

Abstract
We report here a high-throughput method for the modification of bacterial artificial chromosomes (BACs) that uses a novel two-plasmid approach. In this protocol, a vector modified in our laboratory to hold an R6Kγ origin of replication and a marker recombination cassette is inserted into a BAC in a single recombination step. Temporal control of recombination is achieved through the use of a second plasmid, pSV1.RecA, which possesses a recombinase gene and a temperature-sensitive origin of replication. This highly efficient protocol has allowed us to successfully modify more than 2,000 BACs, from which over 1,000 BAC transgenic mice have been generated. A complete cycle from BAC choice to embryo implantation takes about 5 weeks. Marker genes introduced into the mice include EGFP and EGFP-L10a. All the vectors used in this project can be obtained from us by request, and the transgenic animals are available through the Mutant Mouse Regional Resource Center (NINDS/GENSAT collection). CNS anatomical expression maps of the mice are available to the public at http://www.gensat.org.
INTRODUCTION
Transgenic animals generated from traditional constructs—e.g., pBR-type plasmids, cosmids and λ vectors, which hold between 10- and 50-kb DNA—often show incorrect temporal and cell-specific transgene expression. The average mammalian gene is ~30 kb in length, but sequences that control the proper expression of the gene may be positioned far upstream of the start site, far downstream of the poly(A) tail or even within introns. Therefore, transgene expression that mirrors expression of the native gene requires the entire transcription unit and surrounding intergenic regulatory DNA to be present on the construct used to generate the animal. Vectors with larger cloning capacity, such as bacterial artificial chromosomes (BACs), P1-derived artificial chromosomes (PACs) and yeast artificial chromosomes (YACs), have the ability to hold longer stretches of DNA but require more skill to engineer than do traditional constructs [1,2]. The largest of these ‘large’ vectors, YACs, can, in theory, hold any mammalian gene (i.e., inserts up to 2,000 kb), but in practice YACs tend to be unstable and are often chimeric. Although they matured later than YAC technologies, BAC-based cloning strategies became popular because BAC constructs possess high clonal stability and low chimerism and are relatively easy to handle [3–6]. Originally designed to allow cloning of large genomic DNA fragments for a variety of purposes[7–11], the average BAC holds inserts of ~200 kb, a size that is usually sufficient to cover an entire transcriptional unit and its associated regulatory elements[3,12]. What makes this vector the best choice for large-scale transgenic experiments is the ease with which it can be modified to incorporate DNA insertions, deletions or substitutions[13–17]. Almost all current BAC modification protocols use homologous recombination strategies that complement the RecA deficiency of the BAC host strain. Controlled recombination is performed by using exogenous RecA[13,16] or RecA-like proteins from a variety of sources, e.g., recE/recT (from Rac prophage) [18] and Redα(exo), Redβ(bet) and Redγ(gam) (from bacteriophage-λ)[19–21]. The protocol laid out in this paper is a modification of our previous protocol in which we use the pLD53.SC-AB plasmid to modify BACs[16]. pLD53.SC-AB contains an R6Kγ origin of replication (R6Kγ ori)[22], an ampicillin-resistance gene for positive selection, a sacB gene for negative selection[23], a recombination cassette and a recA gene. The original protocol involved the cloning of two homology arms, two recombination steps and the growth of bacteria on agar and sucrose to eliminate those carrying unresolved BACs. The present protocol describes a method for modifying BACs that is about 2 weeks shorter than that previously published[16]. Using this rapid method, The GENSAT Project at The Rockefeller University has modified more than 2,000 BACs and have generated over 1000 BAC transgenic mice since 2002. Scientific publications that have cited The GENSAT Project or made use of GENSAT BAC transgenic mice can be found in the following link http://www.gensat.org/gensat_papers_report.jsp. Key steps in the BAC modification process are described below.
Experimental design
Choosing a BAC
The first step in our BAC modification protocol is the selection of a BAC clone that contains a gene of interest. (See Fig. 1 for an overview of the modification process.) Depending on the size of the gene, there are usually half a dozen or more BACs that cover the coding sequence. Public access to genome databases such as the University of California Santa Cruz (UCSC) Genome Browser, NCBI Clone Registry and the Ensembl Genome Browser have greatly improved the identification of appropriate BAC clones. As part of our selection criteria, we choose a BAC only if it is listed in at least two of the aforementioned databases. An ideal clone will have the gene of interest flanked by at least 50 kb of DNA at both the 5′ and 3′ ends. The inclusion of large regions of genomic sequence around the transgene helps to support proper expression in vivo. If the gene is asymmetrically located within the insert, i.e., if there is very little 5′ or 3′ flanking DNA present, then expression may not be accurate. Genes larger than 150 kb, e.g., mouse genes encoding cadherin-2 (220 kb), ataxin-1 (410 kb) and synaptotagmin-I (512 kb), are not good candidates for BAC transgenesis, as regulatory sequences are not likely to be carried on the BAC. There are many commercial sources for BACs, which can be purchased as libraries or as individual clones. The BACs used by our laboratory (most often C57BL/6J mouse, occasionally human) are purchased from the Children’s Hospital Oakland Research Institute (BACPAC Resources at CHORI) and from the RIKEN Bioresource Center. We currently own two mouse libraries, RPCI-23 and RPCI-24, which trace their origin to the Roswell Park Cancer Institute. All BACs possess a chloramphenicol-resistance gene and are maintained in DH10B host bacteria. In general, the choice of an appropriate BAC is an educated guess; at this time we simply do not know where the regulatory elements for most genes reside. For this reason, we choose the BAC that contains the most intergenic DNA between the gene of interest and its neighbors. We have noticed, however, that our rules for picking BACs are less useful when dealing with complex genetic loci containing closely apposed gene families, such as protocadherin and Hox genes. In some cases, genes other than the one to be targeted are present on the BAC. These passenger genes, if expressed in the transgenic organism, can result in an unwanted phenotype. We advise that investigators examine the BAC for the existence of other genes, particularly if the gene to be modified is small (1–10 kb), and then consider experimental needs before deciding to proceed. In the case of small genes, a smaller BAC that will contain fewer passenger genes can be chosen.
Figure 1.

A flowchart of the two-plasmid/one–recombination step approach to BAC modification. Transgenes held by pLD53.SC2 include EGFP and EGFP-L10a (EGPF-ribosomal fusion protein). A complete cycle from BAC selection to injection of the modified BAC takes ~5 weeks to complete.
A two-plasmid approach: separation of recA and the recombination cassette
Key to the success of this BAC modification protocol is the use of two separate plasmids: a conditionally replicating vector, pLD53.SC2, which contains a selectable marker and has an R6Kγ DNA origin of replication (R6Kγ ori), and a pSV1.RecA plasmid, which contains a recA gene and has a temperature-sensitive origin of replication (ts-ori) (Fig. 2)[13]. This represents a major change from our previous protocol in which the recA gene and the recombination cassette were on the same plasmid. The advantage provided by the use of two plasmids is that RecA expression can be controlled independently of the recombination cassette. Although RecA, or another RecA-like protein, is absolutely required for recombination to occur between the pLD53.SC2 plasmid and the BAC, constitutive expression of the gene can cause unwanted rearrangements of BAC DNA. The presence of a ts-ori on the pSV1.RecA plasmid allows this plasmid to be turned on when needed and eliminated after recombination is completed.
Figure 2.

A diagrammatic representation of the two-plasmid/one–recombination step BAC modification protocol. In this figure, the gene-specific A-homology arm has already been cloned into the EGFP-containing pLD53.SC2 (described in the protocol). Homologous recombination between pLD53.SC2/A-box and the BAC (a supercoiled circular structure that is represented as a line here) requires a recombinase supplied by pSV1.RecA. After recombination is complete, incubation of bacteria with chloramphenicol, ampicillin and tetracycline (chlor/amp/tet) results in the selection of bacteria that contain a correctly modified BAC. Free pLD53.SC2/A-box remaining in the BAC host bacteria will also be eliminated because of the lack of pi protein required for replication. A second incubation of the bacteria at 43 °C will result in the elimination of pSV1.RecA, as it cannot replicate at this temperature. The presence of the pLD53.SC2 sequence on the BAC does not interfere with the transgene expression or with viability in the animals generated. Confirmation of successful BAC modification as well as organism genotyping can be carried out by PCR amplification of the areas indicated by pink arrows, 5′ forward cointegrate (5′ FC) and 3′ reverse marker primers (3′ RM), and blue arrows, 5′ forward R6Kγ ori (5FR6) and 3′ reverse cointegrate (3′ RC) primers.
Plasmid specifics
The pLD53.SC2 plasmid has been modified by our laboratory to hold genes for either enhanced green fluorescent protein (EGFP) or EGFP-ribosomal fusion protein (EGFP-L10a). AscI and SwaI cloning sites on the modified plasmid serve as insertion points for a homology arm that will allow the transgene to be inserted into a BAC. The presence of an R6Kγ ori on the plasmid means that replication of this construct can only occur in the presence of a pi protein, which is not generated by the BAC host cells. Amplification and engineering of pLD53.SC2 must therefore be carried out in a bacterial strain that possesses the pir gene that codes for the pi protein. We use PIR1 or PIR2 strains of Escherichia coli for this purpose. For recombination to occur between a BAC and pLD53.SC2, a homology arm and a recombinase enzyme must be present. The BAC host bacterial strain, DH10B, carries a mutated recA gene that is inactive. To compensate for this deficiency, RecA or another RecA-like enzyme must be supplied. In this protocol, RecA is supplied by pSV1.RecA, which possesses a ts-ori that only functions well at 30 °C. Recombination, therefore, can be temporally controlled by increasing or decreasing culture temperature[13]. pSV1.RecA is ultimately lost when the bacteria are grown at 43 °C. Any pLD53.SC2 that has failed to integrate at the end of the experiment is also lost because DH10B does not produce pi protein and the plasmid cannot independently replicate in the DH10B host bacteria. The absence of free plasmid in host cells greatly reduces background. Both plasmids possess antibiotic-resistance genes (pLD53.SC2 has an ampicillin-resistance gene, pSV1.RecA has a tetracycline-resistance gene) that allow for quick and easy bacterial selection.
Genes inserted into the pLD53.SC2 vector
We have used both EGFP and EGFP-L10a as marker genes, depending on the aim of the experiment.
EGFP: EGFP is a chromophore-containing protein that is 238 amino acids in size and fluoresces if excited at 490 nm. If the number of EGFP molecules translated by a cell is sufficient (this depends on the strength of the promoter driving expression and number of copies inserted), epifluorescence can be directly visualized in living or fixed tissue [24]. If intrinsic fluorescence is not strong enough to be seen in a cell, the EGFP signal can be amplified by immunostaining with specific antibodies.
EGFP-L10a: For the purpose of isolating mRNA from specific cell types, we fused the ribosomal protein Rp110a with EGFP and created a fusion reporter that can be expressed under the regulatory control of a chosen gene[25,26]. If a gene of interest is expressed by a particular cell, the EGFP-L10a fusion molecule will be present in the ribosomes of this cell. Through the use of EGFP immunoprecipitation, the ribosomes and the mRNAs contained within can be isolated. This cell-specific mRNA can be studied by DNA microarray or sequencing technology. This approach to studying gene expression is particularly useful when working with cells that are few in number or are located among other cell types and, therefore, difficult to isolate.
BAC modification
The first requirement for homologous recombination is that a small homology arm be cloned into a pLD53.SC2 plasmid that carries either an EGFP or EGFP-L10a gene. Homologous stretches of DNA in both the BAC of interest and the pLD53.SC2 plasmid dictate the location at which recombination, and therefore the plasmid/transgene’s placement, will occur. In most cases, the homology arm chosen for recombination is a 250–500 bp region of DNA located directly upstream of the ATG start site of the gene on the BAC. This site, referred to as an A-homology arm or A-box, is PCR-amplified with primer sequences that are 25–30 bp in length (See Fig. 3 for a diagrammatic representation of an A-box.). For cloning steps, a key step in the design of the forward 5′ A-box primer is the addition of an AscI or MluI enzyme recognition site at the 5′ end of the primer (AscI = 5′-GGCGCGCC-3′ and MluI = 5′-ACGCGT-3′). The reverse primer is a sequence that is located directly upstream of the ATG start site of the coding sequence and the forward primer is located ~250–500 bp further upstream. In rare cases, template sequences that are rich in GC content cannot be amplified because of the formation of secondary structures. In these circumstances, we synthesize an 80–90-bp oligonucleotide that can serve as a homology arm. The A-homology arm is digested with a specific restriction enzyme and cloned into a similarly digested pLD53.SC2 plasmid. The modified pLD53.SC2 now holds a marker flanked by a region homologous to the region directly upstream of the start site of a gene of interest. It is through this physical arrangement that the pLD53.SC2 vector can be used to place the EGFP or EGFP-L10a gene (or any other cDNA) at the start site of the gene of interest on the BAC. The regulatory elements that would normally drive the transcription of the gene of interest now drive expression of a transgene. The endogenous gene on the BAC will not be expressed because a poly(A) tail is attached to the EGFP or EGFP-L10a. We refer to the modified BAC as a cointegrate. The use of nonresolved BACs in the transformation process has proven to greatly reduce the time needed to generate transgenic mice. As expected, the presence of plasmid sequences in the modified BAC has been shown not to interfere with transgene expression or mouse viability. For experiments in which the investigator wishes to avoid incorporation of plasmid sequences into the BAC, our original two-step approach using the pLD53.SC-AB plasmid can be used[16].
Figure 3.

Diagrammatic representation of an A-homology (A-box) arm. The example chosen is the gene Chat (encoding choline acetyltransferase). The 5′ primer used in the amplification of the Chat A-box was 5′ GGCGCGCCAAGGTGCTCTAGTGCTCTGATCCCAG 3′. The first eight nucleotides in this sequence do not correspond to the genomic sequence of Chat but represent an added AscI recognition site sequence 5′-GGCGCGCC-3′. A key step in designing the 5′ primer is the addition of an AscI or MluI enzyme site at the front of the primer. It serves in a later step when the A-homology arm is ligated into an AscI and SwaI-digested pLD53.SC2 vector at its AscI or SwaI cloning sites. If an internal AscI recognition sequence is present within the homology sequence (can be checked with the DNASTAR program), a MluI recognition site, 5′-ACGCGT-3′, should be added to the end of the primer instead. The enzyme MluI is then used in the digestion step. The 3′ primer used for Chat in the homology amplification step was 5′ CCTAGCGATTCTTAATCCAGAGTAGC 3′. This is the reverse-complement of the 3′ sequence highlighted in the figure.
BAC DNA preparation and microinjection
One of the most important steps in creating BAC transgenic organisms is the use of good-quality DNA for pronuclear injection. Although BAC DNA is stable inside its host cell, the constructs are very large (~200,000 bp) and susceptible to mechanical shear and enzymatic degradation once outside a cell. Several protocols have been published on how to prepare BAC DNA for microinjection into fertilized eggs[27–29]. The purification method described here uses both CsCl gradient centrifugation and membrane dialysis. These methods yield intact and clean BAC DNA that, when injected into pronuclei of fertilized mouse oocytes (we use FVB/N zygotes) by a skilled microinjectionist, results in litters in which 17–19% of the pups are positive for the transgene. The location where the BAC inserts in a chromosome is random. The BAC may insert into any one of the 38 autosomes or 2 sex chromosomes. A chance insertion into the Y chromosome will result in all male progeny of the founder being transgenic, whereas all female progeny will be wild type. (Genotyping in this case is easy.) Infrequently, BACs may integrate after the nuclear DNA has replicated. If this happens, the transgene will not be present in all cells and the founder will be mosaic.
Experimental timeline and applications
Although having a basic knowledge of molecular techniques will be helpful to the investigator wishing to generate transgenic animals, this protocol is a step-by-step guide that includes instructions on how to pick a BAC, prepare plasmid and BAC DNA in bulk, transform bacteria, carry out homologous recombination and purify DNA for pronuclear injection (for an overview see Fig. 1). Special emphasis is placed on areas in which the investigator is most likely to encounter problems. Although it is beyond the scope of this paper to include the actual details for generating transgenic mice, a complete cycle from BAC choice to embryo implantation takes ~5 weeks, with 2.5–3 of these weeks devoted to BAC modification. Some portions of the procedure can be run concurrently. Our laboratory completes 9–10 BAC modification cycles per year, and each technician is usually able to successfully modify about half of the 20 BACs assigned to him or her per cycle. Using the modification method described in this paper, we have successfully engineered more than 2,000 BACs and have produced over 1,000 BAC transgenic mice bearing EGFP and EGFP-L10a transgenes[25,26,30–32]. This BAC modification method can also be used to generate other transgenic lines that carry genes such as td-tomato, lacZ and Cre and Flpe recombinase. In addition, BACs modified with the currently described protocol can be used to generate mice that overexpress mutant or wild-type genes.
Material availability
The plasmids used in this protocol can be obtained from The Rockefeller University by request and modified BACs can be obtained from the Children’s Hospital Oakland Research Institute (BACPAC Resources at CHORI). Transgenic mice generated by the pronuclear injection of modified BACs are available from the Mutant Mouse Regional Resource Centers (NINDS/GENSAT collection). Photographs of nervous and other tissue from EGFP-bearing mice, including confocal images of direct EGFP epifluorescence (Fig. 4) and immunohistochemically stained tissue sections, can be found at http://www.gensat.org, along with additional project details.
Figure 4.

Confocal micrographs of direct epifluorescence captured from tissue sections from six GENSAT EGFP BAC transgenic mice that were generated using the protocol described in this paper. (a) Crym-expressing pyramidal neurons in the cerebral cortex of a P7 mouse. (b) Bnip3-expressing cells lining the intestine of an E15.5 mouse. (c) Id3-expressing glia and blood vessels. (d) Fluorescent fibers in the olfactory nerve layer of an adult Nrp1 mouse. (e) Fcer1g-expressing motor neurons and microglia in the medulla of a P7 mouse. (f) Igsf9-expressing progenitors in the dentate gyrus of the hippocampus. Expression is also seen in interneurons in the molecular layer. Bar in each panel represents 100 μm.
MATERIALS
REAGENTS
Transgene-containing vectors: pLD53.SC2, which contains EGFP, or pLD53.SC296, which contains EGFP-L10a. Both available from the laboratory of Nathaniel Heintz, The Rockefeller University, through Judy Walsh (walshj@rockefeller.edu)
pSV.RecA vector: available from the laboratory of Nathaniel Heintz, The Rockefeller University, through Judy Walsh (walshj@rockefeller.edu)
Swiss Webster female mice (Taconic)
Bacterial artificial chromosomes (BACs in DH10B host bacteria; Invitrogen Corporation, BACPAC Resources at CHORI or Riken Bioresources Center)
PIR1 chemically competent E. coli (Invitrogen, cat. no. C1010-10)
PIR2 chemically competent E. coli (Invitrogen, cat. no. C1111-10)
MAX efficiency DH5α-competent cells (Invitrogen, cat. no. 18258-012)
PCR primers (Invitrogen and Biosynthesis; see REAGENT SETUP)
AscI restriction endonuclease (New England Biolabs, cat. no. R0558L)
EcoRI restriction endonuclease (New England Biolabs, cat. no. R0101L)
MluI restriction endonuclease (New England Biolabs, cat. no. R0198L)
SwaI restriction endonuclease (New England Biolabs, cat. no. R0604L)
T4 DNA ligase (New England Biolabs, cat. no. M0202L)
XmaI restriction endonuclease (New England Biolabs, cat. no. R0180L)
PI-SceI endonuclease (New England Biolabs, cat. no. R0696S)
λ-DNA-HindIII Digest (New England Biolabs, cat. no. N3012S)
Low-range PFG marker DNA ladder (New England Biolabs, cat. no. NO350S)
2-log DNA ladder (New England Biolabs, cat. no. N3200L)
1-Butanol (Fisher Scientific, cat. no. A399)
2-Propanol (Isopropanol, Fisher Scientific, cat. no. A416)
Ammonium acetate (Fisher Scientific, cat. no. A637)
Ampicillin (amp; Sigma-Aldrich, cat. no. A9518; see REAGENT SETUP)
Calcium chloride dihydrate (CaCl2, Fisher Scientific, cat. no. C70–500)
Cesium chloride (CsCl, Fischer Scientific, cat. no. BP1595-500)
Chloramphenicol (chlor; Sigma-Aldrich, cat. no. C0378; see REAGENT SETUP)
Chloroform (Fisher Scientific, cat. no. C298–500)
Ethidium bromide solution (10 mg ml−1; Sigma-Aldrich, cat. no. E1510) ! CAUTION It has mutagenic properties.
Ethanol (Pharmco-AAPER)
EDTA (Sigma-Aldrich, cat. no. E5134)
FailSafe PCR System (Epicentre, cat. no. FS99250)
Glacial acetic acid (Fisher Scientific, cat. no. A38S) ! CAUTION It is corrosive, and both the liquid and mist cause severe burns to all body tissues.
Glycerol (Fisher Scientific, cat. no. BP229)
Herculase Enhanced DNA Polymerase (Stratagene, cat. no. 600262)
Hydrochloric acid (HCl; Fisher Scientific, cat. no. A144S) ! CAUTION It is corrosive, and both the liquid and mist cause severe burns to all body tissues
LB agar, Miller (BD, cat. no. 244510)
LB broth, Miller (BD, cat. no. 244610)
NEBuffer 4 (New England Biolabs, cat. no. B7004S)
Agarose, NuSieve ‘GTG agarose’ (Cambrex Bio Science, cat. no. 50080)
P1 buffer (Qiagen, cat. no 19051)
P2 buffer (Qiagen, cat. no 19052)
P3 buffer (Qiagen, cat. no 19053)
PCR primers (Bio-Systhesis)
Pfu DNA polymerase (Stratagene, cat. no. 600135)
Phenol solution (Sigma-Aldrich, cat. no. P4557) ! CAUTION It is corrosive, and both liquid and vapor are combustible. It is toxic and harmful if swallowed, inhaled or absorbed through the skin.
Potassium acetate (Fisher Scientific, cat. no. BP364)
Pulsed-field gel (see REAGENT SETUP)
QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
S.O.C. Medium (Invitrogen, cat. no. 15544-034).
Sodium chloride (NaCl; Fisher Scientific, cat. no. S271-3)
SDS (Fisher Scientific, cat. no. BP166)
Sodium hydroxide (NaOH; Fisher Scientific, cat. no. S318) ! CAUTION It is corrosive and causes burns to any area of contact.
T4 DNA ligase reaction buffer (New England Biolabs, cat. no. B0202S)
TBE see REAGENT SETUP
Tetracycline (tet; Sigma-Aldrich, cat. no. T3258; see REAGENT SETUP)
Trizma base (Tris; Sigma-Aldrich, cat. no. T1503)
Sodium acetate anhydrous (NaOAc; Fisher Scientific, cat. no. S210–500)
Taq DNA polymerase kit (Qiagen, cat. no. 201203)
dH2O (Sigma-Aldrich, cat. no.W1503)
CHEF-DR III system (Bio-Rad)
EQUIPMENT
Databases. Users can choose a BAC clone of interest by searching the following genome databases: University of California Santa Cruz (UCSC) Genome Browser
(http://genome.ucsc.edu/), NCBI Clone Registry
(http://www.ncbi.nlm.nih.gov/projects/genome/clone/) and the Ensembl Genome Browser
DNASTAR Lasergene 8 SeqBuilder software
Millipore membrane filter (0.025-μm pore; Millipore, cat. no. VSWP02500)
Sterile Falcon polypropylene round-bottom tubes (14 ml; Fisher Scientific, cat. no. 14-959-11B)
Sterile Falcon polypropylene conical tubes (15 ml; Fisher Scientific, cat. no. 14-959-49B)
Sterile yellow pipette tips 1 –20μl (USA Scientific, cat. no. 1111-0806)
Sterile blue pipette tips 101 –1000μl (USA Scientific, cat. no. 1111-2821)
Nalgene Oak Ridge PPCO round-bottom centrifuge tube (30-ml; Fisher Scientific, cat. no. 05-529-1C)
Sterile Falcon polypropylene conical tube (50 ml; Fisher Scientific, cat. no. 14-432-22)
Polypropylene wide-mouth bottle with cap (250 ml; Beckman, cat. no. 356011)
Polypropylene bottle with screw-on cap (500 ml; Beckman, cat. no. 355665)
Polypropylene bottle with screw-on cap (1,000 ml; Beckman, cat. no. 355676)
Polypropylene Erlenmeyer flask (2,000 ml; Fisher Scientific, cat. no. 4102-2000)
Corning centrifuge tube (large volume; Corning, cat. no. 430776)
J6-MI Beckman-Coulter centrifuge with a JS-4.2 rotor (Beckman-Coulter)
J-25i Beckman Avanti centrifuge with a JLA-16.250 rotor (Beckman-Coulter)
Cordless Tube Topper sealer (Beckman, cat. no. 358312)
Optima L-90K ultracentrifuge with a NVT65 rotor (Beckman)
Eppendorf refrigerated microcentrifuge 5417R (Eppendorf)
Chroma Spin+TE-400 columns (Clontech, cat. no. 636076) ▴ CRITICAL Do not substitute; these columns produce the best results, by far.
Electroporation system (Gene Pulser II System; Bio-Rad)
Gene pulser/micropulser cuvettes (Bio-Rad, cat. no. 165-2089)
Quick-seal polyallomer tubes (13.5 ml, 16 × 76 mm; Beckman-Coulter, cat. no. 342413)
Pulsed-field gel electrophoresis apparatus (CHEF-DR II System, Bio-Rad, cat. no. 170-3612)
UV/visible spectrophotometer (American Pharmacia Biotech, cat. no. 802106-00)
UV transilluminator (VWR, cat. no. 21474-582)
Barnstead Lab-Line multi-purpose rotator (Alpha Multiservices, cat. no.2314Q)
AlphaImager 2200 Digital Imaging System (Alpha Innotech, cat. no. IS-2200-110-M)
Vacutainer blood collection tubes (Fisher Scientific, cat. no. 02-833-96)
FVB/N and Swiss Webster mice (Taconic)
REAGENT SETUP
Alkaline lysis solution
Mix 20 ml of 10 N NaOH, 100 ml of 10% (wt/vol) SDS and 880 ml of dH2O. Store at room temperature (25 °C) for several months.
KOAc solution (2 M)
Mix together 250 ml of 7.5 M potassium acetate, 115 ml of glacial acetic acid and 635 ml of dH2O. Store at 4 °C for several months.
Injection buffer
Mix 10 mM Tris (pH 7.5), 0.1 mM EDTA and 100 mM NaCl. Adjust the pH to 7.4 with HCl. Store at 4 °C for a maximum of 3 months.
NaCl-saturated butanol
Mix 40 ml of 3 M NaCl in 200 ml of butanol. Store at room temperature.
Glycerol solution (20% (vol/vol))
Add 20 ml of glycerol to 80 ml of H2O and autoclave. Store at 4 °C for 3 months.
Tris (1 M)
Mix 121 g of Tris base and 1,000 ml of H2O. Adjust the pH to 8.0 with HCl. Store at room temperature.
EDTA (0.5 M)
Mix 186.1 g of EDTA and 800 ml of H2O. Adjust the pH to 8.0 with ~20 g of NaOH pellets.. Store at room temperature.
Tris-EDTA buffer (1× TE)
Add 10 ml of 1 M Tris and 2 ml of 0.5 M EDTA. Store at room temperature.
TE solution (10:50)
Mix together 1 ml of 1 M Tris (pH 8.0), 10 ml of 0.5 EDTA (pH 8.0) and 890 ml of dH2O. Store at room temperature
TBE (1×)
The solution consists of 0.089 M Tris base, 0.089 M boric acid and 0.002 M EDTA. Store at room temperature
DNA-loading dye
It consists of 0.25% (wt/vol) bromophenol blue, 0.25% (wt/vol) xylene cyanol and 30% (vol/vol) glycerol. Store at 4 °C
LB broth
The LB broth medium used in this protocol is supplemented with one or two or three antibiotics: chloramphenicol (20 μg ml−1; LB broth-chlor20), ampicillin (30 or 50 μg ml−1; LB broth-amp30 or -amp50) or tetracycline (10 μg ml−1; LB broth-tet10). Store at 4 °C for up to 3 months
LB agar plates
Add 32 g of LB agar, Miller (powder) to 1 liter of H2O; mix and autoclave the solution. To prepare plates, allow medium to cool and supplement with one or a combination of three antibiotics: chloramphenicol (20 μg ml−1; LB agar-chlor20 plate), ampicillin (30 μg ml−1; LB agar-amp30, plate) or tetracycline (10 μg ml−1; LB agar-amp10 plate). Store at 4 °C for up to 2 months
Ampicillin stock solution (100 mg ml−1)
Mix 1,000 mg of ampicillin in 10 ml of dH2O. Sterilize by filtration and store in 2 ml aliquots at −20 °C for 3 months
Tetracycline stock solution (10 mg ml−1)
Mix 500 mg of tetracycline in 50 ml of 50% ethanol/water (vol/vol). Store at −20 °C for 3 months
Chloramphenicol stock solution (20 mg ml−1)
Mix 1,000 mg of chloramphenicol in 50 ml of 100% ethanol. Store at −20 °C for 3 months
PCR primer design
All PCR primers used in this protocol are custom designed. The concentration of primer with which we work is 10 pmol μl−1. The primers used to amplify the ‘A’ homology arm are 25–30 bp in length and chosen by hand. The reverse 3′ primer is a sequence that is located directly upstream of the ATG start site of the gene and the forward 5′ primer is located ~250–500 bp further upstream. For cloning steps, a key step in the design of the forward 5′ A-box primer is the addition of an AscI or MluI enzyme recognition site at the front of the primer (AscI = 5′-GGCGCGCC-3′ and MluI = 5′-ACGCGT-3′). See Figure 3 for further explanation. This same 5′ A-box primer with added enzyme site can be used in non-cloning PCR steps as well. To analyze the modified BAC, two PCRs are run at the same time. The first set of primers consists of a forward 5′ cointegrate primer, which is located 80–100 bp upstream of the A-box, and a reverse 3′ marker primer of the reporter in the construct (reverse 3′ EGFP: 5′ CGCCCTCGCCGGACACGCTGAAC 3′ or reverse 3′ EGFP-L10a: 5′ CAGGGTGTCGCGTGAGACTTTGCTGC 3′). The second set of primers used consists of a forward 5′ R6Kγ ori (forward 5′ R6Kγ ori 5′ CAGGTTGAACTGCTGATCTTCAGATCCTC 3′) and a reverse 3′ cointegrate primer, which is located 80–100 bp downstream of the A-box.
Pulsed-field gel electrophoresis
Add 2 g of agarose to 180 ml of dH2O and mix by swirling. Heat the solution in the microwave for 4 min. Add 20 ml of 5× TBE and further mix by swirling. Pour the 1% (wt/vol) agarose solution into a pulsed-field gel mold and allow it to polymerize. Transfer the gel to the gel box and insert 2 mm of a low-range PFG Marker DNA ladder gel into one well. Ensure that there are no bubbles. Load 10 μl of samples into other wells. Cover the gel with 0.5× TBE and run it at 16 °C in a CHEF-DR III (Bio-Rad) system at 6 V cm−1, with initial and final switch times of 5 and 20 s and an angle of 120°, for 16 h. Remove gel from the machine and place it in a solution of 0.5 mg ml−1 ethidium bromide (10 μl of ethidium bromide stock solution in 200 ml dH2O). Gently agitate on a rotator for 1 h. Pour out the solution and wash the gel in 200 ml of dH2O for 1 h. Photograph the gel with a digital imaging system. ! CAUTION Ethidium bromide has mutagenic properties. Dispose of the solution properly.
PROCEDURE
Amplification and purification of plasmids ● TIMING 2 d
-
1|
Add plasmids to competent cells. Use option A to amplify the pLD53.SC2 vector, which requires the pi protein to replicate and must be grown in PIR1- or PIR2-competent E. coli. Our preference is PIR1. PIR1 cells are able to maintain about 250 copies of the donor vector, whereas PIR2 cells can maintain about 15 copies of the donor vector. Use option B to amplify the pSV1.RecA vector, which has a temperature-sensitive origin of replication and can be grown in most competent bacteria at 30 °C. We use DH5α-competent cells. These cells are commercially available and grow well in culture. Option A and option B should be carried out in parallel.
▴ CRITICAL STEP Your work environment needs to be extremely clean. We usually clean the centrifuge, bench and pipettes with 70% EtOH before working with plasmids.
(A) Transformation of PIR1- or PIR2-competent E. coli with pLD53.SC2
To amplify pLD53.SC2, thaw a single 50-μl aliquot of PIR1 chemically competent E. coli on ice and add 1.0 μl of pLD53.SC2 DNA.
Mix the bacteria and plasmids by gently tapping the tube with your finger and incubate the mixture for 30 min on ice.
-
Heat-shock the bacteria and plasmid mixture by placing the tubes in a 42 °C water bath for 50 s. Immediately place back on ice for 1 min.
▴ CRITICAL STEP Use a timer for this step. Heat shocking the bacteria longer than this time will reduce transformation efficiency
Add 400 μl of S.O.C. medium to each tube and incubate for 1 h at 37 °C with shaking at 225 r.p.m.
To grow pLD53.SC2, which possesses an ampicillin-resistance gene, set up two LB agar-amp30 plates and spread one with 5 μl of the transformed PIR1 cells and the other with 100 μl of the transformed PIR1 cells onto each plate. Incubate both plates overnight at 37 °C. One of the plates will likely provide better colony resolution than the other.
Pick a single colony of transformed PIR1 bacteria from the lower density plate plate (from Step 1A(v)) and inoculate it in 3 ml of LB broth-amp30. Incubate the medium for 8 h at 37 °C.
-
Transfer this culture into a 2,000-ml Erlenmeyer flask containing 500 ml of LB broth-amp30 and incubate it for 14–16 h at 37 °C with rotation at 300 r.p.m.
▴ CRITICAL STEP Bulk preparation of plasmid saves a significant amount of time if your goal is to modify multiple BACs.
(B) Transformation of DH5α-competent cells with pSV1.RecA
To amplify pSV1.RecA, thaw one tube of the DH5α chemically competent bacteria on ice and add 1.0 μl of pSV1.RecA DNA.
Mix the bacteria and plasmids by gently tapping the tube with your finger and incubate it for 30 min on ice.
-
Heat shock the bacteria and plasmid mixture by placing the tubes in a 42 °C water bath for 50 s. Immediately place it back on ice for 1 min.
▴ CRITICAL STEP Use a timer for this step. Heat shocking the bacteria longer than this time will reduce transformation efficiency
Add 400 μl of S.O.C. medium to each tube and incubate for 1 h at 30 °C with shaking at 225 r.p.m.
To grow pSV.RecA, which possesses a tetracycline-resistance gene, set up two LB agar-tet10 plates and spread one with 5 μl of the transformed DH5α cells and the other with 100 μl of the transformed DH5α cells Incubate plates overnight at 30 °C.
Pick a single colony of transformed DH5α cells from the lower density plate and inoculate it in 3 ml of LB broth-tet10. Incubate for 8 h at 30 °C.
-
Transfer this culture into a 2,000-ml Erlenmeyer flask containing 500 ml of LB broth-tet 10 and incubate it for 14–16 h at 30 °C with rotation at 300 r.p.m.
▴ CRITICAL STEP Bulk preparation of plasmid saves a significant amount of time if your goal is to modify multiple BACs.
-
2|
Transfer the bacterial cultures from Steps 1A(vii) and 1B(vii) to independent 1,000-ml polypropylene bottles and harvest the cells by centrifugation at 4,552g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 30 min at 4 °C. Process the pellets in parallel as described in Steps 3–18 below.
-
3|
Discard the supernatant and resuspend each bacterial pellet in 30 ml of P1 buffer.
-
4|
Add 30 ml of P2 buffer to the mixture to lyse the bacteria. Gently mix and incubate for 5 min at room temperature.
-
5|
Add 30 ml of ice-cold P3 buffer to the lysate to neutralize it. Gently mix and incubate for 20 min on ice.
-
6|
Centrifuge the mixture at 20,369g (J-25I Beckman Avanti centrifuge, JLA-16.250 rotor) for 30 min at 4 °C.
-
7|
To precipitate the DNA, transfer the supernatant into a polypropylene 250-ml wide-mouth bottle containing 63 ml of isopropanol and mix it by inverting several times
-
8|
Centrifuge at 4,552g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 30 min at 4 °C.
-
9|
Discard the supernatant and wash the pellet with 70% (vol/vol) ethanol.
-
10|
Air-dry the pellet for 10 min at room temperature and resuspend it in 5 ml of 1× TE buffer.
-
11|
Extract the DNA from the mixture with equal volumes of phenol/chloroform by shaking for 5 min on a rotating platform.
-
12|
Centrifuge at 4,552g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 5 min at room temperature.
-
13|
Transfer the upper, aqueous phase into a 30-ml round-bottom centrifuge tube. Add 2 volumes of 100% ethanol and mix by inverting several times. Collect the DNA by centrifuging at 16,417g (J-25I Beckman Avanti centrifuge, JA-25.50 rotor) for 30 min at 4 °C.
-
14|
Discard the supernatant and wash the pellet with 70% (vol/vol) ethanol.
-
15|
Air-dry the pellet for 10 min at room temperature and resuspend it in 400 μl of 1× TE buffer.
■ PAUSE POINT The resuspended pellet can be stored for several months at 4 °C, if required.
-
16|
Prepare Chroma Spin+TE-400 columns as directed in the manufacturer’s instructions.
▴ CRITICAL STEP Do not substitute with another purification column. These columns produce the best results, by far.
-
17|
Carefully apply no more than 200 μl of DNA into the center of the column. Two columns are required to purify 400 μl of plasmid. Do not allow any sample to flow along the wall of the column, or else the purification of the sample will not be complete. Centrifuge at 700g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 5 min at 4 °C. The buffer in the collection tube contains the purified pLD53.SC2 or pSV1.RecA DNA.
-
18|
Check the DNA by mixing 1 μl of sample with 8 μ1 of 1× TE and 1 μl of 10× DNA-loading dye, and analyze it on a 1.5% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder and a λ-DNA-HindIII digest as molecular weight markers.
■ PAUSE POINT Aliquots of purified pLD53.SC2 (0.5 μg μl−1) and pSV1.RecA (~20 ng μl−1) can be stored for up to 2 years at −20 °C.
Digestion of purified pLD53.SC2 ● TIMING 2 d
-
19|
Before ligation with the A-box, the purified pLD53.SC2 plasmid must be digested. To a 10–20-μl aliquot of pLD53.SC2 (~5.0 μg), add 20 μl of NEBuffer 4, 4 μl of AscI, and dH2O to a total volume of 200 μl. Mix gently and incubate overnight in a 37 °C water bath.
-
20|
Add to this digest 100 μl of 7.5 M ammonium acetate and 750 μl of 100% ethanol. Mix well and place the tube in the −80 °C freezer for 5–10 min.
-
21|
Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R). Discard the supernatant very slowly from the side opposite to the pellet.
-
22|
Wash the pellet with 1.0 ml of 70% (vol/vol) ethanol. Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R) for 5 min at 4 °C and carefully pour off the ethanol. Air-dry the pellet for 10 min at room temperature.
-
23|
To the pellet add 86 μl of dH2O, 10 μl of NEBuffer 3, 1 μl of 100× BSA and 4 μl of SwaI. Mix gently and incubate overnight at 25 °C.
-
24|
Purify the digested vector by running the sample through a 1% (wt/vol) low-melting-temperature agarose gel. Visualize the band under UV light and cut it from the gel. Aliquot 2 mm blocks of the sample into 1.5-ml Eppendorf tubes. This is the digested pLD53.SC2 to be used in Step 49.
■ PAUSE POINT Store aliquots for up to 2 years at −20 °C.
Preparation of BAC DNA and verification by PCR ● TIMING 2 d
-
25|
Select a BAC clone (in DH10B cells) containing your gene of interest from the BAC library and streak on an LB agar-chlor20 plate. Incubate at 37 °C overnight.
-
26|
Pick a single colony and inoculate 5 ml of LB broth-chlor20. Grow the cells overnight at 37 °C with shaking at 300 r.p.m.
-
27|
Harvest the cells by centrifuging at 4,552g at 4 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 20 min.
-
28|
Discard the supernatant and resuspend the bacterial pellet in 200 μl of P1 buffer.
-
29|
Add 200 μl of P2 buffer to lyse the bacteria. Gently mix and incubate for 5 min at room temperature.
-
30|
Add 200 μl of ice-cold P3 buffer to the lysate to neutralize it. Gently mix.
-
31|
Transfer the mixture to a 1.5-ml Eppendorf tube and centrifuge it at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R).
-
32|
Transfer the supernatant to Vacutainer blood collection tubes and extract with equal volumes of phenol/chloroform by shaking for 5 min on a rotating platform. Centrifuge at 4,552g for 10 min at 20 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor).
-
33|
Transfer the top aqueous phase into a 2-ml Eppendorf tube. Add two volumes of 100% ethanol and mix gently. Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in a Eppendorf microcentrifuge model 5417R).
-
34|
Discard the supernatant and wash the pellet with 70% (vol/vol) ethanol. Air-dry the pellet for 10 min at room temperature.
-
35|
Resuspend the pellet in 50 μl of 1× TE buffer. This buffer contains the supercoiled circular BAC DNA.
▴ CRITICAL STEP The BAC DNA should only be stored at 4 °C (maximum 6 months). Do not store the DNA in the freezer. You will ruin it.
-
36|
Confirm the identity of the amplified BAC by PCR. This is particularly important if choosing BACs from a library. For each BAC to be tested, try two different PCR conditions. Depending on the GC content of the templates, one condition will yield better results than the other. Set up reactions as described below. See Table 1 for a description of the primers required.
-
37|
Run the PCR using the parameters detailed in the table below.
-
38|
Check the DNA by mixing 9 μl of the finished reaction with 1 μl of 10× DNA-loading dye and analyze it on a 1.5% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder as a molecular weight marker. Visualize the bands with a UV transilluminator.
? TROUBLESHOOTING
Table 1.
PCR primer sequences
| Target | Primer sequence | Used in Step no. |
|---|---|---|
| νA-box 5′ end | 250–500 bp upstream of the gene ATG start site | 36, 38 |
| A-box 3′ end | Immediately upstream of the gene ATG start site | 36, 38, 55 |
| R6Kγ ori forward | 5′-CAGGTTGAACTGCTGATCTTCAGATC-3′ | 55, 90 |
| EGFP reverse | 5′-CGCCCTCGCCGGACACGCTGAAC-3′ | 90 |
| EGFP-L10a reverse | 5′-CAGGGTGTCGCGTGAGACTTTGCTGC-3′ | 90 |
| Cointegrate 5′ end | 80–100 bp upstream of the A-box | 90 |
| Cointegrate 3′ end | 80–100 bp downstream of the A-box | 90 |
Table 3.
| Component | Amount
|
Final amount/concentration | |
|---|---|---|---|
| Non-GC rich | GC rich | ||
| νBAC DNA (50 ng μl−1) | 1.0 μl | 1.0 μl | 2.5 ng μl−1 |
| PCR buffer (10×) | 2.0 μl | 2.0 μl | 1× |
| 5′ primer for A-box | 0.5 μl | 0.5 μl | 0.25 μM |
| 3′ primer for A-box | 0.5 μl | 0.5 μl | 0.25 μM |
| dNTP (1 mM) | 1.0 μl | 1.0 μl | 0.05 mM |
| Taq DNA polymerase | 0.1 μl | 0.1 μl | 0.025 U μl−1 |
| Sterile dH2O | 14.9 μl | 10.9 μl | |
| Q-Solution (comes with Taq DNA polymerase) | 4.0 μl | 1× | |
| Total volume | 20.0 μl | 20.0 μl | |
Table 4.
| Cycle | Denature | Anneal | Extend |
|---|---|---|---|
| ν1 | 94 °C, 2 min | — | — |
| 2–31 | 94 °C, 30 s | 55 °C, 1 min | 72 °C, 1 min |
| 32 | 72 °C, 10 min | ||
| 33 | — | Hold at 4 °C |
Amplification of an A-homology arm (A-box) by PCR ● TIMING 1–3 d
-
39|
Use a high-fidelity DNA polymerase to PCR-amplify an A-homology arm from the chosen BAC. We use Herculase enhanced DNA polymerase with Herculase buffer or Pfu DNA polymerase with Pfu buffer for this step. Set up reactions as detailed below. The mixture must be divided into 4 aliquots of 50 μl each before being placed in the thermocycler. See Table 1 for description of the primers required.
-
40|
Run the PCR using the parameters detailed in Step 37.
-
41|
Check the DNA by mixing 5 μl of the finished reaction with 4 μl of 1× TE and 1 μl of 10× DNA-loading dye; analyze it on a 1.5% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder as a molecular weight marker. Visualize the bands with a UV transilluminator.
Table 5.
| Component | Amount | Final amount/concentration |
|---|---|---|
| νBAC DNA (50 ng μl−1) | 4.0 μl | 1 ng μl−1 |
| Herculase reaction buffer (10×) | 20.0 μl | 1× |
| 5′ primer for A-box | 5.0 μl | 0.25 μM |
| 3′ primer for A-box | 5.0 μl | 0.25 μM |
| dNTP (1 mM) | 8.0 μl | 0.04 mM |
| Herculase polymerase | 2.0 μl | 0.05 U μl−1 |
| Sterile dH2O | 156.0 μl | |
| Total volume | 200.0 μl |
Digestion of the A-homology arm (A-box) ● TIMING 1 d
-
42|
To the remaining PCR product from Step 40 (approximate volume will be 200 μl), add 100 μl of 7.5 M ammonium acetate and 750 μl of 100% ethanol. Mix well by turning the tube upside down several times and place the tube in the −80 °C freezer for 5–10 min.
-
43|
Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R).
-
44|
Ensure that you can see the pellet. It may be tiny. Discard the supernatant very slowly from the side opposite to the pellet.
-
45|
Wash the pellet with 1.0 ml of 70% (vol/vol) ethanol. Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R) for 5 min at 4 °C and carefully pour off the ethanol. Air-dry the pellet thoroughly for 10 min or longer at room temperature.
-
46|
Add to the pellet 86 μl of dH2O, 10 μl of NEBuffer 4 and 4 μl of AscI (or MluI if the A-box contains an internal AscI site). Mix by gently tapping the tube with your finger and incubate it overnight in a 37 °C water bath.
-
47|
Purify the digested fragments with a QIAquick PCR Purification Kit (or gel elution where necessary). This is the digested A homology arm (A-box).
-
48|
To assess concentration, mix 1 μl of digested A-box fragments with 9 μl of 1× TE and 1 μl of 10× DNA-loading dye. Analyze on a 1.1% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder and a λ-DNA-HindIII digest as molecular weight markers.
■ PAUSE POINT The digested A-box DNA can be stored for several years at −20 °C.
Cloning of the A-homology arm (A-box) into pLD53.SC2 ● TIMING 2 d
-
49|
Warm an aliquot of the digested pLD53.SC2 from Step 24 for 10 min in a 65 °C water bath to melt the agarose in which the sample is stored. Ensure that the agarose is completely melted.
▴ CRITICAL STEP Reagents must be added in the order described in Steps 50–52 or ligation will not work.
-
50|
Transfer 100 ng of the digested A-box from Step 48 (4–8 μl, volume depends on the concentration) to a 1.5-ml Eppendorf tube and add dH2O to adjust the volume to 15 μl.
-
51|
To the digested A-box, add 2 μl of the heated pLD53.SC2 (~100 ng) vector from Step 49 and allow the mixture to cool. Cooling is very important to avoid denaturing the T4 DNA ligase used in Step 52.
-
52|
Thaw a tube of T4 reaction buffer and vortex it well. Add 2 μl of T4 reaction buffer to the tube containing the digested A-box and pLD53.SC2 and then add 1 μl of T4 DNA ligase. Mix by gently tapping the sides of the tube with your finger. Mix thoroughly but gently. Spin briefly and cover with aluminum foil.
-
53|
Incubate the ligation mixture at room temperature for a minimum of 2–3 h. The incubation can run overnight if desired.
-
54|
Thaw a single aliquot of PIR1-competent E. coli on ice. To a new 1.5-ml Eppendorf tube, add 4 μl of the pLD53.SC2/A-box ligation mix and 12–16 μl of PIR1 cells. Mix the tube gently and incubate for 30 min on ice.
▴ CRITICAL STEP The pi protein produced by the PIR1 bacteria is critical for the replication of the pLD53.SC2 plasmid. Do not use another type of bacteria; the experiment will not work.
-
55|
Heat shock the PIR1 bacteria and plasmid mixture step by placing the tube in a 42 °C water bath for 50 s. Immediately place it back on ice for 1 min.
▴ CRITICAL STEP Use a timer for this step. Heat shocking the bacteria longer than this time will reduce transformation efficiency
-
56|
Add 100 μl of S.O.C. medium to the tube and incubate for 1 h in a 37 °C water bath.
-
57|
Spread 100 μl of the transformed PIR1 bacteria containing the pLD53.SC2/A-box vector onto an LB agar-amp30 plate. Incubate the plate overnight at 37 °C.
-
58|
Use PCR to check 8–10 individual PIR1 colonies for the presence of the modified pLD53.SC2/A-box vector. To check a colony, remove half of it with a sterile yellow pipette tip and mix it with 15.9 μl of dH2O to prepare a suspension. Set up reactions as detailed below. See Table 1 for a description of the primers required.
-
59|
Run the PCR using the parameters detailed in Step 37.
-
60|
Check the DNA by mixing 9 μl of the finished reaction with 1 μl of 10× DNA-loading dye and analyze it on a 1.5% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder as a molecular weight marker. Visualize the bands with a UV transilluminator. ? TROUBLESHOOTING
Table 6.
| Component | Amount | Final amount/concentration |
|---|---|---|
| νPIR1 colony suspension | 15.9 μl | |
| PCR buffer (10×) | 2.0 μl | 1× |
| 5′ primer for R6Kγ ori | 0.5 μl | 0.25 μM |
| 3′ primer for A-box | 0.5 μl | 0.25 μM |
| dNTP (1 mM) | 1.0 μl | 0.05 mM |
| Taq DNA polymerase | 0.1 μl | 0.025 U μl−1 |
| Total volume | 20.0 μl |
Preparation of pLD53.SC2/A-box vector ● TIMING 1 d
-
61|
On the basis of the PCR results, pick a single confirmed colony of transformed PIR1 bacteria from the plate prepared in Step 57. Inoculate 20 ml of LB broth-amp30 and incubate overnight at 37 °C with rotation at 300 r.p.m.
-
62|
Harvest the cells by centrifuging at 4,552g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 15 min at 4°C.
-
63|
Discard the supernatant and resuspend the bacterial pellet in 500 μl of P1 buffer.
-
64|
Add 500 μl of P2 buffer to the mixture to lyse the bacteria. Gently mix and incubate the mixture for 5 min at room temperature.
-
65|
Add 500 μl of ice-cold P3 buffer to the lysate to neutralize it. Gently mix.
-
66|
Centrifuge the mixture at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R).
-
67|
Transfer the supernatant to Vacutainer blood collection tubes and extract with equal volumes of phenol/chloroform by shaking for 5 min on a rotating platform. Centrifuge at 4,552g for 10 min at 20 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor).
-
68|
Transfer the upper, aqueous phase into another tube. Add two volumes of 100% ethanol and mix gently. Collect the DNA by spinning at 4,552g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 30 min at 4 °C.
-
69|
Discard the supernatant and wash the pellet with 70% (vol/vol) ethanol. Air-dry the pellet for 10 min at room temperature and resuspend it in 200 μl of 1× TE buffer.
-
70|
Prepare Chroma Spin+TE-400 columns as directed in the manufacturer’s instructions. Carefully and slowly apply the DNA sample from Step 69 to the center of the column. Do not allow any sample to flow along the wall of the column. Centrifuge the column and tubes at 700g for 5 min at 4 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor). The buffer in the collection tube contains the purified pLD53.SC2/A-box DNA.
-
71|
Transfer the pLD53.SC2/A-box DNA into a 1.5-ml Eppendorf tube and add 750 μl of 100% ethanol and 100 μl of 7.5 ammonium acetate. Mix well and place the tube in a −80 °C freezer for 30 min. Centrifuge at 20,617g for 30 min at 4 °C (14,000 r.p.m. in an Eppendorf microcentrifuge model 5417R).
-
72|
Discard the supernatant and wash the pellet with 70% (vol/vol) ethanol. Air-dry the pellet for 10 min at room temperature and resuspend it in 13 μl of 1× TE buffer. This sample contains the pLD53.SC2/A-box DNA.
-
73|
Check the concentration by mixing 1 μl of sample with 9 μl of 1× TE and 1 μl of 10× DNA-loading dye and analyze on a 1.0% (wt/vol) agarose gel. Use a λ-DNA-HindIII digest as a molecular weight marker.
■ PAUSE POINT The DNA can be stored at −20 °C for several years.
Verification of A-box integration into pLD53.SC2 by enzyme digest ● TIMING 1 d
-
74|
To confirm that the A-box has been successfully integrated into pLD53.SC2, compare the enzyme digestion pattern of the modified plasmid with that of the unmodified pLD53.SC2. Add 2 μl of pLD53.SC2/A-box vector DNA or unmodified pLD53.SC2, 15 μl of dH2O, 3 μl of NEBuffer 4, 1 μl of XmaI and 1 μl of AscI (or EcoRI if MluI was used) to an Eppendorf tube. Mix the contents gently and incubate the mixture overnight at 37 °C.
-
75|
Run the entire volume of both samples on a 1.5% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder as a molecular weight marker. The unmodified plasmid will be represented by a single band. A successfully modified pLD53.SC2 will be represented by two bands. The lower band will be the size of the A-box. The higher band will be the size of the unmodified pLD53.SC2. See Figure 5 for examples of digested pLD53SC2/A-box DNA.
Figure 5.
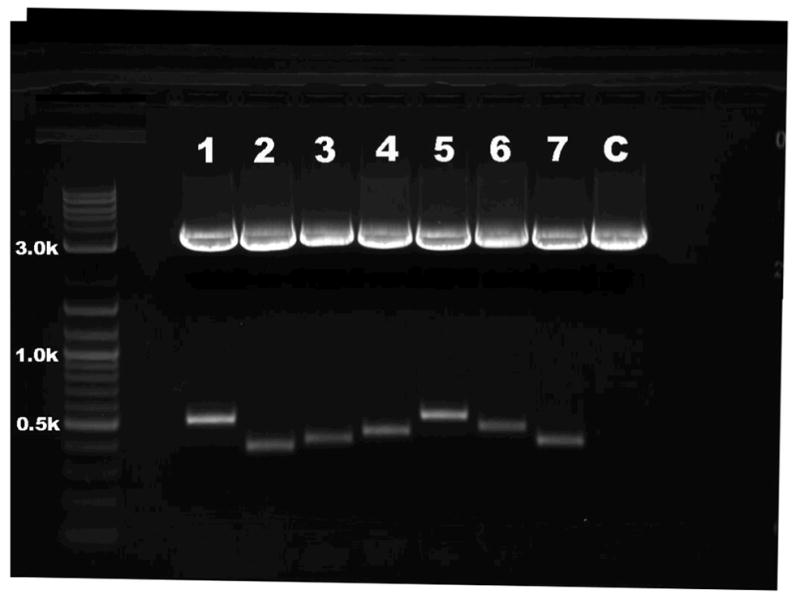
Ethidium bromide–stained agarose gel pattern showing digested pLD53SC2/A-box DNA from seven different genes. DNA was prepared from PCR-positive colonies, and then digested with AscI and XmaI. Samples were analyzed on a 1.5% agarose gel. The seven genes represented include Plekha2 (lane 1), Itgb5 (lane 2), Itga7 (lane 3), Tdo2 (lane 4), Trpc6 (lane 5), Slc39a6 (lane 6) and Sostdc1 (lane 7). The last sample is pLD53SC2 alone as a vector control (C). Fragment sizes were determined by comparison with a 2-log DNA ladder. The lower bands, which range from 395 to 515 bp, are inserts of each gene. The last sample is the vector that does not contain an insert. If the cloning does not work, the lane will contain a single 3,405-bp band representing the unmodified pLD53SC2.
Preparation of BAC host cells chemically transformed with pSV1.RecA ● TIMING 3 d
-
76|
Pick a single colony from the LB agar-chlor20 plate made in Step 25 and inoculate 5 ml of LB broth-chlor20 in a 14-ml Falcon tube. Grow the cells overnight at 37 °C with shaking at 300 r.p.m.
-
77|
Dilute the overnight culture 1:100 with LB broth-chlor20 and incubate for 3–4 h at 37 °C. Check cell density with a spectrophotometer. Grow to an optical density at 600 nm (OD600) of 0.6–0.8.
-
78|
Harvest the cells by centrifugation at 2,560g (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor) for 10 min at 4°C.
-
79|
Discard the supernatant and resuspend the bacterial pellet in 5 ml of ice-cold 50 mM CaCl2. Place the tube on ice for 15 min and repeat Step 78 once.
-
80|
Resuspend the pellet in 300 μl of an ice-cold solution of 50 mM CaCl2 and 20% (vol/vol) glycerol and aliquot 100 μl into three tubes.
■ PAUSE POINT Store two of these tubes at −80 °C. If the pSV.RecA transformation performed in Steps 81–84 does not work, these samples can be used to repeat the process.
-
81|
Thaw an aliquot of pSV1.RecA DNA made in Step 18 and add 2–5 μl of this plasmid DNA to 100 μl of the BAC-competent cells from step 80. Mix the tube gently and incubate for 30 min on ice.
-
82|
Heat shock the bacteria and plasmid mixture by placing the tube in a 42 °C water bath for 50 s. Immediately place the mixture back on ice for 1 min.
▴ CRITICAL STEP Use a timer for this step. Heat shocking the bacteria for longer than this time will reduce transformation efficiency.
-
83|
Add 1.0 ml of S.O.C. medium to the mixture, transfer into a 15-ml Eppendorf tube and incubate for 1 h at 30 °C with shaking at 220 r.p.m.
-
84|
Prepare two LB agar plates containing both chlor20 and tet10. Spread one with 5 μl of the transformed BAC cells and the other with 100 μl of the transformed BAC cells plates. Incubate overnight at 30 °C.
▴ CRITICAL STEP Do not incubate at 37 °C. This temperature will inhibit the replication of pSV.RecA.
-
85|
Pick a single colony from the lower density plate and inoculate it in 5 ml of LB broth containing both chlor20 and tet10. Incubate overnight at 30 °C with shaking at 300 r.p.m.
-
86|
Transfer 1 or 2 ml of overnight culture into 50 ml of LB broth both chlor20 and tet10 in a 250-ml Corning centrifuge tube. Grow cells for 4–5 h with vigorous shaking at 30 °C. Check cell density with a spectrophotometer. Grow cells to an OD600 of 0.6–0.8. Transfer cells into 50-ml Falcon tubes.
▴ CRITICAL STEP Do not incubate at 37 °C. This temperature will inhibit the replication of pSV.RecA.
-
87|
Harvest cells by centrifugation at 2,560g for 10 min at 4 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor).
-
88|
Resuspend the pellet in an equal volume of cold autoclaved 10% (vol/vol) glycerol in water and centrifuge at 2,560g for 10 min at 4 °C. (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor). Repeat this step once.
-
89|
Decant the supernatant as much as possible and gently resuspend the cells to a final volume of 200 μl with cold 10% (vol/vol) glycerol in water.
▴ CRITICAL STEP Glycerol solutions can be difficult to decant. Ensure that the final volume of the dilution of cells is not greater than 200 μl.
-
90|
Dispense 40-μl aliquots into sterile tubes.
■ PAUSE POINT Store for up to 1 year at −80 °C.
Electroporation of BAC host cells with pLD53.SC2/A-box ● TIMING 4 d
-
91|
Thaw 40 μl of pSV1.RecA-transformed BAC-competent cells on ice and add 2 μl of pLD53.SC2/A-box vector DNA (0.5 μg μl−1). Mix the tube gently and incubate for 1 min on ice.
-
92|
Transfer the sample to a cold 0.1-cm cuvette and place it in a BioRad Gene Pulser II electroporation apparatus. The voltage/capacitance/pulse controller parameters used are 1.8 kV/25 μF/200 Ω.
-
93|
After electroporation, add 1 ml of S.O.C. medium to the cuvette and transfer the cell suspension to a 15-ml tube. Incubate the sample for 1 h at 30 °C with shaking at 220 r.p.m.
▴ CRITICAL STEP Do not incubate at 37 °C.
-
94|
Add 5 ml of LB broth containing chlor20 and amp50 and tet10 to the culture in Step 93 and incubate overnight at 30 °C. Only bacteria containing correctly modified BACs and copies of pSV1.RecA will be selected. Unmodified BACs (i.e., those lacking a pLD53.SC2/A-box insert) will be eliminated by exposure to ampicillin. Free pLD53.SC2/A-box remaining in the BAC host bacteria will also be eliminated, as this vector requires the pi protein to replicate.
-
95|
Prepare two LB agar plates containing both chlor20 and amp50. Spread one with 20 μl of the overnight culture and the other with 100 μl of the overnight culture and incubate overnight at 43 °C. One of the plates will likely provide better colony resolution than the other. Free pSV1.RecA remaining in the BAC host bacteria cannot replicate at 43 °C and will be eliminated.
-
96|
Use PCR to check 8–10 individual colonies for the presence of the modified BAC (cointegrate). Successful modification of the BAC is confirmed in two separate PCR reactions. The first reaction (5′ cointegrate PCR) uses a forward 5′ cointegrate primer (a sequence located upstream of the 5′ end of the A-box) and a reverse 3′ marker primer (EGFP or EGFP-L10). The second reaction (3′ cointegrate PCR) uses a forward 5′ primer of R6Kγ ori and a reverse 3′ cointegrate primer (a sequence located downstream of the 3′ end of the A-box). Set up reactions as detailed below. See Table 1 for description of the primers required and Figure 2 for a diagrammatic representation of amplification sites.
-
97|
Run the PCR using the parameters detailed in Step 37.
-
98|
Check the DNA by mixing 9 μl of the finished reaction with 1 μl of 10× DNA-loading dye and analyze it on a 1.2% (wt/vol) agarose gel stained with ethidium bromide. Use a 2-log DNA ladder as a molecular weight marker. Visualize the bands with a UV transilluminator.
? TROUBLESHOOTING
-
99|
Once successfully modified BAC clones are identified, prepare a master LB agar plate that contains chlor20 and amp50. Several different modified BACs can be organized and grown on a single plate if a grid arrangement is used. The bacteria are transferred to the master plate with a sterile yellow pipette tip. Incubate the master plate at 42 °C for 8 h.
■ PAUSE POINT This plate can be used for amplification, rechecks, replica printing, etc. and can be stored for 1 week at 4 °C.
-
100|
From the master LB agar plate that contains chlor20 and amp50 plate, pick a tiny portion of a confirmed colony and use it to inoculate 3 ml of LB broth that contains chlor20 and amp50. Incubate for 8 h at 37 °C with shaking at 300 r.p.m.
-
101|
Transfer 15–50 μl of inoculated broth (volume depends on the cell density) into a 2,000-ml Erlenmeyer flask containing 500 ml of LB broth that contains chlor20 and amp50 and incubate for 14–16 h at 30 °C with shaking at 300 r.p.m. These bacteria contain the successfully modified BACs (cointegrates). The bacteria can be immediately processed as described below for DNA microinjection, or saved for later use.
▴ CRITICAL STEP BACs are normally maintained in low copy numbers in their host bacteria. A large number of cells must be harvested to obtain sufficient quantities of BAC DNA.
■ PAUSE POINT To save the bacteria, add 200 μl of 100% glycerol to 800 μl of culture and mix very well. The bacteria will remain viable for 1 year if stored at −80 °C.
Table 7.
| Component | Amount
|
Final amount/concentration | |
|---|---|---|---|
| 5′ Cointegrate PCR | 3′ Cointegrate PCR | ||
| νDH10B colony suspension | 15.9 μl | 15.9 μl | |
| PCR buffer (10×) | 2.0 μl | 2.0 μl | 1× |
| 5′ forward primer for cointegrate | 0.5 μl | — | 0.25 μM |
| 3′ reverse primer for marker | 0.5 μl | — | 0.25 μM |
| 5′ forward primer for R6Kγ ori | — | 0.5 μl | 0.25 μM |
| 3′ reverse primer for cointegrate | — | 0.5 μl | 0.25 μM |
| dNTP (1 mM) | 1.0 μl | 1.0 μl | 0.05 mM |
| Taq DNA polymerase | 0.1 μl | 0.1 μl | 0.025 U μl−1 |
| Total volume | 20.0 μl | 20.0 μl | |
Purification of modified BAC DNA for microinjection ● TIMING 3–4 d
-
102|
Transfer the bacterial culture to a 1,000-ml polypropylene bottle and harvest the cells by centrifuging at 4,552g for 30 min at 4 °C (J6-MI Beckman-Coulter centrifuge, JS-4.2 rotor). Remove all traces of supernatant.
-
103|
Resuspend the cells in 40 ml of 10 mM EDTA in H2O (pH 8.0), by pipetting up and down several times.
-
104|
Add 80 ml of alkaline lysis solution to the cells and mix by swirling very gently. Incubate for 5 min at room temperature.
-
105|
Add 60 ml of cold 2 M potassium acetate solution. Mix by swirling very gently and incubate for 5 min on ice. Transfer the mixture to a polypropylene 250-ml wide-mouth bottle and centrifuge at 20,369g for 30 min at 4 °C (J-25I Beckman Avanti centrifuge, JLA-16.250 rotor).
-
106|
Transfer supernatant to a 500-ml polypropylene bottle, add 180 ml of isopropanol and mix by gently swirling. Centrifuge at 4,552g for 30 min at 4 °C (J6-MI centrifuge, JS-4.2 rotor). Decant the supernatant.
-
107|
Dissolve the DNA pellet in 18 ml of TE (10:50) and transfer the mixture to a 30 ml round-bottom centrifuge tube. Add 9 ml of 7.5 M KOAc and mix. Incubate for 30 min at −80 °C.
-
108|
Thaw the solution and centrifuge at 4,355g for 10 min at 4 °C (J-25I Beckman Avanti centrifuge, JA-25.50 rotor).
-
109|
Transfer supernatant to a 30-ml round-bottom centrifuge tube and add 2.5 volumes of 100% ethanol. Centrifuge at 20,369g for 30 min at 4 °C to precipitate the DNA (J-25i Beckman Avanti centrifuge, JLA-16.250 rotor).
■ PAUSE POINT The DNA in ethanol can be stored for 2–3 d at −20 °C.
-
110|
Pour off the ethanol and gently resuspend the pellet (while still moist) in 4.4 ml of TE buffer. Meanwhile, in a 50-ml Falcon tube, mix 10.2 g of CsCl with 4.4 ml of TE buffer. Transfer the 4.4-ml DNA solution to the 50-ml Falcon tube that contains the CsCl solution and mix gently until the CsCl has dissolved.
-
111|
Add 0.2 ml of an ethidium bromide solution (10 mg ml−1) to the DNA/CsCl mixture and mix very gently by inverting the tube 2–3 times. Spin at 4,552g for 10 min at 4 °C to remove debris (JM-4.2 rotor).
! CAUTION Ethidium bromide has mutagenic properties. Always wear gloves when handling it.
-
112|
Remove the supernatant and transfer it into a Beckman Quick-Seal tube using a syringe and 18-G needle. Seal tubes carefully with the Cordless Tube Topper and place them in a NVT65 rotor. Use of this particular rotor is important. Centrifuge at 341,650g (Optima L-90K ultracentrifuge) for a minimum of 8 h at 18 °C. An overnight spin is acceptable.
▴ CRITICAL STEP It is very important to balance the weight of the tubes to be centrifuged. Weigh them very carefully to ensure that they do not differ by more than 0.02 g.
-
113|
Remove tubes from the rotor carefully, taking care not to disturb the gradient. Use a 23-gauge needle to poke a hole in the top of the tube. Using a long-wave UV light, carefully remove the illuminated band of DNA with an 18-gauge needle with the needle bevel up. Usually there are two bands. Choose the bottom band. The top band is degraded DNA, whereas the lower band is intact circular BAC DNA. The band can usually be removed in a volume of ~200 μl. Do not take more than 200 μl or you may take a portion of the top degraded band. Transfer the DNA to a 15 ml Falcon tube and bring the total volume up to 2 ml with 1× TE buffer.
-
114|
Add an equal volume of NaCl-saturated butanol to the DNA solution and mix gently. Let the mixture sit for 30 s to allow separation. Remove and discard the top layer. Extract the solution 4–5 times until there is no more pink color.
-
115|
Transfer the DNA solution to a 30-ml round-bottom centrifuge tube and add 1 ml of dH2O to the solution followed by 2.5–3.0 volumes of 100% ethanol. Mix and incubate the solution for 30 min at −20 °C. Centrifuge the solution at 16,417g for 30 min at 4 °C to precipitate the DNA (J-25i Beckman Avanti centrifuge, JA-25.50 rotor).
-
116|
Pour off the ethanol and resuspend the DNA in 0.5 ml of 0.3 M sodium acetate. Transfer the solution to a 1.5-ml Eppendorf tube and add 1 ml of 100% ethanol. Centrifuge the solution at 20,617g for 30 min at 4 °C (Eppendorf Refrigerated Microcentrifuge model 5417R).
-
117|
Discard the supernatant and fill the tube with 70% (vol/vol) ethanol. Allow the tube to sit for 5 min at room temperature. Centrifuge the solution at 20,617g for 10 min at 4 °C (Eppendorf Refrigerated Microcentrifuge model 5417R).
-
118|
Pour off the ethanol and air-dry the pellet for 10 min at room temperature. Resuspend the DNA gently in 20–40 μl of 1× TE buffer. The volume will depend on pellet size. Incubate the solution for 20 min at 37 °C. This is the modified BAC DNA.
-
119|
Check the DNA on a pulse field gel to determine its quality and concentration. See Figure 6 for an example of an optimal pulse field gel result.. ? TROUBLESHOOTING
▴ CRITICAL STEP The BAC DNA should only be stored at 4 °C. Freezing the DNA will destroy it. The tube should be kept away from light.
■ PAUSE POINT BAC DNA can be stored at 4 °C for up to 6 months
-
120|
Before microinjection of oocytes, BACs holding an EGFP or EGFP-L10a transgene should be linearized with the use of the endonuclease PI-SceI. Linearizing BAC DNA has been demonstrated to increase integration into genomic DNA by threefold[33]. Add 100–200 ng of BAC DNA, 2 μl of PI-SceI, 5 μl of NE 10× buffer and dH2O to an Eppendorf tube to produce a final volume of 50 μl. Gently mix and incubate for 5–6 h at 37 °C.
-
121|
Place 20 ml of injection buffer into a sterile Petri dish and float a 25-mm, 0.025-μm-pore Millipore filter on top with the shiny side up. Load the 50 μl of linearized BAC DNA from Step 120 onto the filter and cover the Petri dish with a lid. Allow to sit for 4–6 h at room temperature.
-
122|
Transfer the DNA-containing solution on top of the filter to a microcentrifuge tube and add enough injection buffer to return the solution to the original volume of 50 μl.
-
123|
Check the DNA on a pulsed-field gel to determine the concentration (see REAGENT SETUP).
■ PAUSE POINT The DNA should be stored at 4 °C until injected into oocytes. The lag time between dialysis and use should be no more than 7 d.
▴ CRITICAL STEP The BAC DNA should only be stored at 4 °C. Freezing the DNA will destroy it.
-
124|
Before injection, dilute the DNA with an appropriate amount of injection buffer so that the concentration is 0.2–1 ng μl−1.
-
125|
Check the diluted DNA on pulse field gel again to double confirm the concentration (see REAGENT SETUP). ? TROUBLESHOOTING
-
126|
For each BAC modified, inject ~200 fertilized oocytes obtained from FVB/N parents. Transfer ~40 injected zygotes into pseudopregnant Swiss Webster female mice (from Taconic). ? TROUBLESHOOTING
Figure 6.

Pulsed-field gel patterns of linearized modified BAC DNA. The DNA was digested and run on a 1% agarose pulse field gel at 16 °C in 0.5× TBE in a CHEF-DR III (Bio-Rad) system at 6 V cm−1, with initial and final switch times of 5 and 20 s and an angle of 120° for 16 h. The gel was stained with 0.5 mg ml−1 ethidium bromide for 1 h and was then destained in H2O for 1 h. Fragment sizes were determined by comparison with a low-range PFG marker DNA ladder. Lane M: low-range (2.03–194 kb) PFG marker DNA ladder. The nine genes modified include Bsx (lanes 1 and 2), Gdf10 (lanes 3 and 4; very light band), Kcnip2 (lanes 5 and 6), Cmbl (lane 7), Cyp26b1 (lane 8), Plcxd2 (lane 9), Med12 (lane 10), Cx3cr1 (lane 11) and Fam102b (lane 12). The concentrations of our samples are determined by comparison with six DNA standards on the right side of the gel. The lanes, from left to right, represent 1, 2, 4, 8, 16, and 24 ng DNA.
● TIMING
Steps 1–18, amplification and purification of plasmids: 2 d
Steps 19–24, digestion of the purified pLD53.SC2: 2 d
Steps 25–38, preparation of BAC DNA and verification by PCR 2 d
Steps 39–41, amplification of an A-homology arm (A-box) by PCR: 1–3 d
Steps 42–48, digestion of the A-homology arm (A-box): 1 d
Steps 49–60, cloning of A-homology arm (A-box) into pLD53.SC2: 2 d
Steps 61–73, preparation of pLD53.SC2/A-box vector: 1 d
Steps 74 and 75, verification of A-box integration into pLD53.SC2 by enzyme digest: 1 d
Steps 76–90, preparation of BAC host cells chemically transformed with pSV1.RecA: 3 d
Steps 91–101, electroporation of BAC host cells with pLD53.SC2/A-box: 4 d
Steps 102–126, purification of modified BAC DNA for microinjection: 3–4 d
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| ν36–38 | Failed PCR while trying to confirm BAC identity or homology arm amplification | PCR conditions are not optimal | Use FailSafe PCR system that includes 12 different buffer choices. If PCR still does not work, change primers. If it still does not work, choose a different BAC |
| 58–60 | Failed amplification of pLD53.SC2/A-box with R6Kγ ori and A-box 3′ primers in PIR1 bacterial colonies | The ligation of the A-homology arm with the pLD53.SC2 did not work | Repeat homology arm ligation and transformation from Step 49 to 60. If it still does not work use PIR2 chemically competent E. coli |
| 96–98 | Failed PCR amplification of DNA from chlor/amp-resistant colony (BAC modification did not work) | Your pSC53.SC2/A-box plasmid preparation could be contaminated with unknown amp-resistant plasmids Your DH10B BAC competent cells transformed with pSV1.RecA could be contaminated with unknown amp-resistant plasmids |
Transform DH5α competent cells with pSC53.SC2/A-box. If your DNA is clean, nothing should grow on a chlor/amp plate. If colonies appear, remake the pSC53.SC2/A-box DNA from Steps 61 to 73 Electroporate the cells with dH2O instead of vector. If you still get colonies on a chlor/amp plate make the competent cells from Steps 76–90 |
| 119 | BAC DNA yield is low as determined by pulsed-field gel | Volume of bacteria used to inoculate LB broth at Step 94 was not optimal Shaking is too strong during DNA preparation |
Change the inoculation volume of bacteria at Step 101 Be very gentle during DNA preparation |
| 123–125 | BAC DNA is viscous after linearization | DNA concentration is too high | Dilute DNA with injection buffer to a concentration that allows ease of flow (no lower than 0.125 ng μl−1). If DNA remains too sticky or particulate at the most dilute concentration, prepare again from Steps 120 to 125. In our experience, the ease with which DNA can be injected is linked to the quality of the DNA. Taking time to produce clean and non-sticky DNA assures that the highest number of transgenic mice will be produced |
| 126 | No transgenic mice are produced after zygote injection with modified BAC | BAC DNA is of poor quality Problem with injection buffer A lethal passenger gene may be present in the BAC (rare) |
Prepare DNA again from Steps 100 to 125 Prepare the injection buffer again. Do not deviate from the recipe Repeat with a different BAC that does not hold the lethal gene |
ANTICIPATED RESULTS
This protocol has been used to modify more than 2,000 BACs and generate more than 1,000 transgenic mice. The efficiency of BAC recombination using this system can vary from 70% to 100%. Our current success rate in generating transgenic mice from modified BAC DNA using the procedure described is 17–19%. Although the expression pattern observed in BAC transgenic mice most often mirrors expression patterns observed in in situ hybridization and immunohistochemical studies, occasionally, we observe higher or lower expression that other studies have reported for a particular gene. Greater expression may be the result of multiple BAC copies being present in the genome. Lesser expression may be due to the fact that important regulatory elements may be missing from a BAC. Examples of expression observed in six different BAC transgenic mouse lines generated in our laboratory are shown in Figure 4. Individual cell types can be clearly identified and include projection neurons (Fig. 4a,e), interneurons (Fig. 4f), neuronal progenitors (Fig. 4f), astroglia (Fig. 4c), microglia (Fig. 4e), vascular cells (Fig. 4c) and epithelial cells (Fig. 4b). Cellular processes such as neuronal axons can also be seen (Fig. 4d). These images show direct EGFP epifluorescence; no antibodies were used.
Acknowledgments
We acknowledge C. Wang and S. Mehta for their valuable contribution to the development of the protocol described in this report and we thank S. Mehta and M. Almahariq for critical reading of the paper. We thank Z.D. Barrera for assistance with Figure 2 and J. Walsh for her untiring help with plasmid distribution to the scientific community. This work was supported by an NIH/NINDS contract N01 NS-7-2370, the Simons Foundation; N.H. is a Howard Hughes Medical Institute investigator.
Footnotes
AUTHOR CONTRIBUTIONS
N.H. conceptualized and directed the project. N.H. and S.G. developed the high-throughput method of BAC modification that uses a single recombination step. S.G. designed and conducted experiments. S.G. analyzed data; L.K. improved quality, condensed and summarized the protocol and wrote the paper; L.K. performed confocal microscopy.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Monaco AP, Larin Z. YACs, BACs, PACs and MACs: artificial chromosomes as research tools. Trends Biotechnol. 1994;12:280–286. doi: 10.1016/0167-7799(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 2.Giraldo P, Montoliu L. Size matters: use of YACs, BACs and PACs in transgenic animals. Transgenic Res. 2001;10:83–103. doi: 10.1023/a:1008918913249. [DOI] [PubMed] [Google Scholar]
- 3.Shizuya H, et al. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ioannou PA, et al. A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nat Genet. 1994;6:84–89. doi: 10.1038/ng0194-84. [DOI] [PubMed] [Google Scholar]
- 5.Marra MA, et al. High throughput fingerprint analysis of large-insert clones. Genome Res. 1997;7:1072–1084. doi: 10.1101/gr.7.11.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley JM, et al. High throughput direct end sequencing of BAC clones. Nucleic Acids Res. 1999;27:1539–1546. doi: 10.1093/nar/27.6.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osoegawa K, de Jong PJ. BAC library construction. Methods Mol Biol. 2004;255:1–46. doi: 10.1385/1-59259-752-1:001. [DOI] [PubMed] [Google Scholar]
- 8.Mozo T, et al. A complete BAC-based physical map of the Arabidopsis thaliana genome. Nat Genet. 1999;22:271–275. doi: 10.1038/10334. [DOI] [PubMed] [Google Scholar]
- 9.Hoskins RA, et al. A BAC-based physical map of the major autosomes of Drosophila melanogaster. Science. 2000;287:2271–2274. doi: 10.1126/science.287.5461.2271. Misc: Erratum in Science 288:1751 (2000) [DOI] [PubMed] [Google Scholar]
- 10.Osoegawa K, et al. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000;10:116–128. [PMC free article] [PubMed] [Google Scholar]
- 11.McPherson JD, et al. A physical map of the human genome. Nature. 2001;409:934–941. doi: 10.1038/35057157. [DOI] [PubMed] [Google Scholar]
- 12.Shizuya H, Kouros-Mehr H. The development and applications of the bacterial artificial chromosome cloning system. Keio J Med. 2001;50:26–30. doi: 10.2302/kjm.50.26. [DOI] [PubMed] [Google Scholar]
- 13.Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- 14.Heintz N. BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci. 2001;2:861–870. doi: 10.1038/35104049. [DOI] [PubMed] [Google Scholar]
- 15.Lalioti M, Heath JA. A new method for generating point mutations in bacterial artificial chromosomes by homologous recombination in Escherichia coli. Nucleic Acids Res. 2001;29:E14. doi: 10.1093/nar/29.3.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohtsuka M, Kimura M, Tanaka M, Inoko H. Recombinant DNA technologies for construction of precisely designed transgene constructs. Curr Pharm Biotechnol. 2009;10:244–251. doi: 10.2174/138920109787315033. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 19.Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998;180:2063–2071. doi: 10.1128/jb.180.8.2063-2071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee EC, et al. A highly efficient Escherichia coli–based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 22.Metcalf WW, et al. Conditionally replicative and conjugative plasmids carrying lacZ alpha cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 23.Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI. Positive selection procedure for the entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol. 1985;164:918–921. doi: 10.1128/jb.164.2.918-921.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiedenmann J, Oswald F, Nienhaus GU. Fluorescent proteins for live cell imaging: opportunities, limitations, and challenges. IUBMB Life. 2009;61:1029–1042. doi: 10.1002/iub.256. [DOI] [PubMed] [Google Scholar]
- 25.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chrast R, Scott HS, Antonarakis SE. Linearization and purification of BAC DNA for the development of transgenic mice. Transgenic Res. 1999;8:147–150. doi: 10.1023/a:1008858014473. [DOI] [PubMed] [Google Scholar]
- 28.Gong S, Yang WX. Modification of bacterial artificial chromosomes (BACs) and preparation of intact BAC DNA for generation of transgenic mice. Curr Protoc Neurosci. 2005;Chapter 5(Unit 5.21) doi: 10.1002/0471142301.ns0521s31. [DOI] [PubMed] [Google Scholar]
- 29.Conner DA. Transgenic mouse production by zygote injection. Curr Protoc Mol Biol. 2004;Chapter 23(Unit 23.9) doi: 10.1002/0471142727.mb2309s68. [DOI] [PubMed] [Google Scholar]
- 30.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 31.Geschwind D. GENSAT: a genomic resource for neuroscience research. Lancet Neurol. 2004;3:82. doi: 10.1016/s1474-4422(03)00658-6. [DOI] [PubMed] [Google Scholar]
- 32.Hatten ME, Heintz N. Large-scale genomic approaches to brain development and circuitry. Annu Rev Neurosci. 2005;28:89–108. doi: 10.1146/annurev.neuro.26.041002.131436. [DOI] [PubMed] [Google Scholar]
- 33.Abe K, Hazama M, Katoh H, Yamamura K, Suzuki M. Establishment of an efficient BAC transgenesis protocol and its application to functional characterization of the mouse Brachyury locus. Exp Anim. 2004;53:311–320. doi: 10.1538/expanim.53.311. [DOI] [PubMed] [Google Scholar]