

Abstract
The steroid 5α-reductase (SRD5A) family of enzymes produces steroid hormones that regulate male sexual development. Now, Cantagrel et al. (2010) identify a member of this family, SRD5A3, as a polyprenol reductase with a crucial role in N-linked protein glycosylation and pinpoint SRD5A3 mutations as the cause of a rare Mendelian disease.
Four genes in the human genome have the SRD5A acronym, but only two of them, SRD5A1 and SRD5A2, encode a bona fide steroid 5α-reductase. The other two genes, SRD5A3 and SRD5A2L2, are posers, claiming the name despite having little or no functional ability to reduce steroid substrates. What then are the true substrates of these two pretenders? In this issue of Cell, Cantagrel et al. (2010) ingeniously combine analytical chemistry with genetics in humans, mice, and yeast to uncover the enzymatic and biological function of steroid 5α-reductase SRD5A3. They demonstrate that SRD5A3 encodes a polyprenol reductase that is essential for N-linked glycosylation of proteins in yeast and mammals (Figure 1A), a completely unexpected function.
Figure 1. Steroid 5α-Reductase and N-Linked Protein Glycosylation.
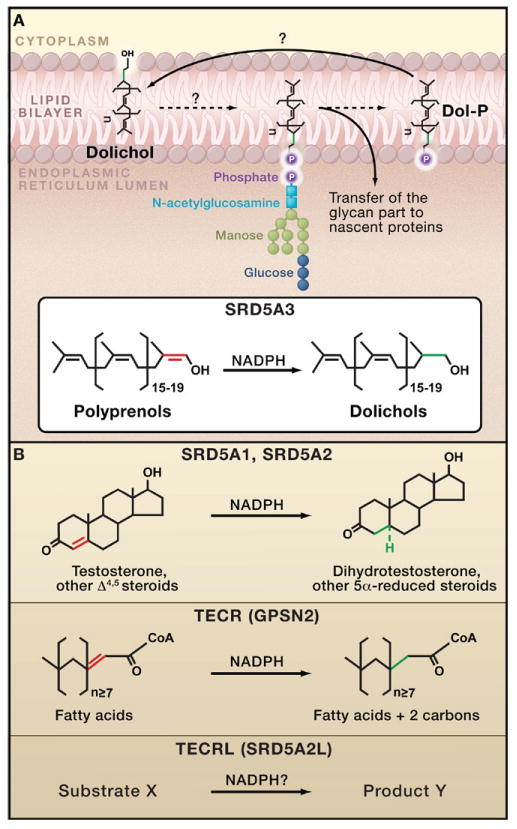
(A) The addition of a 14 sugar oligosaccharide to nascent proteins (N-linked glycosylation) occurs in the membrane of the endoplasmic reticulum (ER), where dolichol-phosphate (Dol-P) serves as a lipid carrier for the glycan unit. The steroid 5α-reductase SRD5A3 catalyzes the reduction of the terminal double bond of polyprenols to generate dolichols, the precursor of Dol-P (Cantagrel et al., 2010). Mutations in SRD5A3 cause a congenital disorder of glycosylation leading to severe developmental anomalies in humans. (B) The other four human steroid 5α-reductase enzymes also reduce the double bond of various lipid substrates using the NADPH cofactor. SRD5A1 and SRD5A2 are required for the proper sexual development of males by catalyzing the synthesis of dihydrotestosterone from testosterone. Although the trans-2,3-enoyl-CoA reductase (TECR) is known to operate on fatty acids, the substrate(s) for the trans-2,3-enoyl-CoA reductase-like (TECRL) enzyme is unknown.
SRD5A1 and SRD5A2 reduce the Δ4,5 bond of steroid substrates using NADPH as a cofactor (Figure 1B) (Russell and Wilson, 1994). It seems that any gene with even a remote sequence similarity to SRD5A1 and SRD5A2 (such as SRD5A3, SRD5A2L, and others that do not use the SRD5A prefix) has been included in this family (Langlois et al., 2010). However, aside from one study on the enzymatic activity of SRD5A3 (Uemura et al., 2008), experimental data on these proteins has been lacking, and it is unclear whether steroids are indeed substrates of SRD5A3 and SRD5A2L.
For most steroid hormones including progestins and glucocorticoids, reduction of the Δ4,5 bond inactivates transcriptional signaling by the hormone. One critical exception to this general rule is the reduction of the Δ4,5 bond in testosterone to produce dihydrotestoster-one (Figure 1B). This reaction is of great importance during the sexual development of males because only dihydrotestosterone (not testosterone) directs the formation and growth of the external genitalia and prostate (Wilson, 1978).
Surprisingly, Cantagrel et al. (2010) find that steroid 5α-reductase SRD5A3 is not involved in steroid hormone formation or sexual development but instead plays a crucial role in the N-linked glycosylation of proteins. Their study begins with an astute clinical observation. Four children of a large family were born with multiple developmental anomalies of the eyes, heart, and brain. A family history revealed several consanguineous relationships, suggesting that the disease was autosomal recessive. Consistent with this mode of inheritance, genome-wide linkage analysis and DNA sequencing identified a homozygous mutation in the SRD5A3 gene. Analysis of individuals with similar symptoms from unrelated families uncovered six additional mutations in SRD5A3. The numerous phenotypes (i.e., the pleiotropy) arising from these mutations strikingly resembled those of patients with congenital disorders of glycosylation, a group of recessively inherited diseases caused by defects in N-linked protein glycosylation.
N-linked protein glycosylation involves the addition of a 14 sugar glycan to select asparagine residues on a nascent protein to facilitate the proper folding and trafficking of the protein. Occurring in the membrane of the endoplasmic reticulum (ER), N-linked protein glycosylation is a byzantine process that involves many steps (Figure 1A). These include the assembly of a lipid carrier for the oligosaccharide, the flip-flopping of this lipid between leaflets of the ER membrane, and multiple cycles of phosphorylation and dephosphorylation of lipids. The large number of players in the pathway renders the genetics of congenital disorders of glycosylation complex. Moreover, major deficiencies in N-linked glycosylation have dire consequences in many organ systems because the majority of proteins, both secreted and membrane-bound, are substrates for this crucial modification.
The similarity of the phenotypes observed for individuals with mutations in SRD5A3 and patients with congenital disorders of glycosylation suggested to Cantagrel and colleagues that SRD5A3 encodes a key enzyme in the pathway of N-linked protein glycosylation. But which enzyme? This is a difficult question to address given the intricacy of the pathway and complexity of its chemistry. In the end, Cantagrel et al. used many types of cutting-edge mass spectrometry to show that SRD5A3 reduces the terminal double bond of polyprenols to form dolichols (Figure 1A). Furthermore, this reduction is required for assembling the complex glycolipids that donate polysaccharides during N-linked glycosylation. Specifically, dolichols in the ER membrane are phosphorylated and tagged with a glycan unit, which is subsequently transferred to an asparagine residue by the activity of oligosaccharyl transferases (Figure 1A).
At this point, Cantagrel and colleagues had discovered the molecular basis of a genetic disease and uncovered the function of an SRD5A family member, but this was just the start. They go on to demonstrate that SRD5A3 is also required for N-linked glycosylation in many different organisms. Deletion of the Srd5a3 gene in mice disrupted protein glycosylation and resulted in death of mouse embryos. Interestingly, deletion of Srd5a3 in mice also boosted the expression of enzymes in the mevalonate pathway, which synthesize the building blocks of polyprenols, the substrates of SRD5A3 (Figure 1A).
In the model plant Arabidopsis thaliana, mutations in the steroid 5α-reductase gene DET2 cause dwarfism and render male plants sterile. Furthermore, these defects are rescued by human SRD5A1 or SRD5A2 (Li et al., 1997). This conservation of function across the great evolutionary divide between primates and plants was unique when reported. Cantagrel and colleagues now describe similarly impressive conservation. They show that human SRD5A3 rescues glycosylation defects in the budding yeast Saccharomyces cerevisiae. Moreover, this property is unique to SRD5A3 because none of the other members of the steroid 5α-reductase family accomplish this task, including the trans-2,3-enoyl-CoA reductase (TECR). Indeed, Cantagrel et al. could not detect any polyprenol reductase activity for TECR (GPSN2), confirming an earlier study, which had identified fatty acids as the substrates of this steroid 5α-reductase family member (Figure 1B) (Moon and Horton, 2003).
The work of Cantagrel and coworkers is creatively exhaustive and nails down every aspect of a difficult research problem from yeast cultures to the clinic. Where could they possibly go from here? For one, their studies in mice with mutations in Srd5a3 suggest a regulatory crosstalk between the mevalonate pathway and N-linked protein glycosylation, and it will be interesting to determine the mechanism underlying this interplay. In addition, the substrate and physiological function of SRD5A2L are still unknown. Will this steroid 5α-reductase function in glycosylation or hormone production, or will it play a completely distinct role from that of its namesake?
References
- Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, Lehle L, Hombauer H, Adamowicz M, Swiezewska E, et al. Cell. 2010 doi: 10.1016/j.cell.2010.06.001. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois VS, Zhang D, Cooke GM, Trudeau VL. Gen Comp Endocrinol. 2010;166:489–497. doi: 10.1016/j.ygcen.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Li J, Biswas MG, Chao A, Russell DW, Chory J. Proc Natl Acad Sci USA. 1997;94:3554–3559. doi: 10.1073/pnas.94.8.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YA, Horton JD. J Biol Chem. 2003;278:7335–7343. doi: 10.1074/jbc.M211684200. [DOI] [PubMed] [Google Scholar]
- Russell DW, Wilson JD. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- Uemura M, Tamura K, Chung S, Honma S, Okuyama A, Nakamura Y, Nakagawa H. Cancer Sci. 2008;99:81–86. doi: 10.1111/j.1349-7006.2007.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JD. Annu Rev Physiol. 1978;40:279–306. doi: 10.1146/annurev.ph.40.030178.001431. [DOI] [PubMed] [Google Scholar]