

Abstract
The Akt kinase signaling pathway is frequently deregulated in many human diseases including cancer, autoimmune disease and diabetes. In nephropathy, associated with diabetes, increased Akt signal transduction results in glomerular especially mesangial cell hypertrophy. The mechanism of Akt activation by elevated glucose is poorly understood. The oncogene DJ-1 prevents oxidative damage and apoptosis of dopaminergic neurons in animal models of Parkinson’s disease and in culture. We identified DJ-1 to increase in response to high glucose in renal glomerular mesangial cells concomitant with an increase in phosphorylation of Akt in a time-dependent manner. Plasmid-derived overexpression as well as downregulation of DJ-1 by siRNA showed the requirement of this protein in high glucose-stimulated Akt phosphorylation. The tumor suppressor protein PTEN acts as a negative regulator of Akt activation. Interestingly, DJ-1 was associated with PTEN and this interaction was significantly increased in response to high glucose. High glucose-induced increase in DJ-1 promoted phosphorylation of the PRAS40, a negative regulator of TORC1 kinase activity, resulting in activating and inactivating phosphorylation of S6 kinase and 4EBP-1, respectively. Furthermore, DJ-1 increased protein synthesis and hypertrophy of mesangial cells. Our results provide evidence for a unique mechanism whereby DJ-1 induces Akt/PRAS40/TORC1-mediated hypertrophy in response to high glucose.
Keywords: Akt kinase, Kidney, Diabetes, mTOR
1. Introduction
DJ-1, also known as CAP1/RS/PARK7, was originally identified as a putative oncogene, which cooperates with Ras to transform cells and increased resistance to apoptosis [1]. It is a ubiquitously expressed single domain dimeric protein with high levels found in brain [2]. There are significant structural similarities between DJ-1 and the bacterial proteases Pfp1/PH1704. Unlike the bacterial homolog however, due to a distortion in the catalytic site, DJ-1 does not possess any protease activity [2].
DJ-1 displays diverse biological functions in many cells. Its most extensively studied biological action is its anti-apoptotic function in neurons of patients with Parkinson’s disease (PD). In fact deletion and point mutations in the coding sequence have been associated with recessive forms of PD [2, 3]. The anti-apoptotic effect of DJ-1 in neurons is elicited by its response to oxidative stress as evidenced by a shift to a more acidic pI both in PD and in neuronal cells due to oxidation of the Cys-106 residue present in the pseudocatalytic site to cysteine-sulfinic acid [4].
DJ-1 is upregulated in many cancers [2, 5]. Furthermore, it was found to be a serum biomarker for breast cancer and malignant melanoma [6, 7]. DJ-1 has been shown to be associated with androgen receptor-interacting protein 3 and increased androgen receptor-dependent transcription [2]. It is present in an RNA-binding complex and has been implicated in fertilization [8, 9]. The role of DJ-1 in diseases other than neuronal disorders and cancer has not been described.
Both type 1 and type 2 diabetic patients with nephropathy exhibit similar renal lesions, which include renal hypertrophy, thickening of basement membrane and accumulation of extracellular matrix components [10]. The pathology involves hyperfiltration, microalbuminuria, frank proteinuria and fibrosis, leading to loss of renal function. Irreversible changes such as tubulointerstitial fibrosis and glomerulosclerosis are preceded by the early hypertrophy of tubular and glomerular compartments [11]. Mesangial expansion due to hypertrophy of mesangial cells and matrix protein expression leads to the clinical manifestations of diabetic kidney disease including decreased glomerular filtration rate and albuminuria. Diabetic milieu augments the expression of hormones and growth factors including insulin-like growth factor-1, vascular endothelial growth factor, angiotensin II and transforming growth factor-β. Although these factors contribute to the pathophysiology of diabetic mesangial cell hypertrophy, involvement of specific signaling cascades are implicated in eliciting the hypertrophic effect of high glucose [12, 13]. The potential role of DJ-1 in diabetic kidney disease has not been examined.
In the present study, we show that high glucose increases the expression of DJ-1 in mesangial cells, resulting in the activation of Akt. We demonstrate increased association of DJ-1 with PTEN in the presence of high glucose. DJ-1 regulates high glucose-induced phosphorylation of PRAS40, leading to activation of TORC1. Furthermore, DJ-1-activated Akt/TORC1 signaling induces mesangial cell hypertrophy similar to high glucose.
2. Materials and Methods
2.1. Reagents
D-mannitol, D-glucose, NP40, anti-actin antibody, anti-FLAG antibody and protease inhibitor cocktail were purchased from Sigma. Phospho-Akt, Akt, phospho-PRAS40 (Thr-246), PRAS40, phospho-S6 kinase (Thr-389), S6 kinase, phospho-4EBP-1 (Thr-37/46) and 4EBP-1 antibodies were obtained from Cell Signaling. DJ-1 and PTEN antibodies and pool of three siRNAs against DJ-1 were purchased from Santa Cruz. Anti-HA antibody was obtained from Covance. 35S-methionine was obtained from PerkinElmer. Wild type FLAG-DJ-1 expression vector was a kind gift from Dr. H. Ariga (Hokkaido University, Japan).
2.2. Cell culture
Renal glomerular mesangial cells were grown as described previously [14, 15]. Briefly, cells were maintained in DMEM containing 5 mM glucose and 17% fetal bovine serum with penicillin/streptomycin. At near confluency, monolayers were serum-deprived for 24 hours. Cells were incubated with 25 mM glucose for indicated time periods. As control, 20 mM mannitol was added to 5 mM glucose containing medium [14, 15].
2.3. Immunoblotting and immunoprecipitation
Mesangial cells were harvested and lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 0.1% protease inhibitor cocktail and 1% NP-40) at 4°C for 30 minutes. The cell debri were centrifuged at 4°C for 20 minutes at 10,000xg. The supernatant was collected as cell lysate and protein estimated using BioRad reagent. For immunoblotting, equal amounts of cell lysates were separated by SDS polyacrylamide gel electrophoresis and transferred to PVDF membrane. Membranes were incubated with the indicated antibodies. The proteins were developed by HRP-conjugated secondary antibodies, using enhanced chemiluminiscence technique as described previously [14–17]. For immunoprecipitation, equal amounts of cell supernatant were immunoprecipitated overnight with the indicated antibodies as described [16]. The immunoprecipitates were separated by SDS polyacrylamide gel electrophoresis, transferred to PVDF membrane, and immunoblotted with the indicated antibodies followed by development with chemiluminiscence reagent as described [14–17].
2.4. Transfection
Mesangial cells were seeded at 70% confluency. The following day the cells were transfected with the indicated plasmids, or siRNA targeting DJ-1, using Fugene HD transfection reagent as described [16–18]. Twelve hours post-transfection, the transfection was repeated in the same manner. The transfected cells were treated with either 25 mM glucose or with 5 mM glucose plus 20 mM mannitol as described above.
2.5. Protein synthesis and measurement of hypertrophy
Transfected mesangial cells were incubated with 25 mM glucose or 5 mM glucose plus 20 mM mannitol for 24 hours. The cells were incubated with 35S-methionine and protein synthesis determined as described previously [14, 15]. To measure hypertrophy, mesangial cells were trypsinized, counted using a hemocytometer and lysed. The total protein was determined using BioRad reagent. Hypertrophy was expressed as ratio of total protein to cell number [14].
2.6. Statistics
The significance of the data was determined by t-test or ANOVA followed by Student-Newman-Keuls analysis as described previously [14–17].
3. RESULTS
3.1. High glucose increases expression of DJ-1
DJ-1 is expressed ubiquitously with maximum expression in brain and has been studied in association with early-onset PD and in cancer [2, 5]. Its role in diabetic renal disease has not been investigated. To explore the function of DJ-1 in mesangial cells, which contribute to the complications of diabetic nephropathy, we examined the effect of high glucose concentration (25 mM) that mimics hyperglycemia in diabetic condition. Immunoblot analysis of the lysates of mesangial cells incubated with 25 mM glucose showed increased expression of DJ-1 within 2 hours of stimulation (Fig. 1A). Mesangial cells incubated with 5 mM glucose plus 20 mM mannitol was used as control (Fig. 1A, lane 1). A genetic screen in Drosophila identified DJ-1 as an antagonist of PTEN, the endogenous inhibitor of Akt kinase signaling [5, 19]. Since high glucose increases the level of DJ-1 in a time-dependent manner, we tested Akt phosphorylation at the same time points. High glucose significantly increased the phosphorylation of Akt with kinetics similar to DJ-1 expression (Fig. 1B). Hypertrophy of mesangial cells in response to high glucose is generally detected at 24 hours [14, 15]. We examined whether increased expression of DJ-1 was still detectable at this time point and beyond. As shown in Fig. 1C, high glucose significantly increased the expression of DJ-1 at 24 hours, which was sustained until 48 hours. Increase in DJ-1 was associated with sustained increase in phosphorylation of Akt (Fig. 1D).
Figure 1.
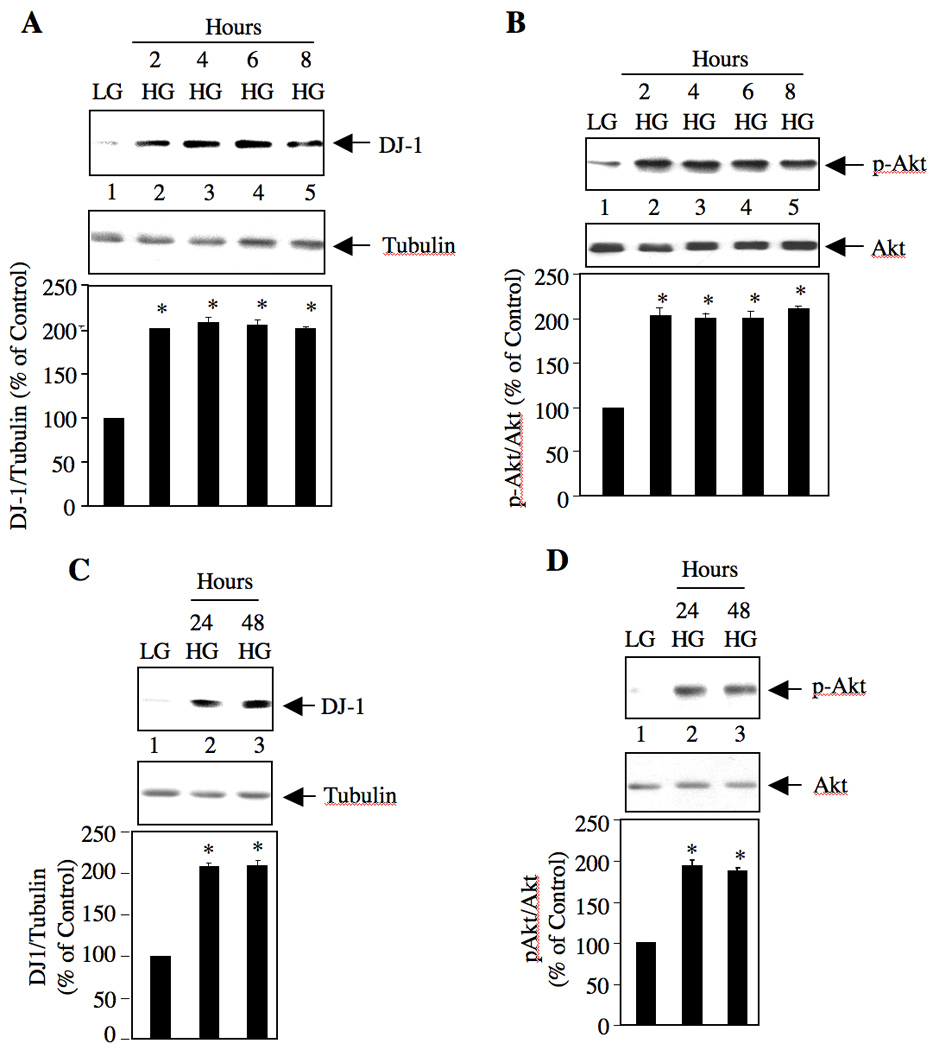
High glucose stimulates DJ-1 expression in mesangial cells in a time-dependent manner. Quiescent mesangial cells were incubated with 25 mM glucose (high glucose; HG) for indicated periods of time. As control, 5 mM glucose plus 20 mM mannitol (LG) was used (lanes 1 in all panels). Cleared cell lysates were immunoblotted with anti-DJ-1 and anti-tubulin (panels A and C), anti-p-Akt and anti-Akt (panels B and D) antibodies as indicated. Bottom panels show the quantification of DJ-1 expression and phosphorylation of Akt. Panel A, n = 4, *p < 0.001 vs LG. Panel B, n = 6, *p < 0.001 vs LG. Panel C, n = 3, *p < 0.001 vs LG. Panel D, n = 5, *p < 0.001 vs LG.
3.2. Association of DJ-1 with PTEN
The mechanism of Akt phosphorylation involves production and sustained presence of phosphatidylinositol 3,4,5-tris-phosphate (PIP3), the level of which is governed by activation states of PI 3 kinase and PTEN [19]. The latter dephosphorylates PIP3 to inhibit Akt kinase phosphorylation/activation. Therefore, reduced activity of PTEN contributes to Akt activation [15]. Many proteins have been identified, which inhibit the function of PTEN by directly binding to it [20, 21]. We reasoned that DJ-1 might also form a complex with PTEN. To test this hypothesis, we expressed HA-tagged PTEN in mesangial cells. Immunoprecipitation of HA-PTEN showed constitutive association between DJ-1 and exogenously expressed PTEN (Supplementary Fig. S1A). Similarly, constitutive association of PTEN was detected in FLAG immunoprecipitates from lysates of mesangial cells transfected with FLAG-DJ-1 (Supplementary Fig. S1B). To examine the effect of high glucose on this association, FLAG-DJ-1-transfected mesangial cells were incubated with 25 mM glucose and analyzed. High glucose increased the association of FLAG-DJ-1 with PTEN at 2 hours and 24 hours, respectively (Fig. 2A and 2B). To confirm whether endogenous DJ-1 associates with PTEN, PTEN was immunoprecipitated from lysates of mesangial cells incubated with high glucose for 2 hours. The immunoprecipitates were immunoblotted with DJ-1 antibody. Fig. 2C shows that high glucose increased the association of PTEN with DJ-1. Similarly, high glucose stimulated complex formation between PTEN and DJ-1 at 24 hours (Fig. 2D). It is possible that increased complex formation may result from the augmented expression of DJ-1 in response to glucose as DJ-1 associates with PTEN under basal conditions (Fig 2). Note that expression of PTEN in the presence of high glucose was reduced (Figs. 2C and 2D middle panels). These results are in consonance with our previous observation [15], which have recently been confirmed by independent investigators [22]. Together, our results indicate that high glucose increases the association of PTEN with DJ-1 in the presence of downregulated PTEN protein levels.
Figure 2.
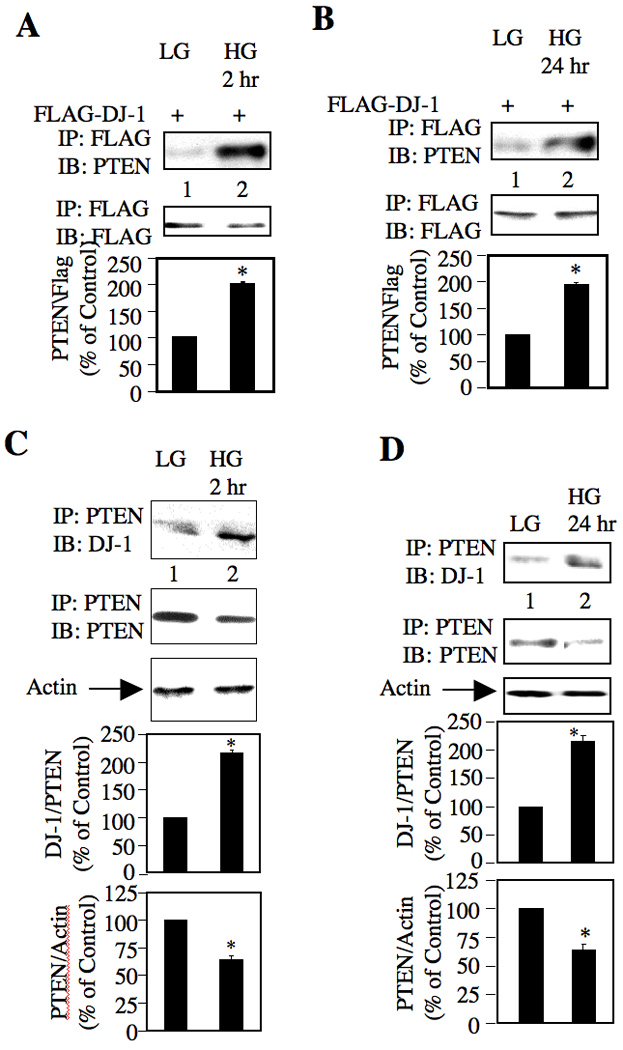
High glucose stimulates association of PTEN with DJ-1. (A and B) Mesangial cells were transfected with FLAG-DJ-1 as described in the materials and methods. Quiescent transfected cells were treated with 25 mM glucose (HG). As control, mesangial cells were incubated with 5 mM glucose plus 20 mM mannitol (LG). The cell lysates were immunoprecipitated with anti-FLAG antibody followed by immunoblotting with anti-PTEN and anti-FLAG antibodies, respectively as described in the materials and methods. (C and D) Lysates of mesangial cells incubated with 25 mM glucose for indicated times were immunoprecipitated with PTEN antibody followed by immunoblotting with anti-DJ-1 and anti-PTEN antibodies, respectively. The bottom panels show quantification of the results. Panel A, n = 3, *p = 0.003 vs LG. Panel B, n = 3, *p = 0.001 vs LG. Panel C, n = 4, *p = 0.002 vs LG for DJ-1 association with PTEN; *p = 0.006 vs LG for PTEN expression. Panel D, n = 3, *p = 0.006 vs LG for DJ-1 association with PTEN; *p = 0.021 vs LG for PTEN expression.
3.3. DJ-1 increases phosphorylation of Akt
We have established above that high glucose increased the expression of DJ-1 concomitant with increased phosphorylation of Akt (Fig. 1), suggesting that DJ-1 may have a regulatory effect on Akt phosphorylation. To confirm this role of DJ-1 in phosphorylation of Akt, we used FLAG-tagged DJ-1. Expression of DJ-1 in mesangial cells in the presence of 5 mM glucose significantly increased phosphorylation of Akt similar to that observed with high glucose alone (Fig. 3A). High glucose did not have any additive effect on DJ-1-induced Akt phosphorylation (Fig. 3A). To confirm this observation, we used siRNA targeted to DJ-1. Expression of siDJ-1 in mesangial cells suppressed DJ-1 protein expression (Fig. 3B), resulting in significant decrease in phosphorylation of Akt (Fig. 3C). These results demonstrate that high glucose enhances expression of DJ-1, which contributes to the increased phosphorylation of Akt.
Figure 3.

DJ-1 regulates high glucose-induced phosphorylation of Akt. (A) Mesangial cells were transfected with FLAG-DJ-1 or vector plasmids as described in the materials and methods. Quiescent transfected cells were treated with 25 mM glucose (HG) for 24 hours. As control, lysates from mesangial cells incubated with 5 mM glucose plus 20 mM mannitol (LG) were used. Cell lysates were immunoblotted with indicated antibodies. (B and C) Mesangial cells were transfected with scrambled (Scr) or siRNA targeting DJ-1 (siDJ-1) as described in the materials and methods. In panel C, the transfected cells were incubated with 25 mM glucose (HG) for 24 hours. The cell lysates were immunoblotted with indicated antibodies. The bottom panels in A and C show quantification of the results. Panel A, n = 4, *p < 0.001 vs LG. Panel C, n = 4, *p < 0.01 vs LG; #p < 0.05 vs high glucose-treated.
3.4 DJ-1 inactivates PRAS40
Phosphorylation and hence activation of Akt results in the phosphorylation of its substrate proteins, which elicit the biological functions of Akt. One such substrate of Akt is PRAS40. PRAS40 acts as a negative regulator of mTOR [23]. Phosphorylation of PRAS40 by Akt inactivates its function, resulting in mTOR activation. We have recently shown that mTOR contributes to renal hypertrophy in rodents with diabetes [14, 24]. Since we showed that the enhanced expression of DJ-1 by high glucose results in Akt phosphorylation (Fig. 3), we examined the role of DJ-1 in PRAS40 phosphorylation. As expected, high glucose enhanced phosphorylation of PRAS40 (Fig. 4A). Expression of DJ-1 significantly increased the phosphorylation of PRAS40 analogous to that found with high glucose alone (Fig. 4A). High glucose did not have any additive effect on PRAS40 phosphorylation in the presence of DJ-1 (Fig. 4A). To confirm these results, we used siRNA against DJ-1. Expression of siDJ-1 in mesangial cells significantly suppressed the phosphorylation of PRAS40 in response to high glucose concomitant with decrease in expression of DJ-1 protein (Fig. 4B). Together these results provide evidence that DJ-1 regulates high glucose-induced phosphorylation and inactivation of PRAS40.
Figure 4.
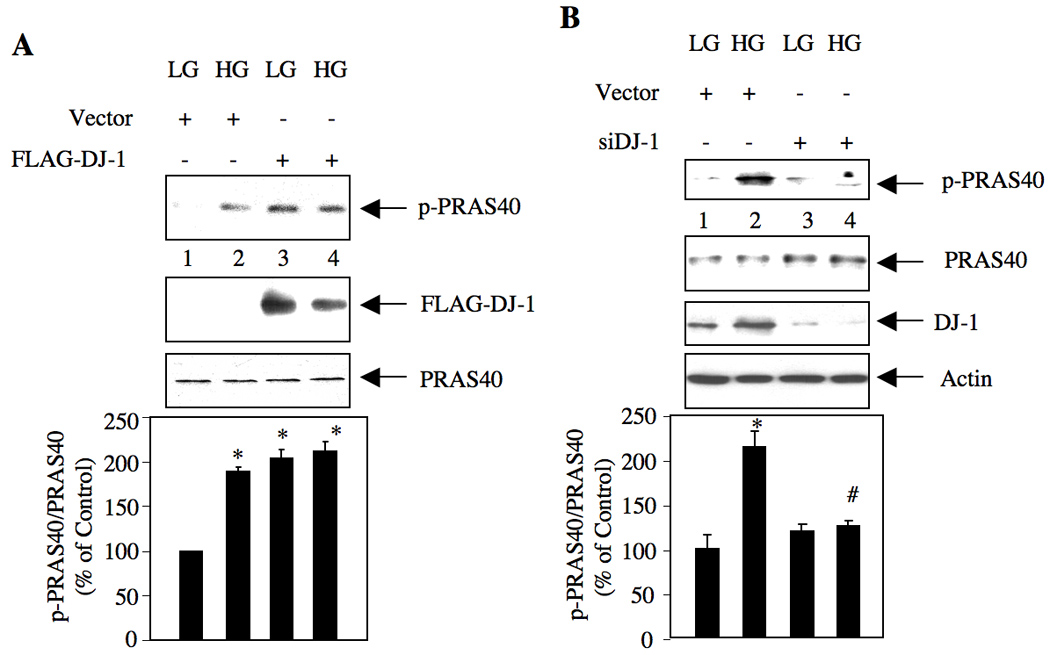
DJ-1 regulates high glucose-induced phosphorylation of PRAS40. (A) Mesangial cells were transfected with FLAG-DJ-1 or vector plasmids as described in the materials and methods. Quiescent transfected cells were treated with 25 mM glucose (HG) for 24 hours. As control, lysates from mesangial cells incubated with 5 mM glucose plus 20 mM mannitol (LG) were used. Cell lysates were immunoblotted with indicated antibodies. (B) Mesangial cells were transfected with scrambled (Scr) or siRNA targeting DJ-1 (siDJ-1) as described in the materials and methods. The transfected cells were incubated with 25 mM glucose (HG) for 24 hours. The cell lysates were immunoblotted with indicated antibodies. The bottom panels show quantification of the results. Panel A, n = 3, *p < 0.001 vs LG. Panel B, n = 4, *p < 0.001 vs LG; #p < 0.001 vs high glucose-treated.
3.5 High glucose-stimulated DJ-1 activates TORC1
PRAS40 binds to raptor, a component of TORC1 [23]. Phosphorylation of PRAS40 appears to dissociate it from raptor, resulting in activation of TORC1. We tested the effect of DJ-1 on TORC1 activation. Phosphorylation of S6-kinase, a substrate of TORC1 was used as an index for TORC1 activation. High glucose increased phosphorylation of S6-kinase (Fig. 5A). Expression of DJ-1 alone was sufficient to augment S6-kinase phosphorylation similar to high glucose (Fig. 5A). Similarly, DJ-1 enhanced phosphorylation of another substrate of TORC1, the mRNA translation repressor 4EBP-1, similar to that in high glucose-treated mesangial cells (Fig. 5B). However, high glucose did not have any additive effect on DJ-1-induced S6 kinase and 4EBP-1 phosphorylation (Figs. 5A and 5B). To confirm these results, siRNA against DJ-1 was used. Downregulation of DJ-1 inhibited high glucose-stimulated phosphorylation of S6 kinase and 4EBP-1 (Figs. 5C and 5D). These results indicate that DJ-1 regulates high glucose-induced TORC1 activation.
Figure 5.
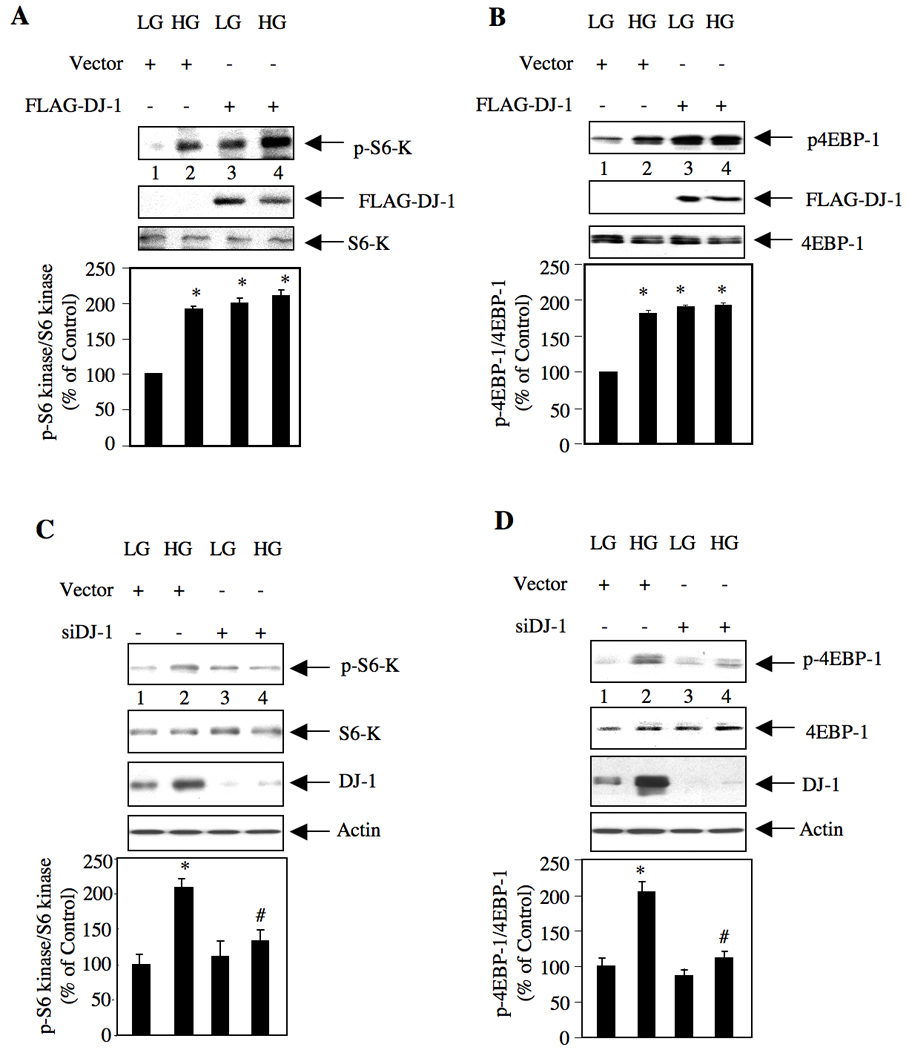
DJ-1 regulates high glucose-induced TORC1 activation. (A and B) Mesangial cells were transfected with FLAG-DJ-1 or vector plasmids as described in the materials and methods. Quiescent transfected cells were treated with 25 mM glucose (HG) for 24 hours. As control, lysates from mesangial cells incubated with 5 mM glucose plus 20 mM mannitol (LG) were used. Cell lysates were immunoblotted with indicated antibodies. (C and D) Mesangial cells were transfected with scrambled (Scr) or siRNA-targeting DJ-1 (siDJ-1) as described in the materials and methods. The transfected cells were incubated with 25 mM glucose (HG) for 24 hours. The cell lysates were immunoblotted with indicated antibodies. The bottom panels show quantification of the results. Panels A and B, n = 3, *p < 0.001 vs LG. Panel C, n = 4, *p < 0.01 vs LG; #p < 0.01 vs high glucose-treated. Panel D, n = 4, *p < 0.001 vs LG; #p < 0.001 vs high glucose-treated.
3.6 DJ-1 regulates protein synthesis and hypertrophy of mesangial cells
We reported recently that Akt-dependent activation of TORC1 regulates high glucose-induced mesangial cell hypertrophy [14]. We have shown above that DJ-1 increases Akt phosphorylation with consequent activation of TORC1 in response to high glucose in mesangial cells (Figs. 3 and 5). Therefore, we tested the involvement of DJ-1 in mesangial cell hypertrophy. Expression of DJ-1 significantly increased protein synthesis similar to that treated with high glucose in mesangial cells (Fig. 6A). Similarly, DJ-1 increased mesangial cell hypertrophy (Fig. 6B). To confirm these observations, when we used siDJ-1 in mesangial cells, downregulation of endogenous DJ-1 significantly decreased high glucose-stimulated protein synthesis (Fig. 6C). Also, inhibition of DJ-1 expression suppressed hypertrophy of mesangial cells in response to high glucose (Fig. 6D). These results demonstrate a contribution of DJ-1 to high glucose-induced mesangial cell hypertrophy.
Figure 6.
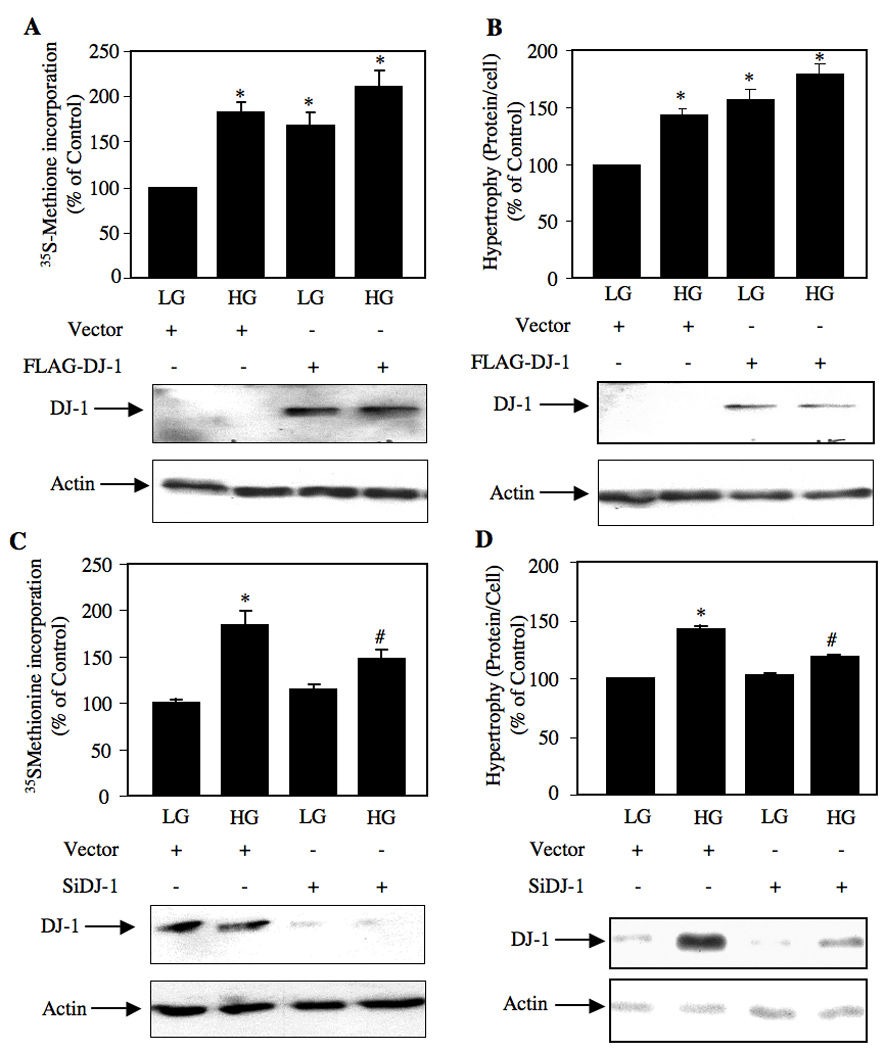
DJ-1 regulates high glucose-stimulated protein synthesis and hypertrophy of mesangial cells. Mesangial cells were transfected with FLAG-DJ-1 or vector plasmids (panels A and B) and scrambled or siDJ-1 (panels C and D) as described in the materials and methods. Quiescent transfected cells were treated with 25 mM glucose (HG) for 24 hours. Control cells were treated with 5 mM glucose plus 20 mM mannitol (LG). (A and C) Protein synthesis was determined as 35S-methionine incorporation as described in the materials and methods [14, 15]. (B and D) Cellular hypertrophy was determined by measurement of total protein per cell as described in the materials and methods [14, 15]. Mean ± SE of triplicate measurements is shown. Panel A, *p < 0.01 vs LG. Panel B, *p < 0.001 vs LG. Panel C, *p < 0.001 vs LG; #p < 0.05 vs high glucose-treated. Panel D, *p < 0.001 vs LG; #p < 0.01 vs high glucose-treated.
4. DISCUSSION
Here we report that high glucose-induced phosphorylation of Akt is mediated by DJ-1. We show that DJ-1 forms a complex with PTEN in response to high glucose. Furthermore, we demonstrate that DJ-1-stimulated Akt kinase phosphorylates and hence inactivates PRAS40. Finally, we show a role of high glucose-stimulated DJ-1 in activation of TORC1, leading to mesangial cell hypertrophy (Fig. 7).
Figure 7.
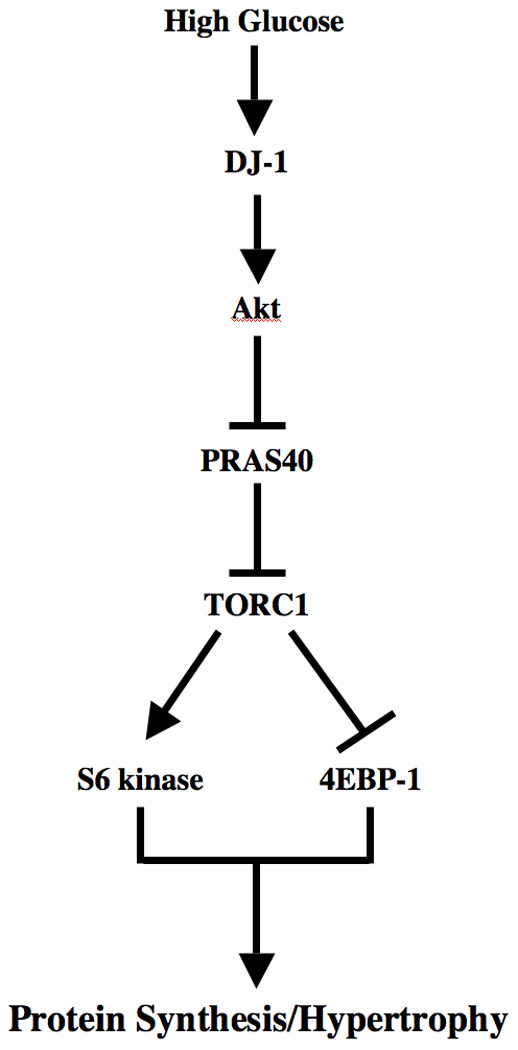
Schematic summarizing our findings.
Increasing evidence indicates that common mechanistic elements like ROS produced by pathologic consequences contribute to many diseases including neuronal disorders associated with aging, cardiovascular complications, cancer and diabetes. A role for DJ-1 has been conclusively established in early-onset PD, where mutation of this gene occurs in 1–2% of recessive forms of the disease [25]. Mice null for DJ-1 locus are normal with no abnormalities in number of dopaminergic neurons in substantia nigra, regulator of the motor function deficiency in PD [26]. However, these mice showed hypersensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), resulting in nigrostriatal dopaminergic dysfunction and motor deficits, which indicate a direct role of DJ-1 in protection against ROS [26, 27]. Furthermore, DJ-1 null mice are sensitive to stroke-induced neural damage demonstrating a role for DJ-1 other than its action on dopaminergic neurons [28]. DJ-1’s cytoprotective effect against ROS was originally related to its direct quenching effect as it lacks any antioxidant enzyme activity. A role of Cys-106, which represents a nucleophilic elbow with potential for oxidation, has been reported to be essential for its neuroprotective function [2, 28]. Also, DJ-1 has been found to increase induction of antioxidant enzyme glutamate cysteine ligase necessary for glutathione biosynthesis [29]. Furthermore, it binds and stabilizes the antioxidant transcriptional regulator Nrf-2 [29].
The inhibitory function of DJ-1 against ROS to protect neuronal cells involved in PD described above does not correlate with the results obtained in the present study. A large body of evidence shows that the pathologic consequences of diabetic nephropathy are regulated by a state of increased oxidative stress [12]. High glucose enhances production of ROS in renal cells including mesangial cells [30]. Recently Gorin et al have shown that Nox4-derived ROS regulate glomerular mesangial cell hypertrophy in response to high glucose [31]. Others and we showed previously that high glucose-stimulated Akt kinase activity is required for hypertrophy of mesangial cells [14, 15, 31]. We show here that 25 mM glucose increased the expression of DJ-1 in a sustained manner for upto 48 hours with concomitant increase in Akt phosphorylation (Fig. 1). A dose response study showed that 18.5 mM glucose was sufficient to increase the expression of DJ-1 and Akt phosphorylation (Supplementary Fig. S2). Furthermore, our results demonstrate a requirement of DJ-1 in the activation of Akt (Fig. 3). These results indicate that increased levels of DJ-1 may not be sufficient to intercept the action of ROS produced in these cells in the presence of high glucose. A corollary of this observation is that the antioxidant property of DJ-1 may be elicited in a cell- and context-dependent manner.
In the fruit fly, the PTEN ortholog was identified as a determinant of eye size [32, 33]. More recently, we demonstrated the requirement for PTEN in renal hypertrophy in streptozotocin-induced diabetic rats and in cultured mesangial cell hypertrophy in response to high glucose [15]. A signaling role of DJ-1 in determining cell size was recently identified in a study by Kim et al involving Drosophila genetics [5]. Overexpression of Drosophila DJ-1A, ortholog of mammalian DJ-1, in the eye suppressed the eye size reduction phenotype induced by dPTEN, indicating an antagonistic function of DJ-1 on the PTEN. This observation was then extended to mammalian cells. Overexpression of DJ-1 (i) completely rescued PTEN-induced fibroblasts from apoptosis, and (ii) enhanced resistance to apoptosis in a PTEN-dependent manner [5]. It was also demonstrated that DJ-1 function required the presence of wild type PTEN. In breast cancer, hyperexpression of DJ-1 correlates negatively with PTEN immunoreactivity and positively with Akt phosphorylation. This correlation was also demonstrated in breast tumor cells in culture where presence of DJ-1 was necessary for PTEN-mediated regulation of Akt phosphorylation [5]. Furthermore, in a model of RNAi-mediated downregulation of DJ-1 in Drosophila eye where the eye size is reduced, expression of the Akt kinase (or, PI 3 kinase) completely suppressed this phenotype, indicating that DJ-1 is upstream of Akt kinase in the signaling cascade [34]. Previously we showed a role for Akt in hypertrophy of mesangial cells involving PTEN [14, 15]. Now we present evidence for the requirement of DJ-1 for high glucose-stimulated Akt phosphorylation and mesangial cell hypertrophy (Figs. 3 and 6).
DJ-1 has been shown to antagonize PTEN tumor suppressor and blocks its effect on apoptosis [5]. Our results indicate that DJ-1 may modulate PTEN by physical interaction and inhibit its activity. In contrast, a recent study showed no direct interaction between PTEN and DJ-1 in a yeast two-hybrid assay, and no coimmunoprecipitation of these two proteins was detected [35]. In contrast to these results, we demonstrate that DJ-1 associates with PTEN in mesangial cells (Figs. 2A, 2B and Supplementary Fig. S1). Furthermore, high glucose significantly increased the association of PTEN with DJ-1 in the presence of reduced PTEN protein levels (Figs. 2A – 2D and Supplementary Fig. S3) [15]. Similar to the PTEN-interacting protein PREX2a, DJ-1 may inhibit PTEN activity [20]. An alternative mechanism may involve oxidation of PTEN by superoxidized DJ-1, which will also result in inhibition of PTEN activity and an increase in Akt phosphorylation [36]. In vitro, DJ-1 was not capable of oxidizing PTEN in the presence of H2O2 [35]. However, in the presence of high glucose, which induces reactive oxygen species in mesangial cells, whether DJ-1 oxidizes PTEN is not known.
Inhibition of DJ-1 in Drosophila dopaminergic neurons significantly suppressed the tyrosine hydroxylase positive neurons with reduced production of dopamine in the brain extracts [34]. Also, downregulation of DJ-1 in this model increased production of ROS suggesting a direct role of ROS in neuron loss. This dopaminergic degeneration phenotype was rescued by overexpression of PI 3 kinase, which was associated with a reduction of the increased ROS to basal levels. Furthermore, in a recent study using DJ-1 null mouse model of MPTP-induced PD, Aleyasin et al demonstrated that reduced Akt phosphorylation is linked to increased loss of substantia nigra neurons [37]. Thus a link between DJ-1 and PTEN/PI 3 kinase/Akt signaling axis in protecting cells in Drosophila and in mammalian models has been established. However, the ROS inhibitory activity of PI 3 kinase in the DJ-1 depletion model of Drosophila does not appear to hold in mesangial cells, as high glucose increases production of ROS concomitant with reduced PTEN activity and increase in PI 3 kinase/Akt signaling and DJ-1 expression [12, 14, 15, 30, 31].
Recent studies in animal models have shown a pivotal role of mTOR in kidney hypertrophy observed in physiological states such as compensatory renal hypertrophy and in disease states such as diabetes [24, 38, 39]. We and others have recently reported that diabetes-induced renal mTOR activation is partly due to high glucose-induced Akt activation [13, 24, 40]. Mammalian mTOR exists in two functionally distinct complexes. TORC1 represents a multiprotein complex with five proteins: mTOR, raptor, deptor, mLST8/GβL and PRAS40 [23, 41]. TORC2 contains all the proteins present in TORC1 with rictor, protor and SIN1 in the absence of raptor [41–44]. Although TORC2 has been shown to phosphorylate Akt at Ser-473, others and we have shown recently that this complex determines the substrate specificity of Akt rather than absolute activity [16, 45]. PRAS40, a component of the TORC1, directly interacts with raptor, the substrate recruiting subunit of the complex. Two important substrates of TORC1, S6 kinase and 4EBP-1, contain a TOS motif, which is necessary for their binding to raptor and for phosphorylation by TORC1 [46]. PRAS40 contains a TOS motif and a KSLP region by which it binds raptor with higher affinity than 4EBP-1 and S6 kinase [23, 47]. Phosphorylation of PRAS40 by Akt dissociates it from the raptor enabling the TORC1 substrates to bind and be phosphorylated [23]. We have recently shown that high glucose in mesangial cells in culture and hyperglycemia in streptozotocin-induced diabetic rats increased phosphorylation of PRAS40 [14]. In the present study, we show a requirement of DJ-1 in high glucose-induced phosphorylation of PRAS40 (Fig. 4).
A study using embryonic fibroblasts and epithelial cells has clarified the role of 4EBP-1 in the regulation of cell size and proliferation, where the mRNA translational repressor forces cell proliferation, indicating that S6 kinase regulates cell size [48]. However, recently we have demonstrated that both S6 kinase and 4EBP-1 act downstream of Akt kinase and contribute to increased protein synthesis necessary for mesangial cell hypertrophy [14, 16]. In line with this observation, we demonstrate increased phosphorylation of S6 kinase and 4EBP-1 by DJ-1 similar to that observed with high glucose (Fig. 5). Thus our results show activation of S6 kinase and inactivation of 4EBP-1 increase protein synthesis leading to mesangial cell hypertrophy (Fig. 6).
Originally DJ-1 was identified as an RNA-binding protein [9]. In a recent significant study, many specific DJ-1-interacting mRNAs were identified by immunoprecipitation and CLIP (cross-linked immunoprecipitation) in dopaminergic neurons. These mRNAs contain GG and/or CC motifs in the untranslated regions [49]. Clustering analysis showed transcripts of three groups among these mRNAs: selenoenzymes (e.g., GPx4), mitochondrial proteins/enzymes (such as cytochrome C oxidases I and II) and components of PTEN/Akt survival pathway. In fact the expression of proteins, whose mRNAs bound DJ-1, were increased in DJ-1 knocked down dopaminergic neuronal cells compared to controls, indicating that mRNA-DJ-1 interaction imposes an inhibition on mRNA translation. According to these results, overexpression of DJ-1 would inhibit expression of those proteins, the mRNAs of which bind DJ-1. Akt1 mRNA was found to interact with DJ-1, indicating its translation inhibition [49]. However, we found no inhibitory effect of DJ-1 overexpression on Akt levels in mesangial cells (Fig. 3A). Similarly, no increase in Akt expression was detected in siDJ-1 expressing mesangial cells (Fig. 3B). Rather, DJ-1 increased, while downregulation of DJ-1 inhibited, Akt phosphorylation in response to high glucose (Fig. 3). Furthermore, in the same study by van der Brug et al [49], S6 kinase and 4EBP-1 mRNAs were identified as binding partners of DJ-1 with proposed inhibition of their expression in the presence of DJ-1. In the present study, we did not notice any change in S6 kinase and 4EBP-1 protein levels regulated by overexpression or downregulation of DJ-1 (Fig. 5). These results indicate that in mesangial cells DJ-1 does not act directly at the level of mRNA transcript to regulate Akt-TORC1-S6 K/4EBP-1 signal transduction pathway.
In summary, we have shown that high glucose increases the expression of DJ-1, presumably involving the tumor suppressor protein PTEN. The functional consequences of Akt phosphorylation show activating and inactivating phosphorylations of S6 kinase and 4EBP-1, respectively, thus demonstrating activation of TORC1. Finally, increased DJ-1 contributes to mesangial cell hypertrophy (Fig. 7). Thus DJ-1 may represent a likely target for the development of therapy to prevent mesangial hypertrophy during the progression of diabetic nephropathy.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank Drs. Brent Wagner and Anthony J. Valente, Department of Medicine, University of Texas Health Science Center at San Antonio for critically reading the manuscript. This work was supported by The Juvenile Diabetes Research Foundation 1-2008-185 and NIH RO1 DK50190 grants to GGC. GGC is a recipient of VA Senior Research Career Scientist Award and is supported by VA Research Service Merit Review grant. NGC is supported by VA Merit Review, Ronald P Williams Orthopedic Oncology Developmental Research Award from Cancer Therapy and Research Center, San Antonio and NIH RO1 AR52425 grants. BSK is supported by grants from NIH (DK 077295 and RC2A 036613) and VA Research Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H. Biochem Biophys Res Commun. 1997;231(2):509–513. doi: 10.1006/bbrc.1997.6132. [DOI] [PubMed] [Google Scholar]
- 2.Kahle PJ, Waak J, Gasser T. Free Radic Biol Med. 2009;47(10):1354–1361. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Science. 2003;299(5604):256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 4.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. Proc Natl Acad Sci U S A. 2004;101(24):9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. Cancer Cell. 2005;7(3):263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Le Naour F, Misek DE, Krause MC, Deneux L, Giordano TJ, Scholl S, Hanash SM. Clin Cancer Res. 2001;7(11):3328–3335. [PubMed] [Google Scholar]
- 7.Pardo M, Garcia A, Thomas B, Pineiro A, Akoulitchev A, Dwek RA, Zitzmann N. Int J Cancer. 2006;119(5):1014–1022. doi: 10.1002/ijc.21942. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Taira T, Niki T, Seino C, Iguchi-Ariga SM, Ariga H. J Biol Chem. 2001;276(40):37556–37563. doi: 10.1074/jbc.M101730200. [DOI] [PubMed] [Google Scholar]
- 9.Hod Y, Pentyala SN, Whyard TC, El-Maghrabi MR. J Cell Biochem. 1999;72(3):435–444. [PubMed] [Google Scholar]
- 10.Fioretto P, Mauer M. Semin Nephrol. 2007;27(2):195–207. doi: 10.1016/j.semnephrol.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziyadeh FN. Am J Kidney Dis. 1993;22(5):736–744. doi: 10.1016/s0272-6386(12)80440-9. [DOI] [PubMed] [Google Scholar]
- 12.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Exp Biol Med (Maywood) 2008;233(1):4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 13.Kasinath BS, Feliers D, Sataranatarajan K, Ghosh Choudhury G, Lee MJ, Mariappan MM. Am J Physiol Renal Physiol. 2009;297(5):F1153–F1165. doi: 10.1152/ajprenal.90748.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dey N, Ghosh-Choudhury N, Das F, Li X, Venkatesan B, Barnes JL, Kasinath BS, Choudhury GG. J Cell Physiol. 2010;225(1):27–41. doi: 10.1002/jcp.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Diabetes. 2006;55(7):2115–2125. doi: 10.2337/db05-1326. [DOI] [PubMed] [Google Scholar]
- 16.Das F, Ghosh-Choudhury N, Mahimainathan L, Venkatesan B, Feliers D, Riley DJ, Kasinath BS, Choudhury GG. Cell Signal. 2008;20(2):409–423. doi: 10.1016/j.cellsig.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 17.Mahimainathan L, Ghosh-Choudhury N, Venkatesan B, Das F, Mandal CC, Dey N, Habib SL, Kasinath BS, Abboud HE, Ghosh Choudhury G. J Biol Chem. 2009;284(41):27790–27798. doi: 10.1074/jbc.M109.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das F, Ghosh-Choudhury N, Venkatesan B, Li X, Mahimainathan L, Choudhury GG. J Cell Physiol. 2008;214(2):513–527. doi: 10.1002/jcp.21236. [DOI] [PubMed] [Google Scholar]
- 19.Cully M, You H, Levine AJ, Mak TW. Nat Rev Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 20.Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, Hibshoosh H, Parsons R. Science. 2009;325(5945):1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. Cell. 2007;128(1):129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. Nat Cell Biol. 2009;11(7):881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. Mol Cell. 2007;25(6):903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Sataranatarajan K, Mariappan MM, Lee MJ, Feliers D, Choudhury GG, Barnes JL, Kasinath BS. Am J Pathol. 2007;171(6):1733–1742. doi: 10.2353/ajpath.2007.070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. Neurology. 2004;62(3):389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- 26.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Proc Natl Acad Sci U S A. 2005;102(14):5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Neuron. 2005;45(4):489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 28.Aleyasin H, Rousseaux MW, Phillips M, Kim RH, Bland RJ, Callaghan S, Slack RS, During MJ, Mak TW, Park DS. Proc Natl Acad Sci U S A. 2007;104(47):18748–18753. doi: 10.1073/pnas.0709379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou W, Freed CR. J Biol Chem. 2005;280(52):43150–43158. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 30.Ha H, Lee HB. Nephrology (Carlton) 2005;10 Suppl:S7–S10. doi: 10.1111/j.1440-1797.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- 31.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. J Biol Chem. 2005;280(47):39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 32.Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C. Genes Dev. 1999;13(24):3244–3258. doi: 10.1101/gad.13.24.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Potter CJ, Tao W, Li DM, Brogiolo W, Hafen E, Sun H, Xu T. Development. 1999;126(23):5365–5372. doi: 10.1242/dev.126.23.5365. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B. Proc Natl Acad Sci U S A. 2005;102(38):13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorner K, Holtorf E, Waak J, Pham TT, Vogt-Weisenhorn DM, Wurst W, Haass C, Kahle PJ. J Biol Chem. 2007;282(18):13680–13691. doi: 10.1074/jbc.M609821200. [DOI] [PubMed] [Google Scholar]
- 36.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. EMBO J. 2003;22(20):5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, Park DS. Proc Natl Acad Sci U S A. 2010;107(7):3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JK, Chen J, Neilson EG, Harris RC. J Am Soc Nephrol. 2005;16(5):1384–1391. doi: 10.1681/ASN.2004100894. [DOI] [PubMed] [Google Scholar]
- 39.Lee CH, Inoki K, Guan KL. Annu Rev Pharmacol Toxicol. 2007;47:443–467. doi: 10.1146/annurev.pharmtox.47.120505.105359. [DOI] [PubMed] [Google Scholar]
- 40.Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, Kashiwagi A. Biochem Biophys Res Commun. 2006;340(1):296–301. doi: 10.1016/j.bbrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 41.Guertin DA, Sabatini DM. Cancer Cell. 2007;12(1):9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Curr Biol. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 44.Wullschleger S, Loewith R, Hall MN. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 45.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. Cell. 2006;127(1):125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 46.Schalm SS, Fingar DC, Sabatini DM, Blenis J. Curr Biol. 2003;13(10):797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Harris TE, Roth RA, Lawrence JC., Jr J Biol Chem. 2007;282(27):20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 48.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. Science. 2010;328(5982):1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan-Mamczarz K, Martindale J, Xie C, Ahmad R, Thomas KJ, Beilina A, Gibbs JR, Ding J, Myers AJ, Zhan M, Cai H, Bonini NM, Gorospe M, Cookson MR. Proc Natl Acad Sci U S A. 2008;105(29):10244–10249. doi: 10.1073/pnas.0708518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.