

Abstract
Pathogenesis of rheumatic heart disease (RHD) remains incompletely understood. Several genes associated with RHD have been described; most of these are involved with immune responses. Single nucleotide polymorphisms in a number of genes affect patients with RHD compared to controls. Molecular mimicry between streptococcal antigens and human proteins, including cardiac myosin epitopes, vimentin and other intracellular proteins is central to the pathogenesis of RHD. Autoreactive T cells migrate from the peripheral blood to the heart and proliferate in the valves in response to stimulation with specific cytokines. The types of cells involved in the inflammation as well as different cytokine profiles in these patients are being investigated. High TNF alpha, interferon gamma, and low IL4 are found in the rheumatic valve suggesting an imbalance between Th1 and Th2 cytokines and probably contributing to the progressive and permanent valve damage. Animal model of ARF in the Lewis rat may further contribute towards understanding the ARF.
Keywords: Autoimmunity, rheumatic Heart disease, susceptibility genes
Rheumatic heart disease (RHD) is still a major health problem in several countries due to the heart lesions that follow a rheumatic fever (RF) episode in 30-45% of patients. The incidence of acute RF (ARF) in some developing countries exceeds 50 per 100,000 children. The worldwide prevalence of RHD is at least 15.6 million cases, and this disease is responsible for around 233,000 deaths/year.[1] RHD results from autoimmune reactions triggered by an untreated S. pyogenes throat infection leading to severe valvular damage in genetically susceptible individuals. Recurrences of ARF play an important role in the worsening of valvular lesionss.[2,3]
In the present review, we focus on genetic susceptibility factors, their role in the development of RF and RHD, and the mechanisms that lead to autoimmune reactions and permanent valvular damage. Animal models of the disease will also be discussed, as will prospective vaccines for the prevention of RF and RHD.
INNATE AND ADAPTIVE IMMUNE RESPONSES : A BRIEF REVIEW
Protection against pathogens in the humans relies on complex interactions between innate and adaptive immunity. Most of the pathogens that enter the body are recognized initially by the innate immune system.[4] This defense mechanism is not antigen-specific and is instead focused on the recognition of a limited number of specific patterns that are shared by groups of pathogens (pathogen-associated molecular patterns – PAMPs) by pattern recognition receptors (PRRs) in the host. These PRRs can be soluble in human serum or cell-associated.[5,6]
The molecules that initiate the complement cascade are examples of soluble PRRs. The complement system is part of the innate immune system and consists of many proteins involved in a cascade of proteolysis and protein complex assembly that culminates in the elimination of invading pathogens.[6] Several components of the bacterial cell surface combine with PRRs such as Ficolin family of proteins, or Mannan binding lectins (MBL). These complexes, in turn combine with serine proteases and lead to complement activation via lectin pathway resulting in opsonophagocytosis of the invading pathogen, apoptosis, or modulation of inflammation.[7–10]
Toll-like receptors (TLRs) are pivotal cell-associated PRRs in the innate immune system. These receptors are capable of recognizing a wide spectrum of organisms, including viruses, bacteria and other parasites, and are classified into different groups based on their localization (cell surface or intracellular) and the type of PAMPs they recognize. TLR activation leads to the production of proinflammatory cytokines that enable macrophages and dendritic cells (DC) to eliminate invading pathogens. DCs can stimulate T cell expansion and differentiation, initiating an adaptive immune response.[4] The molecules produced during the innate immune response act as signals to activate adaptive immune responses. Antigen presenting cells (APCs), such as DCs, are activated and express costimulatory (CD80 and CD86) and MHC molecules on their cell surface that enable these cells to present processed antigens to T cells through the T cell receptor (TCR). Class I MHC molecules, such as HLA-A, -B and -C, present peptides derived from intracellular pathogens to CD8+ T cells, while class II MHC molecules, such as HLA-DR, -DQ and –DP, present peptides derived from extracellular pathogens to CD4+ T cells, which secrete a wide range of cytokines and have both effector and regulatory roles. Cytokines such as TNF-α and IFN-γ act at the site of infection and can affect pathogen survival and control the immune response. Activation of CD4+ T cells not only leads to the expansion of CD4+ effector cells, but also can promote the expansion and differentiation of antigen-specific CD8+ T cells and B cells.[4]
RF AND RHD - GENETIC SUSCEPTIBILITY
The molecules involved with both innate and adaptive immune responses described above are encoded by genes that are associated with RF/RHD [Figure 1] and will be discussed below.
Figure 1.
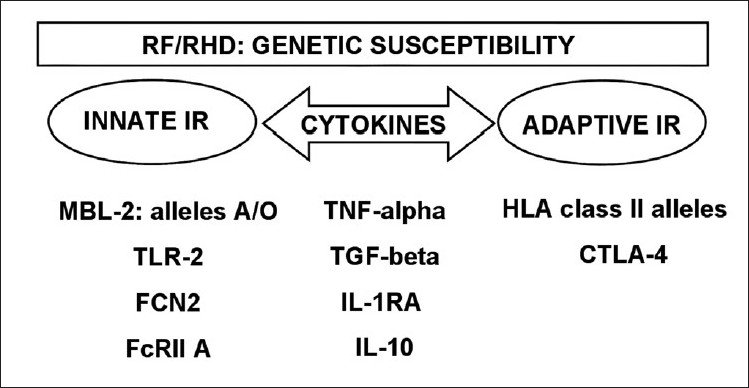
Genes involved with development of Rheumatic Fever and Rheumatic Heart Disease. Several genes controlling innate and/or adaptive immune responses are involved with the development of the disease
Toll like receptor - TLR2 is encoded by a gene located on chromosome 4 in the 4q32 region. A single nucleotide polymorphism (SNP) in exon 3 (2258 G>A) leads to the replacement of arginine with glutamine in codon 753. The genotype 753 Arg/Gln was present more frequently in a Turkish ARF cohort compared with controls.[11] A recent study reported that human cardiac myosin (HCM) binds to TLR2 and TLR8, thus activating human monocytes to release proinflammatory cytokines. These data suggest that pathogenic T cell epitopes from human cardiac myosin may link innate and adaptive responses to promote chronic inflammation in the myocardium.[12]
Polymorphisms in the ficolin genes may yield different serum ficolin protein level.[13] These differences may be important for the pathogenesis of ARF, by causing a prolonged or repeated infections. Polymorphisms in the promoter of the FCN2 gene for L ficolin , a protein that binds to Streptococcus pyogenes, may be important. Haplotype GGA was found more frequently in chronic RHD patients compared to the controls in a study from Brazil.[14]
Similarly, MBL is a member of the lectin pathway of the complement system, as mentioned previously, and plays an important role in innate immune responses by promoting clearance of the bacteria. SNPs have been identified in the promoter region of the MBL2 gene (-550 H/L, -221X/Y and +4P/Q) and in exon 1 (codons 52A/D, 54A/B and 57A/D) and have been analyzed in patients with RF/RHD.
Interestingly, different alleles were associated with different clinical pictures of the disease. RHD patients that developed mitral stenosis (MS) showed an association with the A allele (52A, 54A and 57A) that codes for high production of MBL, and this association correlated with high serum levels of MBL protein in most of the patients studied. Genotypes associated with high production of the protein (YA/YA and YA/XA) were more frequent in patients with acute and chronic carditis, when compared to the controls.[15] In contrast, the majority of RHD patients that developed aortic regurgitation (AR) showed an association with the O allele (52D, 54B and 57D). The O allele codes for low production of MBL, and most of the patients studied had low serum levels of MBL.[16] These results suggest that the MBL2 gene could play a role in the development of valvular lesions.
A receptor for the Fc fragments of immunoglobulin G (FcgRIIA) protects the host against foreign antigens by removing antigen-antibody complexes from circulation.[17] The gene for this receptor which is expressed on mononuclear phagocytes, neutrophils, and platelets, may exhibit single nucleotide polymorphism in the exon 4. These alleles are expressed in a codominant manner and differ in their ability to bind to human IgG2.[18] In Caucasian Turkish children, the 131R/R genotype was associated with a high risk of developing RF and 131H/R with an intermediate risk. Preliminary studies showed that polymorphonuclear leukocytes from subjects homozygous for 131R/R also exhibited reduced uptake of opsonized group B streptococci and other bacteria.[19]
CYTOKINES
Cytokines seem to play a pivotal role in the activation of immunological and inflammatory responses in RF. It has been shown that peripheral blood mononuclear cells (PBMC) from children with RF produce more TNF-α than healthy controls.[20] Moreover, interleukin-6 (IL-6) and TNF-α are considered inducers of the acute phase of RF and are strongly correlated with C-reactive protein.[21,22]
TNF-α is a proinflammatory cytokine that has been associated with the severity of different autoimmune and inflammatory diseases. The gene that encodes this cytokine is located within the MHC region on chromosome 6p21.3. This region is highly polymorphic, and the TNF-alpha gene also contains a large number of polymorphisms.[23] Some of these were investigated in RF/RHD patients in different countries. An SNP in the promoter region of TNF-alpha (-308A) was associated with susceptibility to RHD in Mexico, Turkey, Brazil, and Egypt.[21–26] The TNF-alpha -238G allele was also associated with RHD in Mexican and Brazilian patients.[24,25] The TNF-alpha gene has a proinflammatory effect and is probably associated with the exacerbation of the inflammatory response in RF/RHD patients who present with high serum TNF-α levels[20,22,27,28] and large numbers of TNF-α-producing cells in the throat and valves.[29]
Polymorphisms in other cytokine genes have also been investigated and seem to be involved with the disease. These include TGF beta1,[30–31] interleukin-1 receptor antagonist (IL1Ra),[32] interleukin 10,[21] amongst others. In Taiwanese RHD patients, both the -509T and 869T alleles of TGF beta 1 were found to be risk factors for the development of valvular RHD lesions.[30] Similar results were found in Egyptian patients.[31] RHD patients from Egypt and Brazil with severe carditis showed low frequencies of allele 1 for IL1Ra, suggesting the absence of control of the inflammatory process.[21,32]
Interleukin-10 (IL-10) is one of the most important anti-inflammatory cytokines, together with TGF-β and IL-35. It is produced by activated immune cells, especially monocytes/macrophages and T cell subsets, including regulatory T cells (Tr1 and Treg) and Th1 cells. IL-10 acts through a transmembrane receptor complex, and regulates the functions of many different immune cells.[33] The gene encoding human IL-10 is located on chromosome 1. A large number of polymorphisms have been identified in the IL-10 gene promoter. The genotype IL-10 -1082G/A that is overrepresented in RHD Egyptian patients is associated with the development of multiple valvular lesions (MVL) and with the severity of RHD.[21] More recently, polymorphism in cytotoxic T cell Lymphocyte antigen 4 (CTLA-4), which is a negative regulator of T cell proliferation has also been shown in Turkish RHD patients.[34]
HUMAN LEUCOCYTE ANTIGEN
Human leucocyte antigen (HLA) molecules are encoded by the HLA genes (-A, -B, -C, -DR, -DQ and -DP), which are located on the short arm of human chromosome 6. Early studies of susceptibility for RF/RHD pointed out the association of the disease with several HLA class II alleles, mainly those encoded by the DR and DQ genes.
Several HLA class II alleles were described in locations around the world [Figure 2].[35,36]
Figure 2.
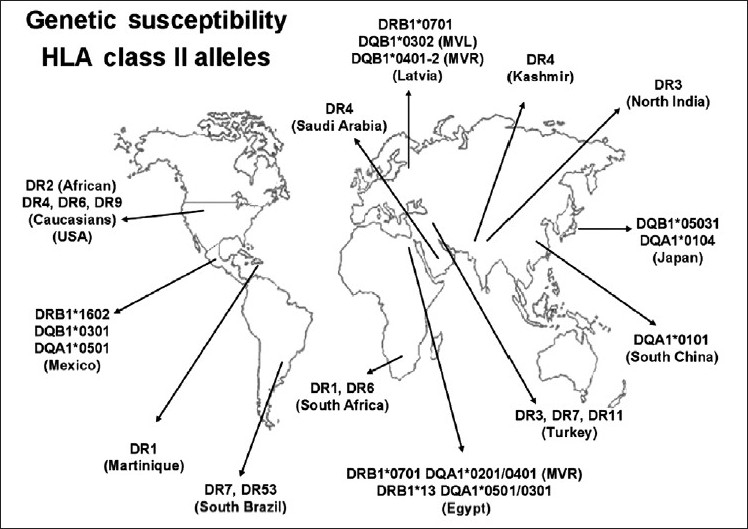
RF/RHD associated HLA class II alleles: distribution around the world Several identified alleles by serology in the 80's and/or molecular biology after the 90's were shown. Americas: (USA, Mexico, Martinique, South of Brazil); Asia: (Paquistan-Kashmir, North India, Latvia, Japan, South China), United Arab Emirates: (Saudi Arabia) Europe-Asia: (Turkey), Africa: (South Africa and Egypt)
The HLA-DR7 allele that was found in Brazilian, Turkish, Egyptian, and Latvian patients could be considered the HLA class II gene that is most consistently associated with RF/RHD. HLA-DR4 and -DR7 are associated with HLA-DR53 [Figure 2]. In addition, the association of HLA-DR7 with some HLA-DQB or -DQA alleles may be related to the development of multiple valvular lesions (MVL) in RHD patients in Egypt and in Latvia.[37–39]
The molecular mechanism by which HLA class II molecules confer susceptibility to autoimmune diseases is not clear. As mentioned above, the role of HLA class II molecules is to present antigens to the TCR, leading to the recruitment of large numbers of CD4+ T cells that specifically recognize antigenic peptides from extracellular pathogens and the activation of adaptive immune responses. Therefore, the associated alleles probably encode molecules that facilitate the presentation of some streptococcal peptides to T cells that later trigger autoimmune reactions mediated by molecular mimicry.
In summary, several alleles of the HLA class II genes appear to be the dominant contributors to the development of RF and RHD. Polymorphisms (SNPs and variable number of tandem repeat sequences of nucleotides) in genes involved with inflammatory responses and host defenses against pathogens that are associated with disease probably contribute to the development of valvular lesions and can determine the type of rheumatic valvular lesions (stenosis, regurgitation, or both) that occur in RHD patients.
RHEUMATIC FEVER AND RHEUMATIC HEART DISEASE ARE MEDIATED BY MOLECULAR MIMICRY BETWEEN S.PYOGENES AND HUMAN PROTEINS
Molecular mimicry between components of B hemolyticus streptococci and human heart tissues is the central problem in the pathogenesis of ARF and RHD. The precise mechanisms are being investigated for many years, and some real progress in the understanding of the pathogenesis is occurring slowly.[40–43]
Both T and B lymphocytes can recognize pathogenic and self antigens via four different types of molecular mimicry: (1) identical amino acid sequences, (2) homologous but non-identical sequences, (3) common or similar amino acid sequences of different molecules (proteins, carbohydrates) and (4) structural similarities between the microbe or environmental agent and its host.[43,44] Autoimmune diseases from molecular mimicry may be facilitated because of the phenomena of epitope spreading and TCR degeneracy. Epitope spreading is the mechanism by which an ongoing immune response leads to reactivity against epitopes that are distinct from the original disease-inducing epitope[43,45] and degeneracy of TCRs, which allows the recognition of a broad spectrum of antigens (self and microbial antigens) by the same T lymphocyte through it`s receptor.[41,42,46]
The M protein is the most important antigenic structure of the S. pyogenes and shares structural homology with γ-helical coiled-coil human proteins such as cardiac myosin, tropomyosin, keratin, laminin, vimentin and several valvular proteins.[40–42,47]
Several studies done in the last 50 years described the presence of cross-reactivity between human proteins and streptococcal antigens recognized by antibodies.[40] Among these human proteins, cardiac myosin and vimentin seem to be the major targets of cross-reactive reactions, along with other intracellular valvular proteins. N-acetyl ß-D-glucosamine, a polysaccharide present in streptococcal cell wall, induces cross-reactivity against laminin, an extracellular matrix alpha helical coiled-coil protein present in the valves.[40] By using affinity purified anti-myosin antibodies, Cunningham΄s group identified a five amino acid (Gln-Lys-Ser-Lys-Gln) epitope of the N-terminal M proteins of serotypes 5 and 6 (M5, M6) as being cross-reactive with cardiac myosin.[40]
The interplay of humoral and cellular immune responses in RHD was recently demonstrated by Cunningham's group through two elegant studies. They showed that, in rheumatic carditis, antibodies that cross-react with streptococcal and human proteins bind to the endothelial surface and upregulate the adhesion molecule VCAM-1[48], leading to inflammation, cellular infiltration and valve scarring.[49] These data suggest that ARF may result from initial antibody mediated damage that later may be perpetuated by cell mediated inflammation.[50]
Although antibodies in the sera of RF/RHD patients cross-react with several human proteins, we demonstrated that rheumatic heart disease lesions are mediated mainly by inflammatory cells and CD4+ T lymphocytes.[51]
Studies performed in the last 25 years showed that CD4+ T cells are the major effectors of autoimmune reactions in the heart tissue in RHD patients.[51–53] The in vitro reactivity of peripheral T cells from RHD patients was evaluated in an early study that showed that these cells were able to recognize a 50- to 54-kDa myocardial protein fraction indicating autoreactivity to heart antigens, which was probably caused by streptococcal infection.[54] The role of T cells in the pathogenesis of RF and RHD was demonstrated through the analysis of heart-tissue infiltrating T cell clones. We demonstrated for the first time that M5 protein peptides (residues 81-96 and 83-103) displayed cross-reactivity with valvular proteins by molecular mimicry.[51] We also showed that valve-infiltrating T cells recognized cardiac myosin peptides by molecular mimicry and epitope spreading mechanisms.[55] These immunodominant M5 epitopes were preferentially recognized by peripheral T lymphocytes from RHD patients, when compared with normal individuals, mainly in the context of HLA-DR7.[56] These results suggested that autoreactive T cells migrate from the periphery to the site of heart lesions. Similarly, Yoshinaga et al.[57] reported that T cell lines derived from heart valve specimens and PBMC from RF and RHD patients react with cell wall and membrane streptococcal antigens. These lymphocytes, however, did not cross-react with the M protein or mammalian cytoskeletal proteins.[57]
Recently, two studies demonstrated mimicry between cardiac myosin and the streptococcal M protein and pointed out different patterns of T cell antigen cross-recognition. One of them focused on peripheral T cell clones from one patient with RHD, which recognized different alpha helical coiled-coil proteins, such as the streptococcal M protein, myosin, laminin and tropomyosin.[58] The other study focused on the reactivity of intralesional T cell clones derived from myocardium and valvular tissue of six RHD patients against cardiac myosin, the streptococcal M5 protein and valve-derived proteins. A high frequency of reactive T cell clones was found (63%). These T cells displayed three patterns of cross-reactivity: (1) cardiac myosin and valve-derived proteins; (2) cardiac myosin and streptococcal M5 peptides; and (3) cardiac myosin, streptococcal M5 peptides and valve-derived proteins.[55]
Using a proteomics approach, we showed that T cells recognize vimentin, further supporting the role of this protein as a putative autoantigen involved in rheumatic lesions. In addition, we identified myocardial and valvular autoantigens that were recognized by heart-infiltrating and peripheral T cells from RF/RHD patients. Novel heart tissue proteins were identified, including disulfide isomerase ER-60 precursor (PDIA3) protein and a 78-kDa glucose-regulated protein precursor (HSPA5).[59] However, their role in RHD pathogenesis and other autoimmune diseases is not clear.
As mentioned above, both epitope spreading and the degeneracy of T cell receptors contributed to the amplification of cross-reactivity that leads to tissue damage.
By using a molecular approach, we evaluated Vb chain usage by TCRs and the degree of clonality of heart-tissue infiltrating T cells. In RHD, the autoreactive T lymphocytes that infiltrate both the myocardium and the valves were identified in oligoclonal expansions by analyzing their TCRs. A high number of T cell oligoclonal expansions were found in the valvular tissue, indicating that specific and cross-reactive T cells migrate to the valves.[49]
An effective immune response depends on cytokine production. CD4+ T helper cells are crucial regulators of the adaptive immune response. Antigen-activated CD4+ T cells become polarized toward a Th1 or Th2 phenotype based on the cytokines they secrete. Th1 cells are involved with cellular immune response and produce IL-2, IFN-γ and TNF-α. Th2 cells mediate humoral and allergic immune responses and produce IL-4, IL-5 and IL-13. A new lineage of CD4+ T cells (Th17) was recently described and is characterized by the production of IL-17. In vitro studies indicated a proinflammatory function for IL-17, and its expression was found to be associated with some inflammatory and autoimmune diseases.[60,61]
The role of cytokines in RF/RHD was first evaluated by examining the sera of patients and peripheral mononuclear cells stimulated by streptococcal antigens. These samples showed increased amounts of proinflammatory cytokines (IL-1, IL-6, TNF-γ and IFN-α).[62] Immunohistochemistry on heart tissue (myocardium and valves) from acute and chronic RHD patients, showed a large number of mononuclear cells able to secrete TNF-γ, IFN-α and the regulatory cytokine IL-10. Importantly, while a significant number of IL-4+ cells were found in the myocardium, these cells were very scarce in valve lesions in RHD patients. It is important to remember that valve damage and not myocarditis is the main problem in ARF. These observations indicated a role for balanced Th1/Th2 cytokines in healing myocarditis and in the induction of progressive and permanent valve damage.[29] IL-17 and Il-23 (Th17 cytokines) were recently analyzed by immunohistochemistry in both myocardium and valvular tissue from RHD patients. We observed a large number of IL-17+ and IL-23+ heart-tissue infiltrating cells (unpublished results), showing that these cytokines also play an important role in the development of heart lesions.
ANIMAL MODELS OF RF/RHD
Humans are unique hosts for S. pyogenes infections.Several studies (in mice, rats, hamsters, rabbits and primates) have been done to find a suitable animal model in which to examine the autoimmune process leading to RF/RHD with little success.[63] In the last decade, a model in Lewis rats has been developed that appears to be useful for the study of RF/RHD. These rats have already been used to induce experimental autoimmune myocarditis and study the pathogenesis of RF/RHD.[64–66]
Immunization of Lewis rats with recombinant M6 protein induced focal myocarditis, myocyte necrosis and valvular heart lesions in three out of six animals. The disease in these animals included verruca-like nodules and the presence of Anitschkow cells, which are large macrophages (also known as caterpillar cells), in mitral valves. Lymph node cells from these animals showed a proliferative response against cardiac myosin, but not skeletal myosin or actin. A CD4+ T cell line responsive to both the M protein and cardiac myosin was also obtained. Taken together, these results confirm the cross-reactivity between the M protein and cardiac myosin triggered by molecular mimicry, as observed in humans, possibly causing a break in tolerance leading to autoimmunity.[67,68]
Similarly, immunizing the Lewis rats with synthetic peptides from the conserved regions of M5 protein, or from B and C regions , or a recombinant M5 proteins have yielded focal myocarditis, infiltration with CD4+T cells, CD68+ macrophage but no typical aschoff`s nodule.[69–72]
Thus the Lewis Rat model could be considered a model of autoimmune valvulitis akin to ARF.
PROSPECTIVE VACCINES AGAINST S. PYOGENES
Many studies have focused on developing a vaccine against S. pyogenes in order to prevent infection and its complications. There are four anti-group A streptococci (GAS) vaccine candidates based on the M protein and eight more candidates based on other streptococci antigens, including group A CHO, C5a peptidase (SCPA), cysteine protease (Spe B), binding proteins similar to fibronectin, opacity factor, lipoproteins, Spes (super antigens) and streptococcal pili.[73]
A multivalent vaccine, currently under phase II clinical trials, combines the amino acid sequences of the N-terminal portion of the M protein from the 26 most common strains of GAS in the US as a recombinant protein.[74–76]
Because the C-terminal portion of the M protein is conserved among the 200 strains identified by their emm-types, vaccines based on this region are expected to provide broad coverage. The first attempt to develop a vaccine based on the C-terminal portion of the M protein was performed by Fischetti et al.[77] This vaccine was able to induce protection against S. pyogenes containing homologous (M6) and heterologous (M14) M protein, demonstrating that the use of conserved region-derived peptides could induce protection against different serotypes.[78]
Conserved epitopes from the M protein have been also studied by a group from Australia, where the incidence of streptococcal infections in aboriginal communities is very high. Two synthetic peptides from the M5 protein (J8 and J14) were selected, and several formulations presented promising results.[79–82] A combination of J8 and the fibronectin-binding repeats region (FNBR) of fibronectin I (SfbI) provided enhanced protection against S. pyogenes in mice.[83]
We developed a vaccine epitope (StreptInCor) composed of 55 amino acid residues of the C-terminal portion of the M protein that encompasses both T and B cell protective epitopes.[84]
The structural, chemical and biological properties of this peptide were evaluated and have shown that StreptInCor is a very stable molecule, an important property for a vaccine candidate.[85] Furthermore, experiments with mice showed that this construct is immunogenic and safe.[86]
The greatest challenge for the development of a GAS vaccine resides in the promotion of immunity without generating cross-reactivity with human tissue. An effective and safe vaccine is still needed, most of all in developing countries.
CONCLUSION
Several genes involved in the control of infection and the immune response play a role in the development of RF and RHD. Some genes are associated with the innate immune response, and others with the adaptive immune response. Many of these genes are responsible for the inflammatory process and autoimmune reactions.
In rheumatic carditis, antibodies that cross-react with streptococcal and human proteins upregulate adhesion molecules, leading to inflammation and increased cellular infiltration. CD4+ T cells that cross-react with heart tissue and streptococcal antigens are the major effectors of heart lesions.
Large numbers of mononuclear cells that infiltrate rheumatic heart lesions produce inflammatory cytokines (TNF-γ and IFN-α) and the imbalance between these cells and the IL-4-producing Th2 cells in the valve tissue might contribute to the progression and maintenance of rheumatic valvular lesions. Th17 cells also play a role in the development of autoimmunity.
Several pathogenic M protein epitopes were identified that can induce cross-reactive responses against human proteins. Both epitope spreading and TCR degeneracy increase the possibility of cross-reactivity between infectious agents and self antigens.
An animal model of Lewis rat displays similar heart lesions to RHD and is considered a good model of the disease.
The development of a vaccine against S. pyogenes is an important goal and, given the number of ongoing studies, will probably be a reality in the near future.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal disease. Lancet Infect Dis. 2005;5:685–94. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Mota CC, Aiello DV, Anderson RH. Chronic rheumatic heart disease. In: Anderson RH, Baker EJ, Penny DJ, editors. Pediatric Cardiology. 3rd ed. Philadelphia: Churchill Livingstone/Elsevier; 2009. pp. 1091–133. [Google Scholar]
- 3.Bland EF, Duckett Jones T. Rheumatic fever and rheumatic heart disease: A 20 years report on 1000 patients followed since childhood. Circulation. 1951;4:836–43. doi: 10.1161/01.cir.4.6.836. [DOI] [PubMed] [Google Scholar]
- 4.Moser M, Leo O. Key concepts in immunology. Vaccine. 2010;28:C2–13. doi: 10.1016/j.vaccine.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 7.Matsushita M. Ficolins: Complement-activating lectins involved in innate immunity. J Innate Immun. 2009;2:24–32. doi: 10.1159/000228160. [DOI] [PubMed] [Google Scholar]
- 8.Sorensen R, Thiel S, Jensenius JC. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immunopathol. 2005;27:299–319. doi: 10.1007/s00281-005-0006-z. [DOI] [PubMed] [Google Scholar]
- 9.Runza VL, Schwaeble W, Männel DN. Ficolins: Novel pattern recognition molecules of the innate immune response. Immunobiology. 2008;213:297–306. doi: 10.1016/j.imbio.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, et al. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J Immunol. 2004;172:1198–202. doi: 10.4049/jimmunol.172.2.1198. [DOI] [PubMed] [Google Scholar]
- 11.Berdeli A, Celik HA, Ozyürek R, Dogrusoz B, Aydin HH. TLR-2 gene Arg753Gln polymorphism is strongly associated with acute rheumatic fever in children. J Mol Med. 2005;83:535–41. doi: 10.1007/s00109-005-0677-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting edge: Cardiac myosin activates innate immune responses through TLRs. J Immunol. 2009;183:27–31. doi: 10.4049/jimmunol.0800861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Messias-Reason I, Kremsner PG, Kun JF. Functional haplotypes that produce normal ficolin-2 levels protect against clinical leprosy. J Infect Dis. 2009;199:801–4. doi: 10.1086/597070. [DOI] [PubMed] [Google Scholar]
- 14.Messias-Reason IJ, Schafranski MD, Kremsner PG, Kun JF. Ficolin 2 (FCN2) functional polymorphisms and the risk of rheumatic fever and rheumatic heart disease. Clin Exp Immunol. 2009;157:395–9. doi: 10.1111/j.1365-2249.2009.03975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafranski MD, Pereira FL, Scherner D, Torres R, Jensenius JC, de Messias-Reason IJ. High-producing MBL2 genotypes increase the risk of acute and chronic carditis in patients with history of rheumatic fever. Mol Immunol. 2008;45:3827–31. doi: 10.1016/j.molimm.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Ramasawmy R, Spina GS, Faé KC, Pereira AC, Nisihara R, Messias RI, et al. Association of mannose-binding lectin gene polymorphism but not of mannose-binding serine protease 2 with chronic severe aortic regurgitation of rheumatic etiology. Clin Vaccine Immunol. 2008;15:932–6. doi: 10.1128/CVI.00324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van de Winkel JG, Anderson CL. Biology of human immunoglobulin G Fc receptors. J Leukoc Biol. 1991;49:511–24. doi: 10.1002/jlb.49.5.511. [DOI] [PubMed] [Google Scholar]
- 18.Yee AM, Phan HM, Zuniga R, Salmon JE, Musher DM. Association between FcgammaRIIa-R131 allotype and bacteremic pneumococcal pneumonia. Clin Infect Dis. 2000;30:25–8. doi: 10.1086/313588. [DOI] [PubMed] [Google Scholar]
- 19.Berdeli A, Celik HA, Ozyürek R, Aydin HH. Involvement of immunoglobulin FcgammaRIIA and FcgammaRIIIB gene polymorphisms in susceptibility to rheumatic fever. Clin Biochem. 2004;37:925–9. doi: 10.1016/j.clinbiochem.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Miller LC, Gray ED, Mansour M, Abdin ZH, Kamel R, Zaher S, et al. Cytokines and immunoglobulin in rheumatic heart disease: Production by blood and tonsillar mononuclear cells. J Rheumatol. 1989;16:1436–42. [PubMed] [Google Scholar]
- 21.Settin A, Abdel-Hady H, El-Baz R, Saber I. Gene polymorphisms of TNF-alpha(-308), IL-10(-1082), IL-6(-174), and IL-1Ra(VNTR) related to susceptibility and severity of rheumatic heart disease. Pediatr Cardiol. 2007;28:363–71. doi: 10.1007/s00246-006-0002-7. [DOI] [PubMed] [Google Scholar]
- 22.Yeðin O, Coþkun M, Ertuð H. Cytokines in acute rheumatic fever. Eur J Pediatr. 1997;156:25–9. doi: 10.1007/s004310050545. [DOI] [PubMed] [Google Scholar]
- 23.Hajeer AH, Hutchinson IV. TNF-alpha gene polymorphism: Clinical and biological implications. Microsc Res Tech. 2000;50:216–28. doi: 10.1002/1097-0029(20000801)50:3<216::AID-JEMT5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 24.Hernández-Pacheco G, Flores-Domínguez C, Rodríguez-Pérez JM, Pérez-Hernández N, Fragoso JM, Saul A, et al. Tumor necrosis factor-alpha promoter polymorphisms in Mexican patients with rheumatic heart disease. J Autoimmun. 2003;21:59–63. doi: 10.1016/s0896-8411(03)00079-9. [DOI] [PubMed] [Google Scholar]
- 25.Ramasawmy R, Faé KC, Spina G, Victora GD, Tanaka AC, Palácios SA, et al. Association of polymorphisms within the promoter region of the tumor necrosis factor-alpha with clinical outcomes of rheumatic fever. Mol Immunol. 2007;44:1873–8. doi: 10.1016/j.molimm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Sallakci N, Akcurin G, Köksoy S, Kardelen F, Uguz A, Coskun M, et al. TNF-alpha G-308A polymorphism is associated with rheumatic fever and correlates with increased TNF-alpha production. J Autoimmun. 2005;25:150–4. doi: 10.1016/j.jaut.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Morris K, Mohan C, Wahi PL, Anand IS, Ganguly NK. Enhancement of IL-1, IL-2 production and IL-2 receptor generation in patients with acute rheumatic fever and active rheumatic heart disease; a prospective study. Clin Exp Immunol. 1993;91:429–36. doi: 10.1111/j.1365-2249.1993.tb05920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narin N, Kütükçüler N, Ozyürek R, Bakiler AR, Parlar A, Arcasoy M. Lymphocyte subsets and plasma IL-1 alpha, IL-2, and TNF-alpha concentrations in acute rheumatic fever and chronic rheumatic heart disease. Clin Immunol Immunopathol. 1995;77:172–6. doi: 10.1006/clin.1995.1140. [DOI] [PubMed] [Google Scholar]
- 29.Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez AP, et al. Rheumatic heart disease: Proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol. 2004;165:1583–91. doi: 10.1016/S0002-9440(10)63415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou HT, Chen CH, Tsai CH, Tsai FJ. Association between transforming growth factor-beta1 gene C-509T and T869C polymorphisms and rheumatic heart disease. Am Heart J. 2004;148:181–6. doi: 10.1016/j.ahj.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 31.Kamal H, Hussein G, Hassoba H, Mosaad N, Gad A, Ismail M. Transforming growth factor-beta1 gene C-509T and T869C polymorphisms as possible risk factors in rheumatic heart disease in Egypt. Acta Cardiol. 2010;65:177–83. doi: 10.2143/AC.65.2.2047051. [DOI] [PubMed] [Google Scholar]
- 32.Azevedo PM, Bauer R, Caparbo Vde F, Silva CA, Bonfá E, Pereira RM. Interleukin-1 receptor antagonist gene (IL1RN) polymorphism possibly associated to severity of rheumatic carditis in a Brazilian cohort. Cytokine. 2010;49:109–13. doi: 10.1016/j.cyto.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–44. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Düzgün N, Duman T, Haydardedeoðlu FE, Tutkak H. Cytotoxic T lymphocyte-associated antigen-4 polymorphism in patients with rheumatic heart disease. Tissue Antigens. 2009;74:539–42. doi: 10.1111/j.1399-0039.2009.01347.x. [DOI] [PubMed] [Google Scholar]
- 35.Guilherme L, Ramasawmy R, Kalil J. Rheumatic fever and rheumatic heart disease: Genetics and pathogenesis. Scand J Immunol. 2007;66:199–207. doi: 10.1111/j.1365-3083.2007.01974.x. [DOI] [PubMed] [Google Scholar]
- 36.Bryant PA, Robins-Browne R, Carapetis JR, Curtis N. Some of the people, some of the time: susceptibility to acute rheumatic fever. Circulation. 2009;119:742–53. doi: 10.1161/CIRCULATIONAHA.108.792135. [DOI] [PubMed] [Google Scholar]
- 37.Guedez Y, Kotby A, El-Demellawy M, Galal A, Thomson G, Zaher S, et al. HLA class II associations with rheumatic heart disease are more evident and consistent among clinically homogeneous patients. Circulation. 1999;99:2784–90. doi: 10.1161/01.cir.99.21.2784. [DOI] [PubMed] [Google Scholar]
- 38.Stanevicha V, Eglite J, Sochnevs A, Gardovska D, Zavadska D, Shantere R. HLA class II associations with rheumatic heart disease among clinically homogeneous patients in children in Latvia. Arthritis Res Ther. 2003;5:R340–6. doi: 10.1186/ar1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visentainer JE, Pereira FC, Dalalio MM, Tsuneto LT, Donadio PR, Moliterno RA. Association of HLA-DR7 with rheumatic fever in the Brazilian population. J Rheumatol. 2000;27:1518–20. [PubMed] [Google Scholar]
- 40.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guilherme L, Kalil J. Role of autoimmunity in rheumatic fever. Fut Rheumatol. 2008;3:161–7. [Google Scholar]
- 42.Guilherme L, Kalil J. Rheumatic fever and rheumatic heart disease: Cellular mechanisms leading autoimmune reactivity and disease. J Clin Immunol. 2010;30:17–23. doi: 10.1007/s10875-009-9332-6. [DOI] [PubMed] [Google Scholar]
- 43.Guilherme L, Köhler KF, Kalil L. Makoswki GS. Advances in clinical chemistry. 53th ed. Springfield: Elsevier; 2011. Rheumatic Heart Disease: Mediation by complex immune events; pp. 31–50. [PubMed] [Google Scholar]
- 44.Peterson LK, Fujinami RS. Molecular mimicry. In: Shoenfeld Y, Gershwin ME, Meroni PL, editors. Autoantibodies. Burlington: Elsevier; 2007. pp. 13–9. [Google Scholar]
- 45.Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol. 1993;11:729–66. doi: 10.1146/annurev.iy.11.040193.003501. [DOI] [PubMed] [Google Scholar]
- 46.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;359:155–7. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 47.Fischetti VA. Streptococcal M protein. Sci Am. 1991;264:58–65. doi: 10.1038/scientificamerican0691-58. [DOI] [PubMed] [Google Scholar]
- 48.Guilerherme L, Kali J, Cunningham M. Molecular mimicry in autoimmune pathogenesis of rheumatic heart disease. Autoimmunity. 2006;39:31–9. doi: 10.1080/08916930500484674. [DOI] [PubMed] [Google Scholar]
- 49.Guilherme L, Dulphy N, Douay C, Coelho V, Cunha-Neto E, Oshiro SE, et al. Molecular evidence for antigen-driven immune responses in cardiac lesions of rheumatic heart disease patients. Int Immunol. 2000;12:1063–74. doi: 10.1093/intimm/12.7.1063. [DOI] [PubMed] [Google Scholar]
- 50.Cunnigham MW. Autoimmunity and molecular mimicry in the pathogenesis of post streptococcal heart disease. Front Biosci. 2003;8:s533–43. doi: 10.2741/1067. [DOI] [PubMed] [Google Scholar]
- 51.Guilherme L, Weidebach W, Kiss MH, Snitcowsky R, Kalil J. Association of human leukocyte class II antigens with rheumatic fever or rheumatic heart disease in a Brazilian population. Circulation. 1991;83:1995–8. doi: 10.1161/01.cir.83.6.1995. [DOI] [PubMed] [Google Scholar]
- 52.Raizada V, Williams RC, Jr, Chopra P, Gopinath N, Prakash K, Sharma KB, et al. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cells antibodies. Am J Med. 1983;74:225–37. doi: 10.1016/0002-9343(83)91124-5. [DOI] [PubMed] [Google Scholar]
- 53.Kemeny E, Grieve T, Marcus R, Sareli P, Zabriskie JB. Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clin Immunol Immunopathol. 1989;52:225–37. doi: 10.1016/0090-1229(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 54.El-Demellawy M, El-Ridi R, Guirguis NI, Abdel Alim M, Kotby A, Kotb M. Preferential recognition of myocardial antigens by T lymphocytes from rheumatic heart disease patients. Infect Immun. 1997;65:2197–205. doi: 10.1128/iai.65.6.2197-2205.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faé KC, da Silva DD, Oshiro SE, Tanaka AC, Pomerantzeff PM, Douay C, et al. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol. 2006;176:5662–70. doi: 10.4049/jimmunol.176.9.5662. [DOI] [PubMed] [Google Scholar]
- 56.Guilherme L, Oshiro SE, Faé KC, Cunha-Neto E, Renesto G, Goldberg AC, et al. T cell reactivty against streptococcal antigens in the periphery mirrors reactivity of heart infiltrating T lymphocytes in rheumatic heart disease patients. Infect Immun. 2001;69:5345–51. doi: 10.1128/IAI.69.9.5345-5351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshinaga M, Figueiroa F, Wahid MR, Marcus RH, Suh E, Zabriskie JB. Antigenic specificity of lymphocytes isolated from valvular specimens of rheumatic fever patients. J Autoimmun. 1995;8:601–13. doi: 10.1016/0896-8411(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 58.Faé KC, Kalil J, Toubert A, Guilherme L. Heart infiltrating T cell clones from a rheumatic heart disease patient display a common TCR usage and a degenerate antigen recognition pattern. Mol Immunol. 2004;40:1129–35. doi: 10.1016/j.molimm.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 59.Faé KC, Diefenbach da Silva D, Bilate AM, Tanaka AC, Pomerantzeff PM, Kiss MH, et al. PDIA 3, HS PA5, and Vimentin, proteins identified by 2DE in the valvular tissue, are target antigens of peripheral and heart infiltrating T cells from chronic rheumatic heart disease patients. J Autoimmun. 2008;31:136–41. doi: 10.1016/j.jaut.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 60.Stockinger B, Veldhoen M, Martin B. Th17 T cells: Linking innate and adaptive immunity. Semin Immunol. 2007;19:353–61. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 61.Dong C. Diversification of T-helper-cell lineages: Finding the family root of il-17 producing cells. Nat Rev Immunol. 2006;6:329–33. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 62.Guilherme L, Faé K, Oshiro SE, Kalil J. Molecular pathogenesis of rheumatic fever and rheumatic heart disease. Expert Rev Mol Med. 2005;7:1–15. doi: 10.1017/S146239940501015X. [DOI] [PubMed] [Google Scholar]
- 63.Unny SK, Middlebrooks BL. Streptococcal rheumatic carditis. Microbiol Rev. 1983;47:97–120. doi: 10.1128/mr.47.1.97-120.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kodama M, Matsumoto Y, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250–62. doi: 10.1016/0090-1229(90)90039-s. [DOI] [PubMed] [Google Scholar]
- 65.Wegmann KW, Zhao W, Griffin AC, Hickey WF. Identification of myocarditogenic peptides derived from cardiac myosin capable of inducing-experimental allergic myocarditis in the Lewis rat. J Immunol. 1994;153:892–900. [PubMed] [Google Scholar]
- 66.Li Y, Heuser JS, Kosanke SD, Hemric M, Cunningham MW. Cryptic epitope identified in rat and human cardiac myosin S2 region induces myocarditis in the Lewis rat. J Immunol. 2004;172:3225–34. doi: 10.4049/jimmunol.172.5.3225. [DOI] [PubMed] [Google Scholar]
- 67.Cunningham MW. T cell mimicry in inflammatory heart disease. Mol Immunol. 2004;40:1121–7. doi: 10.1016/j.molimm.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 68.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal M protein. Infect Immun. 2001;69:4072–8. doi: 10.1128/IAI.69.6.4072-4078.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lymbury RS, Olive CO, Powell KA, Good MF, Hirst RG, Labrooy JT, et al. Induction of autoimmune valvulitis in lewis rats following immunization with peptides from the conserved region of group A streptococcal M protein. J Autoimmun. 2003;20:211–7. doi: 10.1016/s0896-8411(03)00026-x. [DOI] [PubMed] [Google Scholar]
- 70.Gorton D, Blyth S, Gorton JG, Govan B, Ketheesan N. An alternative technique for the induction of autoimmune valvulitis in a rat model of rheumatic heart disease. J Immunol Methods. 2010;355:80–5. doi: 10.1016/j.jim.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 71.Gorton D, Govan B, Olive C, Ketheesan N. B- and T-cell responses in group A Streptococcus M-protein- or peptide-induced experimental carditis. Infect Immun. 2009;77:2177–83. doi: 10.1128/IAI.01514-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cunningham MW, Antone SM, Smart M, Liu R, Kosanke S. Molecular analysis of human cardiac myosin-cross-reactive B- and T-cell epitopes of the group A streptococcal M5 protein. Infect Immun. 1997;65:3913–23. doi: 10.1128/iai.65.9.3913-3923.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steer AC, Batzloff MR, Mulholland, Carapetis JR. Group A streptococcal vaccines: facts versus fantasy. Curr Opin Infect Dis. 2009;22:544–52. doi: 10.1097/QCO.0b013e328332bbfe. [DOI] [PubMed] [Google Scholar]
- 74.Hu MC, Walls MA, Stroop SD, Reddish MA, Beall B, Dale JB. Immunogenicity of a 26-valent group A streptococcal vaccine. Infect Immun. 2002;70:2171–7. doi: 10.1128/IAI.70.4.2171-2177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McNeil SA, Halperin SA, Langley JM, Smith B, Warren A, Sharrat GP, et al. Safety and immunogenicity of 26-valent group a streptococcus vaccine in healthy adult volunteers. Clin Infect Dis. 2005;41:1114–22. doi: 10.1086/444458. [DOI] [PubMed] [Google Scholar]
- 76.Dale JB, Penfound T, Chiang EY, Long V, Shulman ST, Beall B. Multivalent group A streptococcal vaccine elicits bactericidal antibodies against variant M subtypes. Clin Diagn Lab Immunol. 2005;12:833–6. doi: 10.1128/CDLI.12.7.833-836.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bessen D, Fischetti VA. Passive acquired mucosal immunity to group A streptococci by secretory immunoglobulin A. J Exp Med. 1988;167:1945–50. doi: 10.1084/jem.167.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bessen D, Fischetti VA. Synthetic peptide vaccine against mucosal colonization by group A streptococci.I. Protection against a heterologous M serotype with shared C repeated region epitopes. J Immunol. 1990;145:1251–6. [PubMed] [Google Scholar]
- 79.Batzloff MR, Yan H, Davies MR, Hartas J, Lowell GH, White G, et al. Toward the development of an antidisease, transmission-blocking intranasal vaccine for group A Streptococcus. J Infect Dis. 2005;192:1450–5. doi: 10.1086/466528. [DOI] [PubMed] [Google Scholar]
- 80.Olive C, Hsien K, Horváth A, Clair T, Yarwood P, Toth I, et al. Protection against group A streptococcal infection by vaccination with self-adjuvanting lipid core M protein peptides. Vaccine. 2005;23:2298–303. doi: 10.1016/j.vaccine.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 81.Vohra H, Dey N, Gupta S, Sharma AK, Kumar R, McMillan D, et al. M protein conserved region antibodies opsonise multiple strains of Streptococcus pyogenes with sequence variations in C-repeats. Res Microbiol. 2005;156:575–82. doi: 10.1016/j.resmic.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 82.Simerska P, Abdel-Aal AB, Fujita Y, Moyle PM, McGeary RP, Batzloff MR, et al. Development of a Liposaccharide-Based Delivery System and Its Application to the Design of Group A Streptococcal Vaccines. J Med Chem. 2008;51:1447–52. doi: 10.1021/jm701410p. [DOI] [PubMed] [Google Scholar]
- 83.Olive C, Schulze K, Sun HK, Ebensen T, Horváth A, Toth I, et al. Enhanced protection against Streptococcus pyogenes infection by intranasal vaccination with a dual antigen component M protein/SfbI lipid core peptide vaccine formulation. Vaccine. 2007;25:1789–97. doi: 10.1016/j.vaccine.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 84.Guilherme L, Faé KC, Higa F, Chaves L, Oshiro SE, Freschi de Barros S, et al. Towards a vaccine against rheumatic fever. Clin Dev Immunol. 2006;13:125–32. doi: 10.1080/17402520600877026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guilherme L, Alba MP, Ferreira FM, Oshiro SE, Higa F, Patarroyo ME, et al. Anti-group A streptococcal vaccine epitope: Structure,stability and its ability to interact with HLA class II molecules. J Biol Chem. 2011;286:6989–98. doi: 10.1074/jbc.M110.132118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guilherme L, Postol E, Freschi de Barros S, Higa F, Alencar R, Lastre M, et al. A vaccine against S.pyogenes: Design and experimental immune response. Methods. 2009;49:316–21. doi: 10.1016/j.ymeth.2009.03.024. [DOI] [PubMed] [Google Scholar]