tris–HCl, pH 8.0], and 7 M urea. Immunoblotting was performed as previously described [42].
Generation and characterization of novel antibodies
Anti-progranulin antibodies used in this study included a rabbit anti-C-terminus polyclonal antibody (rabbit anti-C) and two monoclonal antibodies (mAbs 407 and 2126) developed for this purpose.
To generate monoclonal antibodies, a His-tagged human progranulin expression plasmid was created by PCR (primers given in Supplementary Table 2) which was then used to generate a HEK293 cell line stably expressing Hisprogranulin. His-progranulin secreted into the media was purified using a Nickel column per manufacturer’s instructions. To generate antibodies that recognize a reduced form of progranulin, purified native His-progranulin containing multiple disulfide bridges was reduced with DTT (100 mM) and run on SDS-polyacrylamide gels. The portion of the gel containing denatured His-progranulin protein was excised, homogenized in phosphate-buffered saline, emulsified with incomplete Freund’s adjuvant, and injected subcutaneously into BALB/c mice. To generate antibodies that recognize native progranulin, His-progranulin protein was emulsified with incomplete Freund’s adjuvant without DTT treatment and injected into BALB/c mice. Subcutaneous boosts (25 μg protein/mouse) were injected on days 21, 35, and 49, followed by intraperitoneal injection of immunogens without adjuvant on day 63. On day 66, the spleen was removed and spleen lymphocytes were fused to Sp2 myeloma cells to produce hybridomas. Resulting hybridoma supernatants were screened by ELISA and immunoblotted against purified progranulin protein in reduced or non-reduced forms.
To generate the rabbit polyclonal antibody, a C-terminal (Ct) peptide corresponding to amino acid residues 523–590 (CLRREAPRWDAPLRDPALR) of progranulin was synthesized and used to immunize rabbits. The antiserum was purified by His-progranulin affinity chromatography using the sulfoLink kit (Pierce, IL) for conjugating the protein to the resin.
We used several methods to verify the specificity of our antibodies for progranulin, including deglycosylation, knockdown, and overexpression experiments. For deglycosylation of the highly glycosylated progranulin protein, we used PNGase F (New England Biolabs) according to standard manufacturer protocols. For knockdown experiments, small interfering RNAs targeted against GRN or sense sequence control (Qiagen) were transfected into HEK293 cells with the siLentFect transfection reagent (BioRAD, Hercules, CA) according to manufacturer instructions. Seventy-two hours after transfection, cells were washed twice with phosphate-buffered saline followed by lysis in cold RIPA buffer with sonication. Cell lysates were cleared by centrifugation at 100,000×g for 30 min at 4°C, and samples were immunoblotted for progranulin expression. Similar transfection and sample preparation procedures were followed for overexpression experiments except that in this case a plasmid containing the His-tagged progranulin construct was transfected into cells.
Enzyme-linked immunosorbent assay
Sandwich ELISAs were used for the measurement of progranulin from tissue extracts. Specifically, mAB 2161 (recognizing native progranulin, see “Generation and characterization of novel antibodies”) was used as the capturing antibody, and our rabbit anti-progranulin Ct polyclonal antibody was used as the detection antibody. A 384-well plate was coated with 10 μg/ml of mAb 2161 overnight at 4°C, followed by blocking (2 h, 37°C). A 30 μl of human brain extract (RIPA fraction, see “Sequential biochemical fractionation”) was incubated with the capture antibody-coated plates (2 h, 37°C), followed by washing with PBS–Tween. Anti-progranulin Ct rabbit polyclonal antibody (1 μg/ml) was added to the plate and incubated for 1.5 h at 37°C. Samples were washed, and a HRP-conjugated goat anti-rabbit antibody was added and incubated for 1 h. The plate was washed, and TMB solution was added (room temperature × 10 min). Finally, the reaction was stopped with 0.1 M phosphoric acid, and the ELISA plate was read by optical densitometry at 450 nm. ANOVA was used to compare the progranulin expression across groups, and Tukey’s test was employed for between-group comparisons.
Immunohistochemistry
Cases were examined by routine neuropathologic diagnostic techniques as described [13, 21, 36]. Frozen brain tissues adjacent to the areas evaluated for mRNA and protein studies were fixed in 70% ethanol with 150 mM sodium chloride, paraffin-embedded, and cut into 6-μm sections. Immunohistochemistry was performed by means of the BioGenex Super Sensitive MultiLink IHC Detection System Kit (BioGenex Laboratories, San Ramon, CA) with 3,3-diaminobenzidine as the chromogen. Primary antibodies used were: rabbit polyclonal anti-TDP-43 (Protein-Tech Group, Chicago, IL; 1:8,000), mouse anti-HLA-DR mAb (DakoCytomation, Glostrup, Denmark; 1:5,000), and rabbit polyclonal anti-glial fibrillary acidic protein MAb (DakoCytomation, Glostrup, Denmark; 1:10,000). Sections stained for TDP-43 and HLADR were pre-treated by boiling in citrate antigen unmasking solution (Vector Laboratories Burlingame, CA).
TDP-43 pathology severity and microglial proliferation in the frontal cortex were graded using a 4-point arbitrary ordinal scale (0 none, 1 rare/mild, 2 occasional/moderate, 3 numerous/severe) by a neuropathologist (MM-L) blinded to clinical and pathological diagnoses as well as GRN status. White and gray matters were scored individually. Medians and interquartile ranges for each group were calculated based on the grouped data.
Results
Quantification of GRN transcript levels in peripheral blood
We isolated total peripheral blood RNA from neurologically normal controls and FTD patients harboring GRN mutations (cases 9–11, Table 1). Using QRT-PCR, we showed that GRN mutation carriers had a significant reduction in mRNA transcripts compared with neurologically normal controls (P = 0.03, Supplemental Fig. 1). This corroborates previous work demonstrating that GRN+ FTLD-TDP patients show decreased GRN mRNA levels in lymphoblastoid cell lines [5, 16] and in mRNA isolated directly from whole blood [15].
Quantification of progranulin transcript levels in brain
Next, we examined mRNA isolated from four regions of the brain. Two regions (frontal cortex and temporal cortex) show marked histopathological involvement in FTLD-TDP, while two regions (occipital cortex and cerebellum) are relatively histopathologically spared [23]. We measured GRN transcript levels by QRT-PCR in brain samples from GRN+ FTLD-TDP (cases 1–7, Table 1), GRN− FTLD-TDP, and neurologically normal controls (Fig. 1). Of note, we performed this analysis without GRN mutation c.26C>A, as it has been shown to result in protein haploinsuffiency, but not mRNA haploinsufficiency [35], unlike the other GRN mutations in our study. Surprisingly, we did not see a decrease in GRN transcript level in mutation carriers compared with individuals without mutations. Rather, in frontal cortex (Fig. 1a), the brain region most affected in FTLD-TDP, patients with GRN mutations showed a significant elevation in GRN transcript relative to normal controls (ANOVA P = 0.02, normal vs. GRN mutant P\0.05). In temporal cortex, occipital cortex, and cerebellum (Fig. 1b–d), no significant differences were seen in mRNA transcript levels. An increase in GRN transcript abundance in the frontal cortex but not in other brain regions was true at the level of individual GRN mutation-bearing patients as well (Supplementary Fig. 2). Moreover, similar results were obtained using a second set of GRN primers directed at the opposite (5′) end of the transcript (Supplementary Fig. 3).
Fig. 1.
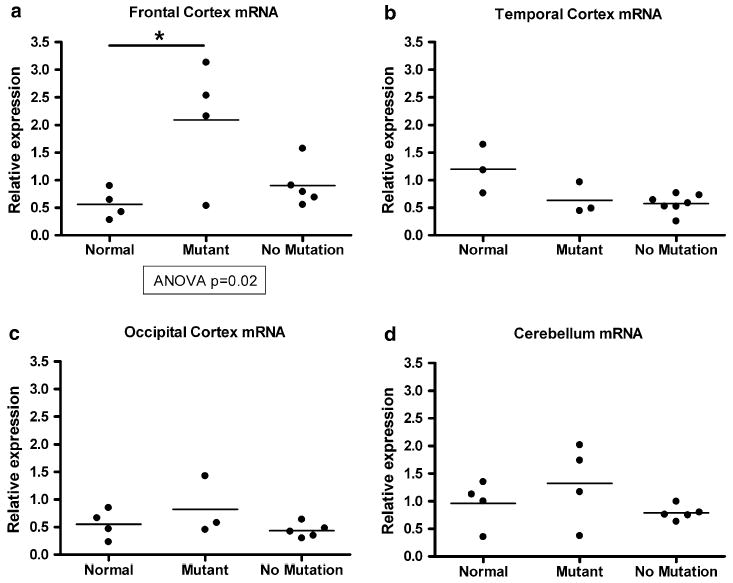
Quantification of GRN mRNA transcript levels from brain samples in normal controls (normal), FTLD-TDP with GRN mutations (mutant), and FTLD-TDP without GRN mutations (no mutation). a Quantitative reverse transcription PCR (QRT-PCR) shows that GRN mRNA is increased in frontal cortex samples from FTLD-TDP cases with GRN mutations; increase relative to normal control is significant (P < 0.05). b–d QRT-PCR shows equivalent amounts of temporal cortex, occipital cortex, and cerebellar GRN mRNA in normal controls and FTLD-TDP with and without GRN mutations. QRT-PCR reactions were performed in duplicate, standardized to the geometric mean of two housekeeping genes (cyclophilin A and β-actin), and normalized to control. Individual cases are shown as circles, and the mean for each group is shown as a bar. ANOVA was used to evaluate progranulin mRNA levels across groups, followed by Tukey’s test for pairwise comparisons
The results were replicated on a larger number of individuals, using a different methodology. We had previously performed global mRNA expression profiling from frontal cortex and cerebellum in 11 neurologically normal individuals, as well as 17 FTLD-TDP cases with (n = 7) and without (n = 10) GRN gene mutations [14]. Within the U133A microarray platforms used, GRN is represented by three probes. We queried the expression of these probes for the subset of GRN+ FTLD-TDP versus neurologically normal controls, as previously described [14]. Microarray data from this cohort corroborated our QRT-PCR results (Fig. 2), although, as others have seen for array data and likely resulting from increased technical noise [44], the magnitude of change was smaller. Specifically, frontal cortex samples from GRN+ FTLD-TDP had increased levels of GRN transcript compared with both normal controls and GRN− FTLD-TDP (Fig. 2a). One of the three probes (Probe 3) showed a statistically significant difference (P = 0.04) between normal controls and GRN mutants after correction for multiple-hypothesis testing. For cerebellar samples, GRN+ FTLD-TDP, GRN− FTLD-TDP, and normal controls all showed comparable amounts of GRN transcript (Fig. 2b).
Fig. 2.
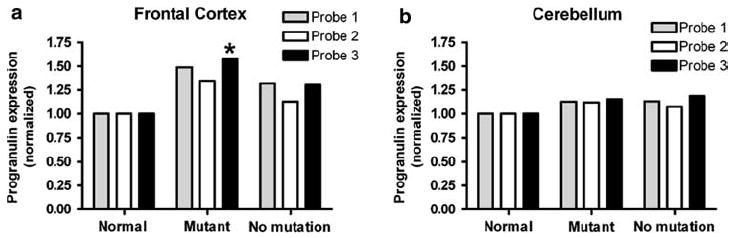
Microarray analysis corroborates QRT-PCR results.a Microarray analysis demonstrates that frontal cortex from GRN mutants (mutant, n = 6) have elevated GRN mRNA relative to controls (normal, n = 8) and FTLD-TDP without GRN mutations (no mutation, n = 10). GRN is represented by three different probes on the Affymetrix U133A microarray used. For probe 3 (*), GRN mutation cases show a significant elevation (P = 0.04) relative to normals. P value corrected for multiple-hypothesis testing by the Benjamini–Hochberg method. b Cerebellar samples from normal controls (n = 8), FTLD-TDP with GRN mutations (n = 5), and FTLD-TDP without GRN mutations (n = 8) show equivalent amounts of GRN mRNA by microarray
In summary, we did not observe a reduction in GRN transcripts in brains of GRN+ FTLD-TDP cases. Instead, GRN mutant samples from frontal cortex, the brain region most affected in FTLD-TDP, exhibited increased levels of GRN mRNA expression, and we obtained the same result regardless of method used.
Determination of allele-specific expression in GRN mutants
Given the increased amounts of total GRN transcript observed in GRN mutation cases, we considered two alternative hypotheses. First, the mutant allele could show partial expression, or, second, there could be increased transcript abundance for the normal allele. To evaluate these hypotheses, we reverse-transcribed mRNA from frontal cortex and cerebellar samples of GRN mutants and then sequenced cDNA surrounding the mutation. For all cases evaluated with coding-region GRN mutations (cases 1, 3–6 in Table 1) sequencing showed that only the normal allele is expressed (Fig. 3). For Case #6 with one mutation and two variants, only the variant alleles were detected, indicating that the c.1009C>T mutation is in trans to both variants (data not shown). Thus, the elevated levels of GRN mRNA transcript in cases with GRN mutations reflect expression of the normal allele.
Fig. 3.
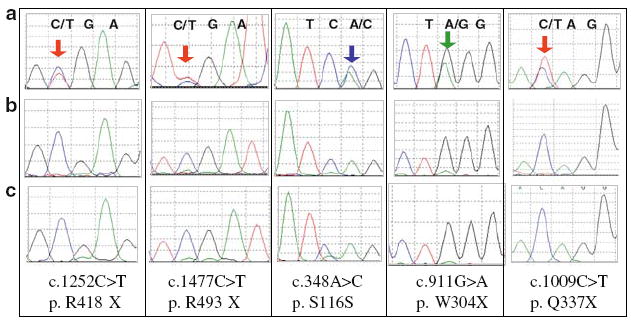
Sequencing of cDNA reveals that increased mRNA transcript levels originate from the normal allele. Brain mRNA from GRN mutants was reverse-transcribed, and the resulting cDNA was sequenced. Row A depicts genomic DNA for each GRN mutant; mutant peaks are indicated by colored arrows. Row B depicts cDNA for each GRN mutant, and Row C depicts normal cDNA for comparison. For all GRN mutants tested, mRNA primarily represents the normal allele indicating that the increased transcript levels result from the normal allele
Quantification of progranulin protein in brain
To answer the question of whether increased levels of GRN mRNA translate into increased progranulin protein levels, we performed immunoblots and quantitated progranulin levels by ELISA on brain samples previously analyzed for mRNA levels (Supplementary Table 1). For reliable immunoblotting and ELISA of progranulin protein, we generated a rabbit polyclonal antibody to a C-terminal peptide, and two novel mAbs (mAbs 407 and 2161) that are progranulin specific.
As shown in Fig. 4a, mAb 407 preferentially recognizes a reduced (DTT treated) form of progranulin, mAb 2161 preferentially recognizes a non-reduced native form, and the rabbit anti-progranulin Ct antibody recognizes both forms. All three antibodies are specific for progranulin, as demonstrated by deglycosylation experiments (Fig. 4b). Because progranulin is a highly glycosylated protein, one would expect bands recognized by progranulin-specific antibodies to decrease in molecular weight after treatment with the deglycosylating enzyme PNGase, which was indeed the case.
Fig. 4.
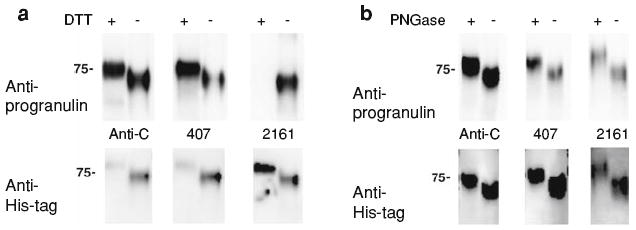
Novel antibodies recognize reduced and non-reduced forms of progranulin. We generated antibodies recognizing different forms of human progranulin. a Purified His-tagged human progranulin treated (+) or untreated (−) with DTT, a reducing agent, was separated on a 7.5% SDS-PAGE gel, and blotted with antibodies indicated (top). The blot was then stripped and re-blotted with anti-His antibody (bottom). As shown in the top, the rabbit polyclonal anti-progranulin Ct antibody (anti-C) does not discriminate between native (no DTT treatment) and reduced (DTT treatment) forms of progranulin, while mAb 407 (407) preferentially recognizes reduced progranulin, and mAb 2161 (2161) recognizes native progranulin. The native non-reduced form of progranulin runs faster relative to the reduced form of progranulin due to the compact conformation of the native protein. b Purified His-tagged human progranulin was deglycosylated with PNGase F (+) or mock-treated (−), and proteins were separated on a 7.5% SDS-PAGE gel. Immunoblotting with rabbit anti-C, mAb 407, or mAb 2161 demonstrates that in each case deglycosylation results in a decrease in the apparent molecular weight of progranulin, consistent with the nature of progranulin as a highly glycosylated protein. As before, immunoblots were performed with the labeled antibody (top), followed by stripping and re-blotting with anti-His antibody (bottom). Because of preferential binding for reduced or non-reduced progranulin, DTT-treated progranulin was used for rabbit anti-C and mAb 407 immunoblots in (b), while non-treated progranulin was used for mAb 2161 immunoblotting
To identify progranulin from human brain samples, we conducted immunoblotting using brain tissue sequentially extracted with buffers of increasing extraction strengths, and we showed that progranulin was recovered primarily in the RIPA fractions (Supplementary Fig. 4). Using mAb 407 and the RIPA fractions, we found decreased amounts of progranulin in GRN+ FTLD-TDP for brain regions relatively unaffected by FTLD-TDP (occipital cortex and cerebellum, Fig. 5c, d). However, in brain regions with more histopathological involvement in disease (frontal cortex and temporal cortex, Fig. 5a, b), GRN mutants showed more inter-individual variability. We, therefore, developed a sandwich ELISA for accurate quantitation of progranulin protein from human brain samples.
Fig. 5.
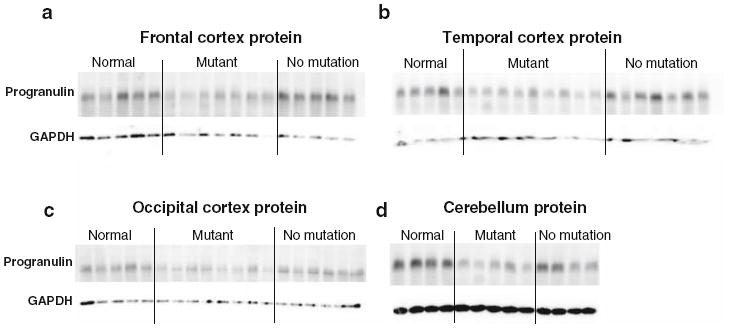
Immunoblot of progranulin protein from brain samples in normal controls (normal), FTLD-TDP with GRN mutations (mutant), and FTLD-TDP without GRN mutations (no mutation). Representative immunoblot of progranulin protein from frontal cortex (a), temporal cortex (b), occipital cortex (c), and cerebellum (d) using mAb 407. In brain regions relatively unaffected by disease (occipital cortex and cerebellum), GRN mutants show uniformly decreased progranulin levels relative to neurologically normal controls, while brain regions that are histopathologically affected in FTLD-TDP (frontal cortex and temporal cortex) show more individual to individual variation in progranulin protein levels among GRN mutants. Each lane represents a separate patient or control sample. Immunoblots were repeated four times. Human brain lysates (RIPA fraction) were separated on a 7.5% SDS-PAGE gel
Because progranulin from biosamples, such as plasma, CSF, or brain extracts is in a native non-reduced form, we developed our sandwich ELISA using mAb 2161 as the capture antibody and the rabbit anti-progranulin Ct antibody (recognizing both non-reduced and reduced forms) as the detection antibody. Highly purified His-progranulin was used to generate the standard curves, and the EC50 for our sandwich ELISA was 10.98 ng/mL (Supplementary Fig. 5). Using this assay, we measured progranulin protein levels in the brains of normal controls, GRN+ FTLD-TDP, and GRN− FTLD-TDP. In occipital cortex and cerebellum (Fig. 6c, d), the two brain regions least affected by FTLD-TDP, GRN mutants showed a clear decrease in progranulin levels compared with normal controls and to GRN− FTLD-TDP. In frontal cortex and temporal cortex (Fig. 6a, b), however, GRN mutants did not show a significant difference in progranulin levels compared with normals, although GRN+ FTLD-TDP and GRN− FTLD-TDP differed significantly, with GRN+ FTLD-TDP showing lower levels of protein expression.
Fig. 6.
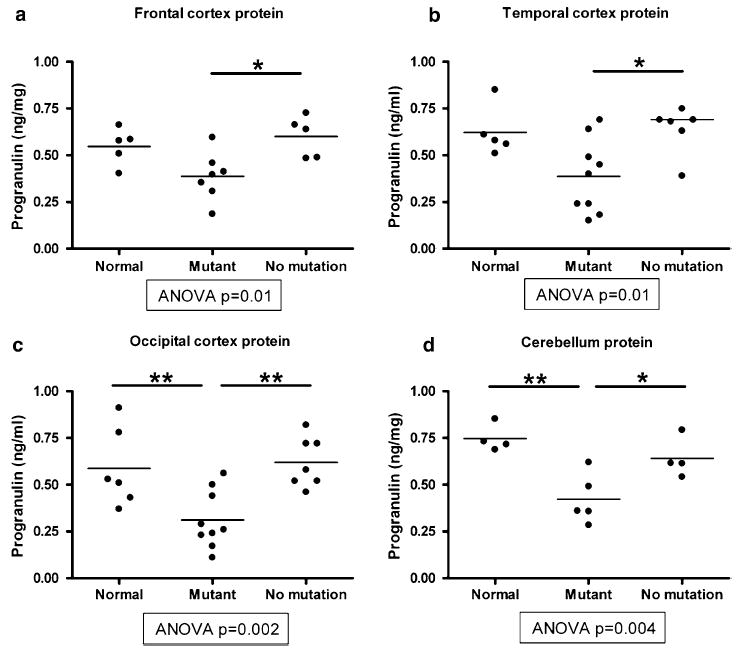
Sandwich ELISA quantification of progranulin protein levels from brain samples in normal controls (normal), FTLD-TDP with GRN mutations (mutant), and FTLD-TDP without GRN mutations (no mutation). Progranulin in human brain lysates (RIPA fraction) was captured with mAb 2161 and detected with rabbit anti-progranulin Ct antibody and HRP-conjugated goat anti-rabbit antibody. a, b In brain regions with more histopathological involvement in FTLD-TDP (frontal and temporal cortex), ELISA shows no significant difference in progranulin protein levels between normal controls and GRN mutants, although GRN mutants did differ from FTLD-TDP without GRN mutations (see bars with * comparing FTLD-TDP cases ± GRN mutations). c, d In brain regions that are relatively histopathologically spared in FTLD-TDP (occipital cortex and cerebellum), ELISA shows significantly decreased progranulin protein levels in GRN mutants relative to both normal controls and FTLD-TDP without GRN mutations (see bars with * or ** comparing GRN mutants to normal controls or FTLD-TDP cases without GRN mutations). Individual cases are shown as circles, and the mean for each group is shown as a bar. ANOVA was used to evaluate progranulin protein levels across groups, followed by Tukey’s test for pairwise comparisons *P < 0.05, **P < 0.01
Taken together, regions that are more histopathologically affected in FTLD-TDP (frontal cortex and temporal cortex) show less difference in progranulin expression between normal controls and GRN mutants, compared with regions that are relatively histopathologically spared (occipital cortex and cerebellum). However, unlike mRNA transcript levels, protein levels remain lower for GRN mutants than for controls in all four brain regions.
Microglia in FTLD-TDP
Although our sequencing results indicate that the increase in GRN mRNA transcript levels seen in GRN mutants is due to the increased expression of the normal allele, they do not answer the question of what cell types are responsible for the increase in GRN transcript. Because GRN is known to be highly expressed in immune system cells [18], including microglia [4], we examined sections of brain immediately adjacent to the areas analyzed for mRNA and protein for the presence of microglia. Previous studies have shown that microglial activation is a feature of many neurodegenerative diseases including Alzheimer’s disease [33] and FTD [49].
As shown in Fig. 7, microglia were present in histopathological sections from GRN+ FTLD-TDP and GRN− FTLD-TDP. However, in frontal cortex samples from GRN mutation cases, microglial infiltration appeared more robust. When severity was assessed on a 4-point scale (0 none, 1 rare/mild, 2 occasional/moderate, 3 numerous/ severe) by a neuropathologist blinded to disease and GRN gene status, the median score for microglial staining was 1.7 in GRN mutants compared to 1.4 in GRN− FTLD-TDP in the gray matter (Fig. 7d). In the white matter, moderate to severe microglial infiltration was present in FTLD-TDP with and without GRN mutations, although the mutants showed slightly greater involvement. This robust microglial response was not present in normal control frontal cortex samples, nor was it present in cerebellar samples from normal controls or FTLD-TDP cases with or without GRN mutations.
Fig. 7.
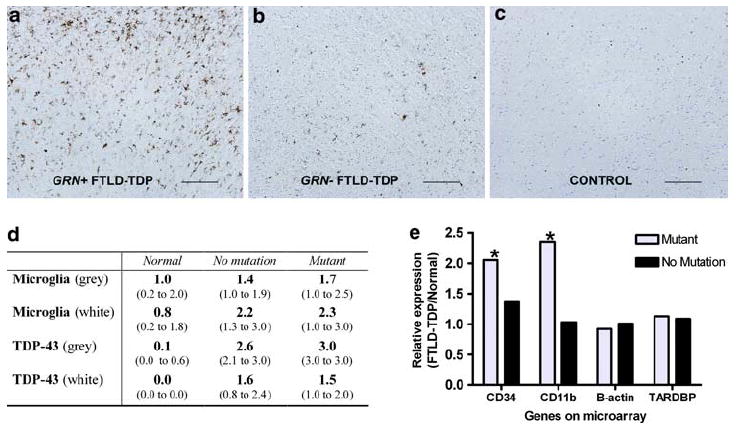
Microglial infiltration in GRN mutants. a–c Gray matter sections from frontal cortex are shown for cases of (left to right) FTLD-TDP with GRN mutations (GRN+), FTLD-TDP without GRN mutations (GRN–), and normal controls. Cases stained for microglia with HLA-DR. Scale bar represents 200 μm. d Scoring of immunohistochemical staining for TDP-43 pathology and microglial infiltration was performed for frontal cortex sections by a neuropathologist blinded to disease and GRN mutation status. Scores ranged from 0 (no microglial staining) to 3 (most robust microglial staining). GRN+ FTLD-TDP showed more microglial infiltration than GRN– FTLD- TDP and normals in both gray and white matter. Values shown are median score and interquartile range (parentheses) calculated from grouped data. e Microarray expression results are shown for two microglial markers (CD34 and CD11b) as well as a housekeeping gene (β-actin) and the gene encoding TDP-43 (TARDBP). Significantly increased mRNA expression was seen in frontal cortex samples from GRN mutants for CD34 (*P = 0.02) and CD11b (*P = 0.05). Bars represent relative expression for FTLD-TDP with (GRN mutant) and without (no mutation) GRN mutations, normalized to control. P values are corrected for multiple-hypothesis testing
Based on these immunohistochemical findings, we returned to our mRNA global expression profiling results and queried the expression of several microglial marker genes in cases with and without GRN mutations. Both CD34, a marker of bone marrow-derived hematopoietic cells that labels proliferating brain microglia [3, 19], and CD11b, a marker of monocyte/macrophage lineage cells expressed in microglia [1] showed increased mRNA expression in frontal cortex samples from GRN+ FTLD-TDP compared with GRN− FTLD-TDP and to normal controls (Fig. 7e). CD34 and CD11b expression was significantly increased in GRN mutants (corrected P = 0.02 and P = 0.05, respectively) compared with normal controls. We did not see significant differences in either CD34 or CD11b expression comparing normal controls and GRN− FTLD-TDP. In cerebellar samples, CD34 and CD11b expression did not differ significantly among normal controls, GRN+ FTLD-TDP, and GRN− FTLD-TDP (data not shown).
Thus, the infiltration of brain regions affected in FTLD-TDP by GRN-expressing microglia may partially explain the high levels of GRN transcripts in frontal cortex of GRN mutants.
Discussion
The 2006 discovery that GRN mutations are a major cause of FTLD-TDP [5, 16] opened new avenues for research linking these genetic defects to disease phenotypes. To date, over 60 different GRN mutations have been described (AD and FTD mutation database; http://www.molgen.ua.ac.be/FTDMutations), many of them resulting in either demonstrable or predicted premature termination. This feature of GRN mutations, along with autosomal dominant inheritance patterns, suggested progranulin haploinsufficiency as an upstream disease mechanism in FTLD-TDP, a hypothesis supported by subsequent in vitro studies of lymphoblastoid cell lines derived from GRN mutation carriers [5, 16, 22]. Progranulin haploinsufficiency as the first step in a pathogenic cascade is widely accepted now [17] despite an extreme paucity of studies that have evaluated affected versus unaffected brain regions in FTLD-TDP [29].
The present study addresses the question of progranulin expression in FTLD-TDP brain specifically and, in doing so, demonstrates several surprising findings. First, we show that, contrary to expectations based on GRN haploinsufficiency in FTLD-TDP caused by GRN mutations, and contrary to our findings of reduced GRN mRNA in blood samples, GRN mutants do not show decreased GRN mRNA levels in brain samples. Indeed, in severely affected frontal cortex, GRN mutants show elevated GRN mRNA levels. In comparison, in the only prior study of GRN mRNA brain expression [29], one GRN mutation case showed equivalent amounts of GRN mRNA in frontal cortex, and lower amounts of GRN mRNA in cerebellum, compared with normal controls. Second, we show that progranulin protein levels do not fully correspond to elevated GRN mRNA in GRN mutants for reasons that remain enigmatic.
In the work presented here, we used a regional sampling approach, which by definition involves sampling of mixed populations of glial, microglial, and neuronal cells. We chose this approach to minimize the amount of RNA degradation, which is greater with microdissection techniques. Moreover, because both full-length progranulin and its granulin peptide derivatives are secreted, such an approach can capture both intracellular and extracellular protein. This logic is also compelling when one considers possible cellular sources of the elevated levels of GRN mRNA seen in frontal cortex of GRN mutants. In this study, we showed that the normal allele is responsible for the increase in total amount of GRN mRNA. Our immunohistochemical and microarray studies further suggest that microglia may be the cell type primarily responsible for this increase in GRN transcripts. Whether the robust microglial infiltration in GRN mutants is dependent on the same pathways that cause TDP-43 accumulation remains to be determined, and future investigations into the role of microglia in FTLD-TDP caused by GRN mutations would be a valuable addition to the data presented here. In addition, studies specifically evaluating cell-typespecific expression of GRN may add to our understanding of GRN-associated FTLD-TDP.
Why do we not see a complete correspondence between levels of GRN mRNA expression and levels of progranulin protein expression? Several explanations are possible. First, our GRN mRNA results could be unreliable because of RNA degradation or other technical reasons. However, we guarded against this possibility by checking RNA quality both subjectively and objectively, with only samples with an RIN >6 and well-delineated peaks on the Bioanalyzer electrophoregram retained for evaluation. Furthermore, the RNA results were seen with two different methodologies, although the microarray results are less sensitive, as we and others have seen previously with this method [14, 44]. Second, we may have failed to capture all of the full-length progranulin protein. This explanation seems unlikely, however, since here we also obtained the same result with two different methodologies (ELISA and immunoblot) and performed sequential extractions of brain samples in order to capture progranulin in all biochemical fractions of the samples examined here. Third, there may be post-transcriptional changes to the mRNA that affect its translation. Indeed, one recent report implicates a specific microRNA in the post-transcriptional regulation of progranulin expression [38], although others have reported no association in FTLD-TDP with polymorphisms at the microRNA-binding site [40]. Fourth, the literature suggests that full-length progranulin is primarily a precursor protein for granulin peptide cleavage products [7]. As such, elevations in GRN mRNA may result in higher expression of both full-length progranulin and granulin peptides. Therefore, it is quite possible that increased expression of specific granulin peptides, not captured by the antibodies used in this study which are specific for the full-length form of progranulin, is the main result of increased expression of GRN mRNA. Increased expression of granulin peptides might then be expected to have downstream effects in inflammatory or cell-cycle signaling. Such downstream effects are worthy of further investigation, especially as the literature suggests that full-length progranulin and the granulin peptides may at times act similarly, e.g. in regulating neurite outgrowth [47], and at other times act in opposing ways, e.g. in inflammation [25].
In summary, the results presented in this study show that increased GRN mRNA is present in the most severely affected brain areas of GRN mutation-associated FTLD-TDP and that microglial are the most likely cellular source of this message. One limitation of studies using postmortem brain samples is that one cannot rule out the possibility that changes seen are due to non-specific effects such as cell loss and gliosis. It is, therefore, possible that the elevated GRN transcript levels and microglial infiltration seen in our GRN mutation-bearing frontal cortex samples are non-specific. However, one would expect similar degrees of these nonspecific effects in FTLD-TDP cases regardless of the presence of GRN mutations. Yet in the postmortem brain samples studied here, increased GRN expression and increased microglial infiltration were only seen in the GRN mutants, suggesting that increased microglial activation may be a pathogenic feature of GRN mutation-associated FTLD-TDP.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health [K08AG033101-01; AG10124; AG17586; NS44266; AG15116; NS53488], the American Academy of Neurology-ALS Association [Clinician–Scientist Development Award to ACP], and the Burroughs Wellcome Fund [Career Award for Medical Scientists to ACP]. VMYL is the John H. Ware, 3rd, Professor of Alzheimer’s disease research. JQT is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology. Wethank Theresa Schuck for assistance in immunohistochemical studies. We thank Linda Kwong for helpful advice and discussions. Finally, we thank our patients and their families whose generosity made this work possible.
Footnotes
Conflict of interest statement The authors declare that they have no conflicts of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s00401-009-0576-2) contains supplementary material, which is available to authorized users.
Contributor Information
Alice S. Chen-Plotkin, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA chenplot@mail.med.upenn.edu Department of Neurology, University of Pennsylvania School of Medicine, Philadelphia, PA, USA.
Jiping Xiao, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA.
Felix Geser, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA.
Maria Martinez-Lage, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA.
Murray Grossman, Department of Neurology, University of Pennsylvania School of Medicine, Philadelphia, PA, USA.
Travis Unger, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA.
Elisabeth M. Wood, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA
Vivianna M. Van Deerlin, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA
John Q. Trojanowski, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA
Virginia M.-Y. Lee, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, 3rd Floor Maloney, 3600 Spruce Street, Philadelphia, PA 19104, USA
References
- 1.Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol. 1990;30:81–93. doi: 10.1016/0165-5728(90)90055-r. [DOI] [PubMed] [Google Scholar]
- 2.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 3.Asheuer M, Pflumio F, Benhamida S, et al. Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proc Natl Acad Sci USA. 2004;101:3557–3562. doi: 10.1073/pnas.0306431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker CA, Manuelidis L. Unique inflammatory RNA profiles of microglia in Creutzfeldt–Jakob disease. Proc Natl Acad Sci USA. 2003;100:675–679. doi: 10.1073/pnas.0237313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 6.Bateman A, Belcourt D, Bennett H, Lazure C, Solomon S. Granulins, a novel class of peptide from leukocytes. Biochem Biophys Res Commun. 1990;173:1161–1168. doi: 10.1016/s0006-291x(05)80908-8. [DOI] [PubMed] [Google Scholar]
- 7.Bateman A, Bennett HP. Granulins: the structure and function of an emerging family of growth factors. J Endocrinol. 1998;158:145–151. doi: 10.1677/joe.0.1580145. [DOI] [PubMed] [Google Scholar]
- 8.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing (English summary) J Roy Statist Soc Ser B. 1995;57:289–300. [Google Scholar]
- 9.Bhandari V, Palfree RG, Bateman A. Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine-rich granulin domains. Proc Natl Acad Sci USA. 1992;89:1715–1719. doi: 10.1073/pnas.89.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhandari V, Giaid A, Bateman A. The complementary deoxyribonucleic acid sequence, tissue distribution, and cellular localization of the rat granulin precursor. Endocrinology. 1993;133:2682–2689. doi: 10.1210/endo.133.6.8243292. [DOI] [PubMed] [Google Scholar]
- 11.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 12.Bronner IF, Rizzu P, Seelaar H, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. 2007;15:369–374. doi: 10.1038/sj.ejhg.5201772. [DOI] [PubMed] [Google Scholar]
- 13.Cairns NJ, Neumann M, Bigio EH, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen-Plotkin AS, Geser F, Plotkin JB, et al. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppola G, Karydas A, Rademakers R, et al. Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol. 2008;64:92–96. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 17.Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24:186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- 19.Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29:227–234. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Finch N, Baker M, Crook R, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59:952–962. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 23.Geser F, Brandmeir NJ, Kwong LK, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- 24.Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc. [DOI] [PubMed] [Google Scholar]
- 25.He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- 26.He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- 27.Huey ED, Grafman J, Wassermann EM, et al. Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann Neurol. 2006;60:374–380. doi: 10.1002/ana.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Ber I, van der Zee J, Hannequin D, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat. 2007;28:846–855. doi: 10.1002/humu.20520. [DOI] [PubMed] [Google Scholar]
- 29.Leverenz JB, Yu CE, Montine TJ, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130:1360–1374. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114:49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- 32.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGeer EG, Klegeris A, McGeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(Suppl 1):94–97. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 34.McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee O, Wang J, Gitcho M, et al. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29:512–521. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 37.Plowman GD, Green JM, Neubauer MG, et al. The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J Biol Chem. 1992;267:13073–13078. [PubMed] [Google Scholar]
- 38.Rademakers R, Eriksen JL, Baker M, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 40.Rollinson S, Rohrer JD, van der Zee J, et al. No association of PGRN 3′UTR rs5848 in frontotemporal lobar degeneration. Neurobiol Aging. 2009 May 13; doi: 10.1016/j.neurobiolaging.2009.04.009. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 41.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126:2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 42.Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroeder A, Mueller O, Stocker S, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi L, Reid LH, Jones WD, et al. The microarray quality control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shoyab M, McDonald VL, Byles C, Todaro GJ, Plowman GD. Epithelins 1 and 2: isolation and characterization of two cysteine-rich growth-modulating proteins. Proc Natl Acad Sci USA. 1990;87:7912–7916. doi: 10.1073/pnas.87.20.7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sleegers K, Brouwers N, Van Damme P, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–609. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- 47.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Deerlin VM, Wood EM, Moore P, et al. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol. 2007;64:1148–1153. doi: 10.1001/archneur.64.8.1148. [DOI] [PubMed] [Google Scholar]
- 49.Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008;67:1001–1010. doi: 10.1097/NEN.0b013e318188b204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu J, Nathan C, Jin W, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.