

Abstract
All higher eukaryotes utilize protein tyrosine kinases (PTKs) as molecular switches to control a variety of cellular signals. Notably, many PTKs have been identified as proto-oncogenes whose aberrant expression, mutations or co-option by pathogens can lead to human malignancies. Thus, it is obvious that PTK functions must be precisely regulated in order to maintain homeostasis of an organism. Investigations over the past fifteen years have revealed that members of the Cbl family proteins can serve as negative regulators of PTK signaling, and biochemical and cell biological studies have unraveled the mechanistic basis of this regulation. Yet, it is only recently that the field has begun to appreciate the real significance of this novel regulatory apparatus in shaping PTK-mediated signaling in organismic contexts and in human diseases. Here, we discuss recent progress in murine models that are beginning to provide insights into the critical roles of Cbl proteins in physiological pathways, with important implications in understanding how aberrations of Cbl proteins contribute to oncogenesis.
Key words: Cbl, E3 ubiquitin ligase, protein tyrosine kinase, signal transduction, animal models, immunology, hematology, leukemia, stem cell, RING finger
Protein tyrosine kinases (PTKs) function as signaling switches in higher eukaryotes and play essential roles in mediating cellular responses to extracellular cues, including changes in cell proliferation, differentiation, migration and survival.1 It is therefore not surprising that altered expression and/or mutational activation of PTKs can promote oncogenesis while deficiencies of PTKs are linked to serious pathologies. Naturally, use of potentially oncogenic products as basic cellular switches demands well-orchestrated regulatory mechanisms. Among the many PTK regulatory mechanisms, recruitment of E3 ubiquitin ligases has recently emerged as a novel mechanism that links two fundamentally important cellular machineries—the PTKs and the ubiquitin pathway. In mammalian cells, this regulatory process is mediated by members of the Cbl family of proteins.2
Cbl family proteins are evolutionarily-conserved RING finger domain-containing E3 ubiquitin ligases.3–5 A substantial body of cellular and biochemical studies has shown that the negative regulatory functions of Cbl proteins in PTK signaling are dependent on their ability to facilitate the ubiquitination of activated PTKs and many of their downstream components (Fig. 1). The ubiquitin modification serves as a signal for lysosomal sorting and degradation (in the case of many receptors with intrinsic or associated PTK activity),6,7 proteasomal degradation (for several non-receptor signaling components),8 or for proteolysis-independent negative regulation through undefined mechanisms.9
Figure 1.
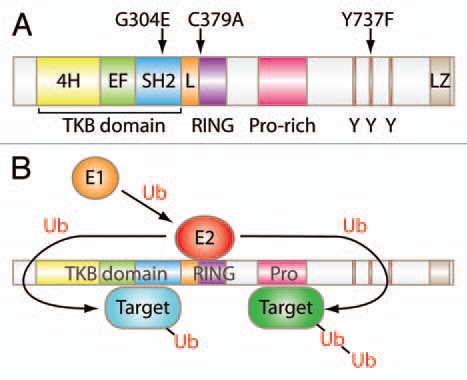
Structure and function of Cbl. (A) Schematic representation of mammalian Cbl. G304E, C379A and Y737F indicate amino acid substitution mutant mouse strains generated for Cbl. TKB, tyrosine kinase-binding domain; 4H, four-helix bundle; EF, EF hand; L, linker; Y, tyrosine residue; LZ, leucine-zipper. (B) Model of Cbl ubiquitin ligase function. A ubiquitin-activating enzyme (E1, orange) activates ubiquitin (Ub) and transfers it to a ubiquitin-conjugating enzyme (E2, red) which interacts directly with a ubiquitin ligase (E3) and transfers Ub to target proteins (blue and green). Either single (mono) or multiple (poly) ubiquitin moieties may be added to target proteins. This figure is for illustration purpose only and is not intended to indicate that targets bound to the TKB domain undergo mono-ubiquitination or those bound to the proline-rich domain undergo poly-ubiquitination.
In mammals, the Cbl family is composed of three members, Cbl (also known as c-Cbl), Cbl-b and Cbl-c (Cbl-3). Rigorous molecular/biochemical studies in model cell systems have shown that the highly-conserved N-terminal Tyrosine Kinase Binding (TKB) domain, composed of a four-helical bundle, a calcium-binding EF hand and a variant SH2 domain, mediates specific binding to cognate phosphotyrosine-containing motifs in PTKs and some non-PTK targets. A short linker region and a RING finger domain immediately C-terminal to the TKB domain together serve to bind to E2 ubiquitin-conjugating enzymes and are essential for the E3 ubiquitin ligase activity of Cbl proteins. High degrees of conservation of the TKB, linker and RING finger domains characterize all Cbl proteins within and across species (Fig. 1).
The carboxyl-half of the Cbl proteins is more divergent. Cbl and Cbl-b contain multiple protein interactions motifs such as the proline-rich regions that bind to SH3 domains of various signaling proteins, tyrosine residues that become phosphorylated and interact with key SH2 domain-containing signaling proteins including Vav guanine nucleotide exchange factors, the p85 regulatory subunit of phosphatidylinositol-3-kinase (PI3K) and Crk family adaptors known to link PTK activation to Ras-related Rap GTPases.
While studies in model cell systems have shown that all mammalian Cbl proteins can mediate similar negative regulatory functions towards PTK signaling, these systems have not been able to provide a clear picture of the relative contributions of individual Cbl family members in the context of myriad of PTKs that have been analyzed using in vitro systems. Evolution of multi-gene families is often associated with a need to create functional redundancy as well as to impart specialized functions to individual family members. Thus, in vivo models are an essential component of efforts geared towards fully understanding the importance of multiple mammalian Cbl proteins in regulating PTK-dependent biological functions. Targeted mutagenesis in mice has emerged as an apt and unbiased approach to gain insights into the in vivo roles of signal transduction pathways under physiological contexts, in particular for members of multi-gene families. While many of the observations in targeted mutant mice confirm the conclusions from in vitro systems, often these models have revealed functional roles not predicted by in vitro studies.
Over the past dozen years, several mouse lines carrying mutations in the Cbl family genes were generated. Interesting patterns emerged from studies of these mice; while previous biochemical analyses focused heavily on Cbl-dependent regulation of receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptors (PDGFRs) as well as non-RTKs including ZAP-70 and Src-family kinases, organ systems most strongly affected by Cbl family gene deletions are hematopoietic, immune and metabolic systems, which are not commonly associated with EGFR or PDGFR. These findings immediately raised questions about the pathways in which Cbl family proteins function in vivo. In this review, we examine the phenotypes of mouse models with Cbl family mutations in an attempt to reconcile in vitro and in vivo findings.
On a C57BL/6 background, Cbl-null mutant mice are born at a sub-Mendelian ratio but live-born mutant mice have normal lifespan (Naramura M, unpublished observations). We and others originally reported mild splenomegaly and skewed thymocyte development in the absence of Cbl.10,11 Other notable phenotypes include changes in energy metabolism12 and reduced fertility of male mice.13 Recent studies in mouse models as well as identification of oncogenic CBL mutations in human myeloid malignancies have pointed to critical functions of Cbl proteins in normal and pathological hematopoiesis. This aspect is further discussed below.
Despite close structural similarities of Cbl and Cbl-b proteins, Cblb-null mice do not share phenotypes with Cbl-null mice. Lack of Cbl-b does not have discernable effects on hematopoiesis or thymocyte development. The most prominent phenotype of Cblb-null mice is hypersensitivity of mature T cells to stimulation through the T-cell antigen receptors (TCRs).14,15 Normally, quiescent mature T cells require signals from both TCRs and co-stimulatory molecules such as CD28 for full activation to mount effective immune responses. However, Cbl-b-deficient T cells become fully activated without co-stimulation; physiological consequences of this defect manifest as impaired immunological tolerance induction, spontaneous and/or induced autoimmune diseases and rejection of transplanted and spontaneous tumors.16,17 Furthermore, Cbl-b functions are required to limit inflammatory responses in various organs including pancreas,18 lung,19 and adipose tissue20 or to limit airway responses to allergic challenges.21
These phenotypic differences between Cbl-null and Cblb-null mice may be attributed to differential tissue distribution of the two proteins. In the immune system, Cbl is highly expressed in the thymus, with the highest expression in the CD4, CD8-double positive compartment while Cbl-b expression is more prominent in mature peripheral T cells.22
While both Cbl- and Cblb-null mutant mice showed recognizable but relatively mild in vivo phenotypes, deficiency of the third member of the family, Cbl-c, is not linked to any significant phenotypes.23 This finding is surprising as Cbl-c is expressed primarily in epithelial cells, yet epithelial tissues developed normally in the absence of Cbl-c.23 Interestingly, transgenic expression of Cbl-c in the mammary epithelium led to a mild delay in the mammary gland development.24 Therefore, it is conceivable that the impact of Cbl-c deficiency was masked by the presence of other family members, namely, Cbl and Cbl-b in Cbl-c single-deficient mice.
Indeed, the redundant and overlapping functions of Cbl family proteins are evident from the robust phenotypes of Cbl, Cbl-b double-deficient mice. Unlike single-deficient mice, simultaneous deletion of Cbl and Cbl-b in the germline leads to early embryonic lethality.22 In contrast, limiting double deficiency to T-cell compartment is compatible with fetal development.22 However, double-deficient T cells develop independent of MHC-restricted thymic selection25 and mice eventually succumb to autoimmune-like vasculitis early in adult life.22 Similarly, targeted double-deficiency in the B-cell compartment led to a failure to acquire tolerance to self antigens and mice developed SLE-like autoimmune diseases.26 For both of these models, common molecular changes include delayed downregulation of cell surface antigen receptors and more persistent activation of downstream signaling pathways.
Recently, we analyzed mice with Cbl, Cbl-b double-deficiency in the hematopoietic stem cells.27 While loss of Cbl alone reportedly led to significant expansion of the hematopoietic stem/progenitor compartment,28 simultaneous loss of both Cbl and Cbl-b markedly enhanced this phenotype and all mice succumbed to aggressive myeloproliferative disease-like leukemia within two to three months after birth.27
Refinement of gene targeting techniques enabled in vivo validation of protein-protein interactions defined by in vitro studies. Mice expressing mutant Cbl family proteins from their endogenous promoters were generated using this approach. So far, such “knock-in” mutants have been reported for the Cbl TKB (G304E, equivalent to G306E in human Cbl),29 and RING finger (C379A, equivalent to C381A in human Cbl),30 domains, as well as one of the C-terminal region tyrosine residues (Y737F, equivalent to Y731F in human Cbl),31 that upon phosphorylation is known to interact with p85 subunit of PI3K, as well as for the Cbl-b RING finger domain (C373A, equivalent to C373A in human Cbl-b).32 Knock-in mutants highlight the importance of specific interactions and also demonstrate the complexity of in vivo analyses based on in vitro biochemistry. For instance, the TKB domain mutation was originally identified in C. elegans as a loss-of-function mutation.33 Numerous in vitro studies demonstrated that the G306E mutation, the human counterpart of the G304E mutation, abrogates interaction between Cbl and its phosphotyrosine-containing targets such as ZAP-70.34 In Cbl-null thymocytes, ZAP-70 activity is elevated,10,11 supporting the physiological relevance of Cbl in the regulation of this tyrosine kinase. However, while the G304E mutant spleens are as enlarged and bone marrow myeloid compartments as expanded as Cbl-null counterparts, ZAP-70 activity in the G304E mutant thymocytes was comparable to cells with wild-type Cbl.29 Molecular basis of this surprising outcome has not been fully clarified. Nevertheless, these results underscore the redundant and flexible nature of the multi-gene Cbl family proteins as signal transduction modifiers in vivo.
In contrast, mice expressing the Cbl RING finger loss-of-function mutant (C379A) exhibited more severe phenotypes than Cbl-null mice.30,35 Most homozygous C379A mutants die between mid-gestation and immediately after birth; heterozygous C379A mutation on a Cbl-null background is tolerated and animals exhibit enhanced thymic deletion30 and develop myeloid leukemia within a year.35 These phenotypes are remarkably reminiscent of Cbl, Cbl-b double-deficiency, albeit somewhat milder; in Cbl, Cbl-b double-deficient mice, germline deletion lead to embryonic death by 10 days of gestation,22 and the myeloproliferative disease progressed more rapidly in hematopoietic compartment-specific Cbl, Cbl-b double-deficient mice with median life span of 67 days.27 Considering that Cbl-b is still present in the C379A mutant model, these observations naturally raise a question whether C379A functions as a dominant-negative (DN) inhibitor of Cbl-b, thus creating a functional equivalency of the Cbl, Cbl-b double deficient cellular environment. This directly relates to the molecular mechanisms of leukemogenesis in patients with CBL mutations. Most CBL mutations associated with myeloid malignancies cluster around the linker and the RING finger domains and in vitro studies with selected mutants have demonstrated their lack of E3 ubiquitin ligase activity (reviewed in ref. 36). Furthermore, in a majority of patients with CBL mutations, the malignant cells derived from patients show a duplication of the mutant CBL allele concurrent with loss of the wild-type allele due to acquired uniparental isodisomy of 11q where the CBL gene resides.36 Whether RING finger-mutant Cbl simply functions as a DN inhibitor of wild-type Cbl-b protein or possesses additional functions is an important issue for determining optimal therapeutic approaches for patients with CBL mutations. Notably, mice with an equivalent RING finger domain mutation in Cbl-b (C373A) do not show comparable changes in the hematopoietic system,35 indicating that a mutant Cbl-b may not be able to override regulations imposed by normal Cbl protein. Future studies should clarify whether this reflects the distinct functions of Cbl and Cbl-b, or may be due to their expression patterns and/or relative levels.
Taken together, mouse models with Cbl family mutations have firmly established the essential roles of Cbl and Cbl-b as E3 ubiquitin ligases in the hematopoietic and immune systems. These models also highlight the need for systematic reassessment of previously defined Cbl-interacting proteins and mechanisms of interaction under physiological conditions. Availability of mice deficient in each Cbl protein as well as the Cbl, Cbl-b double-deficient (and potentially the triple-deficient) strains should facilitate such studies without compounding effects of endogenous Cbl family proteins. In this context, development of models in which multiple Cbl family genes can be targeted for deletion in a tissue-specific manner (at present this is only feasible with Cbl) should further allow dissecting the developmental versus organ/tissue homeostasis roles of Cbl family proteins. These models should also help interrogate the precise mechanisms of PTK regulation by members of the multi-gene Cbl family in physiological contexts as well as in disease states.
Acknowledgements
Works in authors' laboratories are supported by grants from US National Institutes of Health (CA87986, CA99163, CA105489 and CA116552 to H.B.; CA96844 to V.B.) and US Department of Defense Breast Cancer Research Grant (W81 XWH-10-1-0740 to M.N.).
References
- 1.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao N, Dodge I, Band H. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol. 2002;71:753–763. [PubMed] [Google Scholar]
- 3.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 4.Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity. 2004;21:7–17. doi: 10.1016/j.immuni.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt MHH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–918. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 6.Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, et al. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee PS, Wang Y, Dominguez MG, Yeung YG, Murphy MA, Bowtell DD, et al. The Cbl protooncoprotein stimulates CSF-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. EMBO J. 1999;18:3616–3628. doi: 10.1093/emboj/18.13.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao N, Miyake S, Reddi AL, Douillard P, Ghosh AK, Dodge IL, et al. Negative regulation of Lck by Cbl ubiquitin ligase. Proc Natl Acad Sci USA. 2002;99:3794–3799. doi: 10.1073/pnas.062055999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol. 2001;2:870–875. doi: 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- 10.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci USA. 1998;95:15547–15552. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molero JC, Jensen TE, Withers PC, Couzens M, Herzog H, Thien CBF, et al. c-Cbl-deficient mice have reduced adiposity, higher energy expenditure and improved peripheral insulin action. J Clin Invest. 2004;114:1326–1333. doi: 10.1172/JCI21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El Chami N, Ikhlef F, Kaszas K, Yakoub S, Tabone E, Siddeek B, et al. Androgen-dependent apoptosis in male germ cells is regulated through the protooncoprotein Cbl. J Cell Biol. 2005;171:651–661. doi: 10.1083/jcb.200507076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 15.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 16.Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007;117:1029–1036. doi: 10.1172/JCI29472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007;204:879–891. doi: 10.1084/jem.20061699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokoi N, Komeda K, Wang H, Yano H, Kitada K, Saitoh Y, et al. Cblb is a major susceptibility gene for rat type 1 diabetes mellitus. Nat Genet. 2002;31:391–394. doi: 10.1038/ng927. [DOI] [PubMed] [Google Scholar]
- 19.Bachmaier K, Toya S, Gao X, Triantafillou T, Garrean S, Park GY, et al. E3 ubiquitin ligase Cblb regulates the acute inflammatory response underlying lung injury. Nat Med. 2007;13:920–926. doi: 10.1038/nm1607. [DOI] [PubMed] [Google Scholar]
- 20.Hirasaka K, Kohno S, Goto J, Furochi H, Mawatari K, Harada N, et al. Deficiency of Cbl-b gene enhances infiltration and activation of macrophages in adipose tissue and causes peripheral insulin resistance in mice. Diabetes. 2007;56:2511–2522. doi: 10.2337/db061768. [DOI] [PubMed] [Google Scholar]
- 21.Oh SY, Park J, Zheng T, Kim Y, Wu F, Cho SH, et al. Cbl-b regulates airway mucosal tolerance to aeroallergen. Clin Exp Allergy. 2010;41:434–442. doi: 10.1111/j.1365-2222.2010.03593x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naramura M, Jang I, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths EK, Sanchez O, Mill P, Krawczyk C, Hojilla CV, Rubin E, et al. Cbl-3-deficient mice exhibit normal epithelial development. Mol Cell Biol. 2003;23:7708–7718. doi: 10.1128/MCB.23.21.7708-7718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiore F, Estebe B, Gibier P, Orsoni J, Courbard J, Chodosh LA, et al. Abnormal mammary gland development in MMTV-CBLC transgenic mouse. In Vivo. 2009;23:225–228. [PubMed] [Google Scholar]
- 25.Huang F, Kitaura Y, Jang I, Naramura M, Kole HH, Liu L, et al. Establishment of the major compatibility complex-dependent development of CD4+ and CD8+ T cells by the Cbl family proteins. Immunity. 2006;25:571–581. doi: 10.1016/j.immuni.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Kitaura Y, Jang IK, Wang Y, Han Y, Inazu T, Cadera EJ, et al. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity. 2007;26:567–578. doi: 10.1016/j.immuni.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naramura M, Nandwani N, Gu H, Band V, Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci USA. 2010;107:16274–16279. doi: 10.1073/pnas.1007575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rathinam C, Thien CBF, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22:992–997. doi: 10.1101/gad.1651408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thien CBF, Scaife RM, Papadimitriou JM, Murphy MA, Bowtell DDL, Langdon WY. A mouse with a loss-of-function mutation in the c-Cbl TKB domain shows perturbed thymocyte signaling without enhancing the activity of the ZAP-70 tyrosine kinase. J Exp Med. 2003;197:503–513. doi: 10.1084/jem.20021498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thien CBF, Blystad FD, Zhan Y, Lew AM, Voigt V, Andoniou CE, et al. Loss of c-Cbl RING finger function results in high-intensity TCR signaling and thymic deletion. EMBO J. 2005;24:3807–3819. doi: 10.1038/sj.emboj.7600841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thien CBF, Dagger SA, Steer JH, Koentgen F, Jansen ES, Scott CL, et al. c-Cbl promotes T cell receptor-induced thymocyte apoptosis by activating the phosphatidylinositol-3-kinase/Akt pathway. J Biol Chem. 2010;285:10969–10981. doi: 10.1074/jbc.M109.094920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oksvold MP, Dagger SA, Thien CBF, Langdon WY. The Cbl-b RING finger domain has a limited role in regulating inflammatory cytokine production by IgE-activated mast cells. Mol Immunol. 2008;45:925–936. doi: 10.1016/j.molimm.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Yoon CH, Lee J, Jongeward GD, Sternberg PW. Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science. 1995;269:1102–1105. doi: 10.1126/science.7652556. [DOI] [PubMed] [Google Scholar]
- 34.Rao N, Lupher ML, Ota S, Reedquist KA, Druker BJ, Band H. The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J Immunol. 2000;164:4616–4626. doi: 10.4049/jimmunol.164.9.4616. [DOI] [PubMed] [Google Scholar]
- 35.Rathinam C, Thien CBF, Flavell RA, Langdon WY. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell. 2010;18:341–352. doi: 10.1016/j.ccr.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa S, Shih L, Suzuki T, Otsu M, Nakauchi H, Koeffler HP, et al. Deregulated intracellular signaling by mutated c-CBL in myeloid neoplasms. Clin Cancer Res. 2010;16:3825–3831. doi: 10.1158/1078-0432.CCR-09-2341. [DOI] [PubMed] [Google Scholar]