

Abstract
Neurodegenerative diseases are characterized by progressive dysfunction and death of neurons in specific areas of the nervous system. Loss of neurons is often associated with multiple stresses such as abnormal aggregation of misfolded proteins, deficiency of protein degradation system, mitochondrial dysfunction and excessive oxidative products. HDAC6 has recently been suggested to be an integral factor that copes with these stresses. In this mini-review, we summarize our current understanding of how HDAC6 promotes inclusion formation, facilitates autophagic degradation of protein aggregates and dysfunctional mitochondria. Finally, the possibility for HDAC6 to be a potential preventional and therapeutical target of some neurodegenerative diseases is put forward.
Key words: HDAC6, inclusion, autophagy, mitochondrial dysfunction, neurodegeneration
Histone deacetylase 6 (HDAC6) was identified as a member of the class II HDAC enzymes based on its homology of the deacetylase domain to other histone deacytylases.1 It has aroused broad interest since its discovery because of its unique properties: (1) HDAC6 contains a duplication of the catalytic domain that belongs to the class I/II histone deacetylases, while other HDACs contain only one of such domain;2 (2) An additional zinc finger-containing domain, BUZ, which is able to bind polyubiquitin, resides in the C-terminal region of HDAC6;3,4 (3) HDAC6 is identified to be predominantly located in the cytoplasm,5 which makes its functions distinct from other HDACs and is consistent with the fact that the in vivo substrates of HDAC6 are a-tubulin, Hsp90 and cortactin.6–11 The unique feature of HDAC6 to possess a duplicated deacetylase domain and a BUZ domain provides a support for its broad functions in coping with misfolded cellular proteins. In this mini-review, we briefly summarize the state-of-art functions of HDAC6 and propose a model illustrating its dual role in coping with inclusions and dysfunctional mitochondria to avoid neurodegenerative disorders.
HDAC6 and Inclusion Formation
Aggregation and deposition of proteins (often abnormal proteins) are common features of many neurodegenerative diseases, such as Parkinson disease, Alzheimer disease and Huntington disease.12 Understanding the mechanism of how the proteinaceous deposition forms, known as inclusion formation, may be the key to prevent/treat these neurodegenerative disorders. In cultured cells, misfolded proteins tend to accumulate to form aggregates when they are not degraded by the ubiquitin-proteasome pathway. The undegraded proteins accumulate to form a pericentriolar inclusion-like structure called aggresome in a microtubule-dependent manner.13 Aggresomes and inclusions are not only morphologically similar but also containing many common components. Therefore, knowledge about aggresome formation in cultured cells should provide us clues to understand how inclusions form in humans.
Aggresomes, often enriched with ubiquitin, are mainly located at the microtubule-organizing center (MTOC) and their formation requires the microtubules. Kawaguchi and colleagues identified HDAC6 as a multivalent adapter that is capable of binding both polyubiquitinated proteins and dynein motors, and proposed that HDAC6 recruits misfolded protein cargos, which are polyubiquinated, to transport to the aggresomes along the microtubules.14 HDAC6 has also been found to interact with p97/VCP, another ubiquitin-binding protein that is associated with the ubiquitin proteosome system (UPS). The balance of HDAC6 and p97/VCP determines whether the ubiquitinated proteins are to be degraded or to form aggresomes,15 because p97/VCP promotes proteasome-mediated protein degradation whereas HDAC6 stimulates accumulation of ubiquitinated proteins to form aggresomes. However, aggresome formation is impaired in the p97/VCP mutant cells16 suggesting that interaction between HDAC6 and p97/VCP is required for proper aggresome formation. HDAC6 regulates not only aggresome formation under the conditions of proteasome insufficiency as summarized above, but also aggregation of those ubiquitinated proteins whose ubiquitinations are independent from the UPS pathway. K63-linked polyubiquitination was reported to play a proteasome-independent role in protein quality control in contrast to K48-linked polyubiquitination which functions to target substrate to the proteasome.17 HDAC6 is an important regulator to recruit K63-polyubiquitinated misfolded DJ-1 to form aggresomes.18 It is worthwhile to point out that, in addition to the ubiquitinated proteins, HDAC6 also binds to a ubiquitin-like modifier FAT10 to mediate FAT10-containing aggresome formation under the conditions of proteasome impairment.19
The findings that HDAC6 functions in the process of aggresome formation offer an important strategy to elucidate the mechanism(s) of inclusion formation. It has been hypothesized that inclusions are resulted from self-assembly of protein monomers into polymers under certain stress conditions. However, how aggregation-prone proteins form inclusions in vivo and what kinds of factors regulate this process are largely unknown. Du and colleagues have shown now that HDAC6 facilitates oligomeric α-synuclein to form inclusions through physical association with each other in Drosophila Parkinson disease model.20 Inclusion formation in the presence of HDAC6 sequesters toxic α-synuclein oligomers to protect dopaminergic neurons. HDAC6 selectively interacts with the oligomers, but not with purified monomeric α-synuclein. Since HDAC6 has been reported to interact with ubiquitinated proteins through its BUZ domain during aggresome formation,14 it would be very interesting to examine whether the oligomers of α-synuclein are polyubiquitinated in vivo. However, the possibility that HDAC6 may be directly or indirectly associated with a specific oligomeric conformation is not excluded. Inclusions are located not only perinuclearly as most aggresomes are, but also at the neuropil regions, which raises a question whether these HDAC6-dependent inclusion formation also requires the participation of microtubules.
HDAC6 and Inclusion Elimination
Recent studies indicate that cells have evolved a strategy to remove the non-functional proteins, including aggresomes and protein aggregates, through the degradation system such as autophagy.21,22 Again, mechanism(s) of aggresome elimination process should lend insights into how inclusions are eliminated. Autophagy is the primary cellular pathway responsible for degradation of long-lived proteins, macromolecular complexes and damaged organelles.23 A connection between autophagy and HDAC6 has been established through the observation that HDAC6 rescues polyglutamine-induced neurodegeneration in an autophagy-dependent manner.22 In agreement with this report, Iwata and colleagues reported that HDAC6 is required for the recruitment of autophagic components that accumulate around the aggresomes.21 Moreover, an intact microtubule cytoskeleton network is also required for recruiting autophagic machinery to the aggresomes. Therefore, two possibilities exist to explain the functions of HDAC6 in the process of the recruitment of autophagic components to the aggresomes. First, HDAC6 that is in the aggresomes may serve as a signal to recruit autophagic components. Second, HDAC6 that is outside of the aggresomes may function as a link between dynein and ubiquitin-like autophagic components such as Atg8 and Atg12. However, interactions between HDAC6 and Atg8 and/or Atg12 remains to be demonstrated, which is very likely because it has been shown that HDAC6 interacts with other ubiquitin-like proteins such as FAT10.19 It is worthwhile to note that HDAC6 may not be the only protein that regulates the autophagic recruitment. A recent study shows that recruitment of autophagosomes to protein aggregates is mediated by another ubiquitin-binding protein p62 that binds to an autophagosome protein LC3, a process that is not dependent on HDAC6.24 However, the fusion of such autophagosome to lysosomes for final degradation involves HDAC6.24 In summary, HDAC6 likely functions not only in the process of inclusion formation, but also inclusion elimination (Fig. 1). Inclusion formation assisted by HDAC6 is possibly an immediate response of the cells to the misfolded protein stress before a final autophagic degradation can permanently eliminate the stress, in which HDAC6 continues to act as a key regulator.
Figure 1.
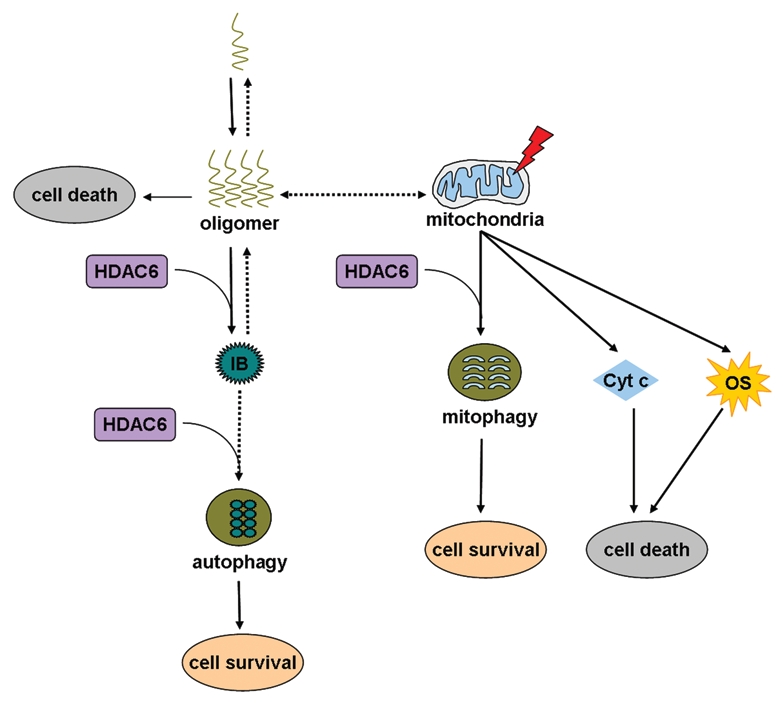
A model of how HDAC6 functions in different pathways to cope with different cellular stresses. Oligomers derived from self-assembly of aggregation-prone proteins are a major cause of several neurodegenerative diseases. An immediate strategy for HDAC6 to cope with the toxic oligomers is to bind to and promote them to form inclusions. The ultimate strategy for HDAC6 to eliminate these nonfunctional protein aggregates is to facilitate autophagic degradation of these aggregated inclusions. Another strategy for HDAC6, when the neurons are challenged by mitochondrial dysfunction, is to promote mitophagy. Mitochondrial dysfunction may lead to oxidative stress and cytochrome c release, which are toxic to the cells. IB, inclusion body; Cyt c, cytochrome c; OS, oxidative stress.
HDAC6 and Mitochondria-Associated Neurodegeneration
Abnormal protein aggregates, especially soluble oligomers that contain a common disease-associated conformational epitope, are important causative factors in neurodegenerative pathogenesis.20 HDAC6 has been shown to be involved in reducing the toxicity of oligomers by promoting inclusion formation (as discussed above). Another decisive cause of many neurodegenerative diseases is mitochondrial dysfunction. Mutation(s) of parkin leads to autosomal recessive juvenile parkinsonism (AR-JP) that is an early-onset Parkinson disease.25 In Drosophila, loss of parkin results in mitochondrial impairment-induced neurodegeneration and energy failure-derived spermatid individualization defect.26 A number of studies have shown that damaged mitochondria and accumulated oxidative stress promote cell death and neurodegeneration.27 HDAC6 has previously been shown to directly interact with Parkin.28 Recently Lee and colleagues report that Parkin-directed clearance of damaged mitochondria through autophagy, i.e., mitophagy, is HDAC6 dependent.29 Parkin mutant cells, associated with familial AR-JP, are defective in mitophagy,29 suggesting that mitophagy is the key to suppress neurodegeneration. Therefore, we propose that the HDAC6 regulated-mitophagy protects neurons by clearing damaged mitochondria (Fig. 1).
In summary, HDAC6 suppresses the development of neurodegenerative diseases including Parkinson disease, Huntington disease and Spinobulbar muscular atrophy,20–22 possibly through different mechanisms. Although the precise mechanisms of HDAC6 in each particular neurodegenerative disease remain to be elucidated, its function in the neuronal stress surveillance and clearance has made HDAC6 to become a potential therapeutic target for many neurodegenerative diseases.
Acknowledgements
This work has been financially supported by the 973 program (2009CB918702), the NSFC (30623005, 31071087 and 30771217).
Abbreviations
- HDAC6
histone deacetylase 6
- MTOC
microtubule-organizing centre
- UPS
ubiquitin proteasome system
- AR-JP
autosomal recessive juvenile parkinsonism
References
- 1.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation and clinical implication. Mol Cell Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hook SS, Orian A, Cowley SM, Eisenman RN. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc Natl Acad Sci USA. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdel A, Curtet S, Brocard MP, Rousseaux S, Lemercier C, Yoshida M, et al. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol. 2000;10:747–749. doi: 10.1016/s0960-9822(00)00542-x. [DOI] [PubMed] [Google Scholar]
- 6.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 7.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 8.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 9.Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 13.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 15.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju JS, Miller SE, Hanson PI, Weihl CC. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. 2008;283:30289–30299. doi: 10.1074/jbc.M805517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, et al. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalveram B, Schmidtke G, Groettrup M. The ubiquitin-like modifier FAT10 interacts with HDAC6 and localizes to aggresomes under proteasome inhibition. J Cell Sci. 2008;121:4079–4088. doi: 10.1242/jcs.035006. [DOI] [PubMed] [Google Scholar]
- 20.Du G, Liu X, Chen X, Song M, Yan Y, Jiao R, et al. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol Biol Cell. 21:2128–2137. doi: 10.1091/mbc.E10-03-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 22.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 24.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 26.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jellinger KA. Recent advances in our understanding of neurodegeneration. J Neural Transm. 2009;116:1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- 28.Jiang Q, Ren Y, Feng J. Direct binding with histone deacetylase 6 mediates the reversible recruitment of parkin to the centrosome. J Neurosci. 2008;28:12993–13002. doi: 10.1523/JNEUROSCI.2860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation and HDAC6-dependent mitophagy. J Cell Biol. 189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]