

Abstract
In the cerebellar cortex, parallel fiber-to-stellate cell (PF-SC) synapses exhibit a form of synaptic plasticity manifested as a switch in the subunit composition of synaptic AMPA receptors (AMPARs) from calcium-permeable, GluA2-lacking to calcium-impermeable, GluA2-containing receptors. Here, we examine the role of stargazin (γ-2), canonical member of the transmembrane AMPAR regulatory protein (TARP) family, in the regulation of GluA2-lacking AMPARs and synaptic plasticity in SCs from epileptic and ataxic stargazer mutant mice. We found that AMPAR-mediated synaptic transmission is severely diminished in stargazer SCs, and that the rectification index (RI) of AMPAR current is reduced. Activity-dependent plasticity in the rectification of synaptic AMPARs is also impaired in stargazer SCs. Despite the dramatic loss in synaptic AMPARs, extrasynaptic AMPARs are preserved. We then examined the role of stargazin in regulating the rectification of extrasynaptic AMPARs in nucleated patches and found, in contrast to previous reports, that wild-type extrasynaptic AMPARs have moderate RI values (average RI = 0.38), while those in stargazer SCs are low (average RI = 0.24). The GluA2-lacking AMPAR blocker, philanthotoxin-433 (PhTx-433), was used as an alternative measure of GluA2 content in wild-type and stargazer SCs. Despite the difference in RI, PhTx-433 sensitivity of both synaptic and extrasynaptic AMPARs remains unchanged, suggesting that the dramatic changes in RI and the impairment in synaptic plasticity observed in the stargazer mouse are not the result of a specific impairment in GluA2 trafficking. Together, these data suggest that stargazin regulates compartment-specific AMPAR trafficking, as well as activity-dependent plasticity in synaptic AMPAR rectification at cerebellar PF-SC synapses.
Introduction
The bulk of fast excitatory synaptic transmission in the CNS is mediated by AMPA-type glutamate receptors (AMPARs), tetramers assembled from GluA1-4 subunits (Bredt and Nicoll, 2003). The GluA2 subunit is a crucial determinant of the biophysical properties of AMPARs (Cull-Candy et al., 2006; Isaac et al., 2007; Liu and Zukin, 2007). GluA2-containing AMPARs are impermeable to calcium and exhibit a linear current–voltage (I–V) relationship (Hollmann et al., 1991; Hume et al., 1991; Verdoorn et al., 1991; Burnashev et al., 1992), whereas GluA2-lacking AMPARs are calcium permeable and exhibit inwardly rectifying I–Vs owing to voltage-dependent block by endogenous intracellular polyamines (McBain and Dingledine, 1993; Bochet et al., 1994; Jonas et al., 1994; Bowie and Mayer, 1995; Geiger et al., 1995; Kamboj et al., 1995; Koh et al., 1995). Although GluA2-containing AMPARs predominate in the CNS, specific populations of cells express varying proportions of GluA2-lacking and GluA2-containing AMPARs (Isaac et al., 2007; Liu and Zukin, 2007), two populations of receptors that can be independently regulated (Tóth and McBain, 1998; Liu and Cull-Candy, 2000).
AMPARs are associated with a family of transmembrane AMPAR regulatory proteins (TARPs), auxiliary subunits that are involved in every aspect of AMPAR function, from surface expression and synaptic targeting to gating and pharmacology (Nicoll et al., 2006; Ziff, 2007; Milstein and Nicoll, 2008; Coombs and Cull-Candy, 2009; Sager et al., 2009; Kato et al., 2010). Recent evidence from work on heterologously expressed receptors indicates that some TARP subtypes display AMPAR subunit selectivity (Kato et al., 2008; Soto et al., 2009), suggesting the existence of TARP subtype-dependent streams for trafficking GluA2-lacking and GluA2-containing AMPARs. It is, therefore, of considerable interest to examine the role of TARPs in a neuronal cell-type that normally expresses a mixed population of GluA2-lacking and GluA2-containing AMPARs.
Cerebellar stellate cells (SCs), which reside in the molecular layer and receive glutamatergic input from parallel fibers (PFs), exhibit a unique compartmentalization of AMPARs: a largely synaptic pool of GluA2-lacking AMPARs and an extrasynaptic pool of GluA2-containing AMPARs. High-frequency PF stimulation has been shown to induce a change in the rectification of synaptic AMPARs, which is explained by an activity-dependent switch between these two pools of receptors leading to linearization of synaptic I–Vs (Liu and Cull-Candy, 2000). Subsequent work showed that this striking form of synaptic plasticity at PF-SC synapses depends on interactions with PICK1, GRIP, and NSF (Gardner et al., 2005; Liu and Cull-Candy, 2005).
In the present study we examined the potential role of TARPs in the regulation of basal AMPAR trafficking and in activity-dependent synaptic plasticity. SCs express high levels of stargazin (TARP γ-2) (Fukaya et al., 2005; Lein et al., 2007), which is disrupted in the epileptic and ataxic stargazer mutant mouse (Letts et al., 1998). We found that stargazin is required for the synaptic delivery of AMPARs and synaptic plasticity in AMPAR rectification in SCs, but it is not necessary for the surface trafficking of extrasynaptic AMPARs. Furthermore, we found that the dramatic differences in rectification index (RI) of both synaptic and extrasynaptic I–Vs in stargazer mice is unlikely to be the result of a change in GluA2 content.
Materials and Methods
Animals.
All experiments were carried out in accordance with animal welfare regulations set out by the University of California, San Francisco Institutional Animal Care and Use Committee (San Francisco, CA). Both stargazer mutant mice (Letts et al., 1998) and GluA2 knock-out (GluA2−/−) mice (Jia et al., 1996) have been previously described. Wild-type mice were primarily acquired from wild-type breeding pairs derived from the stargazer background. Some wild-type mice were acquired from C57BL/6 breeding pairs.
Brain slice preparation.
Transverse cerebellar slices (250 μm thick) were prepared from P16–26 mice. Mice were anesthetized with isoflurane and decapitated and their brains were rapidly removed and placed in an ice-cold, high-sucrose cutting solution consisting of the following (in mm): 87 NaCl, 1.25 NaH2PO4, 25 NaHCO3, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 glucose, and 75 sucrose, saturated with 95% O2/5% CO2. Slices were cut using a DTK-1000 Microslicer (Ted Pella). Freshly cut slices were then placed in an incubating chamber containing artificial CSF (ACSF), containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 Na2PO4, 25 glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2/5% CO2 and allowed to recover at 35°C for ∼30 min. Slices were then maintained for at least 0.5–1 h in ACSF at room temperature (21–22°C) before recording. Coronal slices of hippocampus (250 μm thick) were prepared in an identical fashion.
Whole-cell recording.
Following recovery, slices were transferred to a submersion chamber on an upright Olympus BX51 microscope and perfused with ACSF saturated with 95% O2/5% CO2 at room temperature (21–22°C). Cerebellar SCs were visualized by infrared differential interference contrast microscopy and identified on the basis of the following: (1) their location in the outer third of the molecular layer; (2) the high probability of detecting spontaneous action potentials in cell-attached patch configuration; and (3) their high input resistance (>1 GΩ) following break-through. Cells were recorded using thin-walled borosilicate glass pipettes (World Precision Instruments) with pipette resistance values of 4–6 MΩ and filled with an internal solution consisting of the following (in mm): 140 CsMeSO3, 10 HEPES, 10 EGTA, 2 NaCl, 2 Mg-ATP, 1 QX-314, 5 tetraethylammonium chloride, 1 CaCl2, and 0.1 spermine, pH 7.3, with CsOH. Recordings were conducted using an Axopatch 1D amplifier (Molecular Devices) filtered at 2 kHz and digitized at 10 kHz using a National Instruments digitizer and were acquired using custom procedures in IGOR Pro (Wavemetrics). Recordings of mEPSCs were carried out at 20 kHz to ensure high resolution of decay kinetics. Input resistance (Rin), series resistance (Rs), and leak current were consistently monitored throughout all experiments, and recordings were excluded from the analysis if any of these measures deviated significantly from baseline. All recordings were carried out at room temperature (21–22°C).
Synaptic recording and data analysis.
Miniature EPSCs (mEPSCs) from both wild-type and stargazer SCs were measured in the presence of picrotoxin, d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5), and tetrodotoxin (TTX) at a holding potential of −60 mV. Analysis of mEPSCs was carried out using custom procedures in Igor Pro (Wavemetrics) (Milstein et al., 2007). Amplitudes are expressed as absolute values. Following peak alignment, the decay kinetics of mEPSCs were well fit by double-exponential functions (illustrated in Fig. 1E). However, for the sake of simplicity and ease of comparison with previous work, we calculated a weighted tau (τ) value (Cathala et al., 2005; Milstein et al., 2007), which was performed in the following way:
 |
Figure 1.

The frequency and amplitude of mEPSCs are reduced and decay kinetics are faster at stargazer PF-SC synapses. A, Representative mEPSC traces from a wild-type (left) SC and a stargazer (right) SC at a holding potential of −60 mV. B, Bar plot showing that mEPSC frequency (mean ± SEM) of stargazer (stg) SCs is significantly reduced relative to wild-type (wt) SCs (wt: average mEPSC frequency = 0.55 ± 0.06 Hz, n = 12; stg, average mEPSC frequency = 0.08 ± 0.02 Hz, n = 11; p < 0.0001, Wilcoxon rank sum test). C, Average mEPSC waveform from wild type (left) (averaged over 4368 events from 12 cells) and average mEPSC waveform from stargazer (right) (averaged over 985 events from 11 cells). D, Bar plot showing that the mEPSC amplitude (mean ± SEM) of stargazer SCs is significantly reduced relative to wild type (wt: average mEPSC amplitude = 24.3 ± 1.5 pA, n = 12; stg: average mEPSC amplitude = 16.2 ± 0.8 pA, n = 11; p < 0.0001, Wilcoxon rank sum test). Amplitudes are presented as absolute values. E, Overlay of averaged, peak-aligned, and normalized mEPSC waveforms from wild-type (gray) and stargazer (black) SCs. Dotted lines represent double-exponential fits to the decay of each waveform. F, Bar plot showing that the weighted tau (τw,decay) values of mEPSC waveforms from stargazer SCs is significantly faster than wild type (wt: average mEPSC τw,decay = 1.82 ± 0.13 ms, n = 12; stg: average mEPSC τw,decay = 1.37 ± 0.06 ms, n = 11; p < 0.05, Wilcoxon rank sum test). Open circles represent values from individual cells. Asterisks indicate significance: ***p < 0.0005, *p < 0.05, N.S. Wilcoxon rank sum test).
Parallel fiber stimulation was carried out using a theta glass (Warner Instruments) stimulating electrode, filled with ACSF, placed in the molecular layer 100–200 μm away from the recorded cell with an ISO-Flex stimulus isolator (A.M.P.I.) controlled by a Master-8 pulse stimulator (A.M.P.I.). Synaptic I–Vs were obtained by stimulating at 0.2 Hz with holding potentials of −60, 0, and +40 mV. Approximately 20 raw evoked EPSCs (eEPSCs) were averaged together to obtain an average eEPSC at any given holding potential. To control for the possibility that depolarization could introduce variability in the amplitude of the eEPSC at −60 mV through retrograde inhibition (Beierlein and Regehr, 2006), the averaged eEPSC before the depolarizing step was averaged with the one after. RIs of eEPSCs were calculated using a ratio of slopes from point currents obtained at −60, 0, and +40 mV for I–Vs as follows:
 |
In measuring AMPA-NMDA ratio, an AMPAR-mediated eEPSC was first recorded at −60 mV in the presence of picrotoxin. Following complete block of the eEPSC by the subsequent addition of NBQX (10 μm) the membrane potential was changed to +40 mV to reveal and isolate the NMDAR-mediated eEPSC. The peak amplitude of each current component (averaged over 10–20 eEPSCs) was used in the calculation of AMPA/NMDA ratio (IAMPA/INMDA).
Paired-pulse facilitation (PPF) was determined by delivering 2 pulses at 50 Hz (interstimulus interval = 20 ms) with a frequency of 0.05 Hz, and averaging over ∼20 raw waveforms. PPF was represented as EPSC2/EPSC1 where EPSC1 and EPSC2 are the peak amplitudes of the first and second eEPSCs, respectively.
High-frequency synaptic stimulation for the induction of synaptic plasticity was carried out by first acquiring a 5–10 min baseline eEPSC recording at 0.2 Hz, at a holding potential of −60 mV, followed by 5 bursts of 100 pulses at 50, delivered at a frequency of 0.2 Hz at −60 mV. Following high-frequency stimulation (HFS), eEPSC amplitude at −60 mV was tracked at a frequency of 0.2 Hz, and I-Vs were then obtained after 15 and 30 min In some experiments, only the 15 min time point was included because of a significant change in Rs or loss of the cell.
Experiments in which philanthotoxin-433 (PhTx-433) was used to quantify GluA2-content of synaptic AMPARs were conducted by first determining the synaptic I–V and then acquiring a 5 min baseline before bath applying PhTx-433 for ∼30 min. The amplitude of the eEPSC at a holding potential of −60 mV was monitored at a stimulation frequency of 0.2 Hz. Values for the percentage of remaining current in the presence of PhTx-433 was determined by comparing the average eEPSC amplitude during the last 5 min of the recording with the average during the 5 min baseline.
Nucleated and outside-out patch recording and data analysis.
Nucleated patches from SCs were obtained following break-through by gently applying negative pressure and slowly coaxing the membrane-enveloped nucleus away from the cell and out of the slice. Intact nucleated patches were characterized by maintenance of the gigaohm seal, extremely high Rin, and the appearance of a perfectly spherical smooth bleb at the tip of the pipette. Nucleated patches were immediately positioned within the zone of laminar flow of the ValveLink 8 (AutoMate Scientific) multibarrel manifold tip of a gravity-flow perfusion pencil (internal diameter = 250 μm). Control solution was HEPES-buffered ACSF containing the following (in mm): 150 NaCl, 2.5 KCl, 10 HEPES, 10 glucose, 2 CaCl2, and 1 MgCl2, pH 7.4, with NaOH. Conventional outside-out patches from both cerebellar SCs and hippocampal CA1 pyramidal neurons were acquired in a similar fashion, but in the absence of negative pressure.
Control perfusion solution contained 100 μm picrotoxin, 100 μm d-AP5, 500 nm TTX, and 100 μm cyclothiazide (CTZ), to which 500 μm glutamate (Glu) or 500 μm kainate (KA) was added. Varying concentrations of PhTx-433 were added to the glutamate/CTZ-containing solution for channel block experiments. The amplitude of glutamate/CTZ-evoked currents was determined in one of two ways: either from the peaks of glutamate/CTZ-evoked currents holding steadily at −60 mV, or from point measurements at −60 mV obtained from voltage ramp traces. All values of peak glutamate/CTZ-evoked current were determined from the patch's first exposure to agonist. Experiments in which we determined the kainate/glutamate (KA/Glu) ratios were performed by sequentially applying kainate and glutamate, all in the presence of CTZ, at a constant holding potential of −60 mV. Values for agonist-evoked currents were determined from peak currents. To obtain I–V relationships from glutamate-evoked currents from nucleated patches, we applied voltage ramps from −100 to +100 mV over 100 ms (2000 mV/s) from a holding potential of −60 mV (illustrated in Fig. 5A) at a frequency of 1 Hz. An average trace from the control solution (averaging over 3–4 overlapping traces) was subtracted from an average trace from the glutamate/CTZ solution. The resulting subtracted current was plotted against voltage and normalized to the maximal current amplitude to permit traces to be superimposed. RI values of glutamate/CTZ-evoked currents in nucleated patches were calculated from I–Vs using a ratio of slopes from point currents obtained at −60, 0, and +40 mV, as described for eEPSCs.
Figure 5.

Inward-rectification of extrasynaptic/somatic AMPARs is enhanced in stargazer mice. A, Example of glutamate/CTZ-evoked, AMPAR-mediated current in a nucleated patch from a wild-type SC while applying a fast voltage ramp (left). The patch was held at −60 mV while the voltage ramp ran from −100 and +100 mV over 100 ms (2000 mV/s) and was applied at a frequency of 1 Hz. Control solution contained picrotoxin, d-AP5, TTX, and CTZ. Control current was then subtracted from agonist-evoked current and plotted as a function of voltage and, in this example, with an RI of 0.31 (right). B, A representative conventional outside-out patch from a SC from the same slice (left) with an RI value of 0.26. Bar graph representing average RI from conventional outside-out patches from wild-type SCs (average RI = 0.31 ± 0.07, n = 5) (right). C, Normalized I–V curves from wild-type (left) and stargazer (right) nucleated patches. Curves from individual SCs are shown in gray and the averaged curves are shown in black. D, Bar plot showing the collected RI values for I–V curves from nucleated patches from wild-type (wt) and stargazer (stg) SCs (wt: average RI = 0.38 ± 0.01, n = 51; stg: average RI = 0.24 ± 0.02, n = 38; p < 0.0001, Wilcoxon rank sum test). Open circles represent values from individual cells. Asterisks indicate significance: ***p < 0.0005.
Quantification of PhTx-433 block in nucleated patches was carried out by determining the percentage of the remaining current in the presence of a given concentration of PhTx-433 relative to the steady-state amplitude of glutamate/CTZ-evoked currents. Amplitudes were determined by averaging over ∼10 s of steady-state current. Glutamate/CTZ-evoked currents exhibited a variable degree of desensitization, and PhTx-433 was not applied to patches in which glutamate/CTZ-evoked currents desensitized rapidly and did not reach steady state. Values for the percentage of remaining current were obtained only from a patch's first exposure to PhTx-433.
Drugs.
Picrotoxin (Sigma) was used at a concentration of 100 μm to block GABAA receptors. d-AP5 (Ascent Scientific) was used at 100 μm to block NMDARs. TTX (Tocris Bioscience) was used at 500 nm to block voltage-gated sodium channels. 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) (Tocris Bioscience) was used at 10 μm to block AMPARs. 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (Tocris Bioscience) was used at 10 μm as a partial agonist at AMPARs. Trichlormethiazide (TCM) (Sigma) was used at 400 μm to block AMPAR desensitization, as was CTZ (Ascent Scientific), used at 100 μm. PhTx-433 (Chiralix) was used at varying concentrations from 1 nm to 10 μm to block GluA2-lacking AMPARs.
Statistics.
All data are presented as mean ± SEM. Significance, defined as p < 0.05, was calculated with unpaired or paired nonparametric Wilcoxon rank sum test as appropriate using KaleidaGraph (Synergy Software).
Results
SCs from stargazer mice exhibit severe deficits in AMPAR-mediated synaptic transmission
To determine whether stargazin is required for the maintenance of synaptic AMPARs at single PF-SC synapses, we compared the characteristics of mEPSCs in wild-type and stargazer mice. Whole-cell recordings were obtained from SCs in transverse cerebellar slices in the presence of picrotoxin, d-AP5, and TTX. We found that both the frequency and amplitude of mEPSCs recorded from SCs in stargazer mice were significantly reduced relative to those in wild-type mice. Recording at a holding potential of −60 mV, wild-type SCs exhibited an average frequency of 0.55 ± 0.06 Hz (n = 12), whereas in stargazer SCs the average frequency was 0.08 ± 0.02 Hz (n = 11), representing an 85.3% reduction (p < 0.0001) (Fig. 1A,B). The amplitude of mEPSCs was also significantly reduced but not to the same extent. Wild-type mEPSCs had an average amplitude of 24.3 ± 1.5 pA (n = 12), whereas in stargazer SCs the average amplitude was 16.2 ± 0.8 pA (n = 11), representing a 33.3% reduction (p < 0.0001) (Fig. 1C,D). In addition, we assessed the role of stargazin in modulating the decay kinetics of AMPAR mEPSCs (Fig. 1E,F). We calculated a weighted tau value from averaged mEPSC waveforms (see Materials and Methods) and found that among stargazer mice (1.37 ± 0.06 ms; n = 11) decay was significantly faster than that among wild-type mice (1.82 ± 0.13 ms; n = 12), representing a 32.8% decrease in the weighted tau (p < 0.05) (Fig. 1E). Together, these data show that SCs from stargazer mice exhibit an overall reduction in AMPAR-mediated synaptic transmission, formally consistent with a diminishment in postsynaptic AMPARs and/or a reduction in presynaptic release probability.
We then used the AMPA/NMDA ratio as another metric for assessing the role of stargazin in maintaining synaptic AMPARs at PF-SC synapses. Previous work has shown that although single stimuli can evoke relatively large synaptic AMPAR-mediated EPSCs (Atluri and Regehr, 1998), NMDAR-mediated currents are comparatively small in SCs (Glitsch and Marty, 1999). However, short high-frequency trains have been shown to elicit large amplitude NMDAR-mediated spillover current (Carter and Regehr, 2000; Clark and Cull-Candy, 2002). We found that with sufficient stimulation intensity, we were able to reproducibly elicit a comparatively small but measurable NMDAR-mediated current with single stimuli at positive potentials. AMPA-NMDA ratios were then obtained by comparing isolated AMPAR- and NMDAR-mediated eEPSCs elicited by single stimuli (see Materials and Methods). The AMPA/NMDA ratio of wild-type SCs was 10.7 ± 1.7 (n = 12), whereas the ratio of stargazer SCs was 3.3 ± 0.5 (n = 11); representing a reduction of 69.7% compared with wild-type (p < 0.0001) (Fig. 2A,B).
Figure 2.

AMPAR-mediated synaptic transmission is diminished and inward rectification enhanced at stargazer PF-SC synapses without an effect on paired-pulse ratio. A, Overlay of AMPAR- and NMDAR-mediated eEPSCs from a wild-type (left) and a stargazer (right) SC. The bottom traces were recorded at a holding potential of −60 mV in the presence of picrotoxin. The top traces were at a holding potential of +40 mV in the presence of NBQX and picrotoxin. Stimulus artifacts were removed for clarity. B, Bar plot showing values for AMPA/NMDA ratios (peak IAMPA/peak INMDA). Stargazer (stg) SCs exhibit AMPA/NMDA ratios that are reduced by 69.7% relative to wild-type (wt) SCs (wt: average AMPA/NMDA ratio = 10.7 ± 1.7, n = 12; stg: average AMPA/NMDA ratio = 3.3 ± 0.5, n = 11; p < 0.0001, Wilcoxon rank sum test). C, Representative paired-pulse traces (interstimulus interval = 20 ms) for a wild-type (left) and a stargazer (right) SC at a holding potential of −60 mV. Stimulus artifacts are removed for clarity. D, Bar plot of paired-pulse ratio (EPSC2/EPSC1) represented as mean ± SEM, showing no significant difference between wild type and stargazer (wt: average PPF = 1.78 ± 0.09, n = 12; stg: average PPF = 1.77 ± 0.1, n = 9, N.S.). E, Overlay of eEPSCs in a wild-type (left) and stargazer (right) SC measured at −60, 0, and +40 mV in the presence of picrotoxin and D-AP5, with stimulus artifacts blanked. Insets show normalized I–Vs for each example. F, Bar graph showing collected RI values for wild-type and stargazer mice (wt: average RI = 0.46 ± 0.02 pA, n = 75; stg: average RI = 0.23 ± 0.02 pA, n = 43; p < 0.0001, Wilcoxon rank sum test). Open circles represent values from individual cells. Asterisks indicate significance: ***p < 0.0005, N.S. is not significant.
Together with the mEPSC data, these results indicate that stargazin is required for the majority of AMPAR-mediated synaptic transmission at PF-SC synapses. To address the possibility that these severe synaptic deficits also have a presynaptic component as suggested by electron microscopic studies of stargazer cerebella (Richardson and Leitch, 2005; Leitch et al., 2009), we examined paired-pulse ratio as a way of measuring presynaptic release probability. PF-SC synapses have been shown to exhibit prominent PPF (Atluri and Regehr, 1998; Xu-Friedman and Regehr, 2000). We used pairs of stimuli separated by 20 ms and found no significant difference in PPF (wild type: 1.78 ± 0.09; n = 12; stargazer: 1.77 ± 0.1; n = 9; N.S.) (Fig. 2C,D), indicating that a presynaptic change in release probability is unlikely to contribute to the synaptic defects we observed at PF-SC synapses in the stargazer mouse. This result is consistent with the absence of a presynaptic phenotype at PF-Golgi cell synapses in stargazer mice (Menuz et al., 2008). The dramatic reduction in synaptic strength at PF-SC synapses is thus best explained by a specific defect in the trafficking of postsynaptic AMPARs. In particular, the dramatic reduction in mEPSC frequency and the moderate reduction in mEPSC amplitude likely reflect the preservation of remaining AMPARs in a dramatically diminished number of AMPAR-containing PF-SC synapses.
PF-SC synapses in stargazer mice exhibit changes in rectification index
We next asked whether stargazin is involved in determining the subunit composition of synaptic AMPARs in SCs. Recent evidence suggests that TARP family members are capable of determining subunit composition (Menuz et al., 2008) and have preferential interactions with particular GluA subunits (Kato et al., 2008; Soto et al., 2009). An informative measure of AMPAR subunit composition is the rectification index, which takes advantage of the fact that GluA2-lacking AMPARs exhibit voltage-dependent block by endogenous intracellular polyamines and hence exhibit inwardly rectifying I–V curves (Bowie and Mayer, 1995; Kamboj et al., 1995; Koh et al., 1995). The RI of eEPSCs, recorded with an internal solution containing 100 μm spermine, can be calculated from the I–V curve (see Materials and Methods), and GluA2-content can then be inferred by the degree of rectification. Previous work has shown that under basal conditions synaptic AMPARs at PF-SC synapses are moderately inwardly rectifying (average RI = ∼0.4) (Liu and Cull-Candy, 2000), suggesting an indeterminate mix of GluA2-lacking and GluA2-containing synaptic AMPARs.
Recording in the presence of both picrotoxin and d-AP5, we found that the basal rectification values of wild-type PF-SC synapses are indeed moderately inwardly rectifying with an average RI of 0.46 ± 0.02 (n = 75), generally consistent with previously reported values. In contrast, PF-SC synapses in stargazer mice were significantly more inwardly rectifying with an average RI of 0.23 ± 0.02 (n = 43), representing a 50% decrease in RI relative to that in wild-type mice (p < 0.0001) (Fig. 2E,F). The most straightforward explanation for this dramatic difference in RI is that PF-SC synapses in stargazer mice have a greater proportion of GluA2-lacking AMPARs than wild type, implying that stargazin has a specific role in trafficking GluA2-containing AMPARs to synapses. However, given recent evidence that TARP association alters I–V shape as a result of modulation of the spermine affinity of GluA2-lacking AMPARs (Soto et al., 2007), an alternative explanation is that the change in RI is not the result of a defect in subunit-selective trafficking but reflects a TARP-dependent change in AMPAR pore properties. Yet another possibility is that diminished excitatory drive from upstream granule cells results in reduced activity-dependent GluA2 incorporation (Liu and Cull-Candy, 2002). To distinguish between these various possible explanations, we looked at the phenomenon of activity-dependent subunit switching observed at PF-SC synapses.
Synaptic plasticity is disrupted at PF-SC synapses in stargazer mice
Cerebellar SCs exhibit a unique form of synaptic plasticity manifested as both a depression in the amplitude of AMPAR-mediated eEPSCs at negative potentials and a change in the RI of synaptic AMPARs following high-frequency stimulation or HFS, interpreted as a switch in synaptic AMPAR subunit composition. This phenomenon was shown to be largely postsynaptic as evidenced by the lack of change in synaptic failures (Liu and Cull-Candy, 2000). Variants of this form of activity-dependent change in AMPAR rectification have been demonstrated at PF-SC synapses following activation of extrasynaptic NMDARs (Sun and Liu, 2007) and group I mGluRs (Kelly et al., 2009). More recent work has shown that a single fear-inducing stimulus can also trigger an AMPAR subunit switch at PF-SC synapses in a transcription-dependent manner (Liu et al., 2010). In the prevailing model of this form of plasticity, synaptic GluA2-lacking AMPARs are selectively removed and replaced by extrasynaptic GluA2-containing AMPARs following HFS of PFs (Liu and Cull-Candy, 2000; Cull-Candy et al., 2006). Given the profound deficits in AMPAR-mediated transmission and a possible defect in GluA2 trafficking in the stargazer mouse, we asked whether stargazin is involved in this form of plasticity.
Consistent with previous results, we found that wild-type SCs (Fig. 3A,C) exhibit a modest but significant depression in the absolute amplitude of eEPSCs at −60 mV 15 and 30 min following HFS in the form of 5 bursts of 100 pulses at 50 Hz, delivered at 0.2 Hz (control: 294.2 ± 25.1 pA, n = 33; 15 min: 232.6 ± 26.6 pA, n = 33, p < 0.0001 relative to control; 30 min: 252.4 ± 24.7 pA, n = 22, p < 0.001 relative to control). In stargazer SCs, average eEPSC amplitudes following HFS were also significantly depressed (Fig. 3B,D) (control: 296.6 ± 36.0 pA, n = 16; 15 min: 223.6 ± 36.7 pA, n = 16, p < 0.0001 relative to control; 30 min: 276.2 ± 36.9 pA, n = 10, p < 0.005 relative to control). These data suggest that stargazer SCs are not impaired in their ability to exhibit depression in eEPSC amplitude at −60 mV following HFS of PFs. A caveat, however is that the depression at −60 mV may also have a presynaptic component. Previous work has shown that PF-SC synapses can also exhibit activity-dependent synaptic plasticity characterized by entirely presynaptic forms of long-term depression (Rancillac and Crépel, 2004; Soler-Llavina and Sabatini, 2006). Nevertheless, a change in presynaptic release probability cannot readily account for activity-dependent changes in rectification.
Figure 3.

Synaptic plasticity in rectification is impaired at PF-SC synapses in stargazer mice. A, Representative experiment illustrating synaptic plasticity in the RI of AMPAR-mediated eEPSCs at PF-SC synapses in a wild-type mouse. Traces represent currents at −60, 0, and +40 mV recorded in the presence of picrotoxin and D-AP5. The control trace, measured 5 min before HFS, had an RI of 0.34. Following HFS (5 × 100 pulses at 50 Hz), the RI values were elevated to 0.48 (after 15 min) and 0.50 (after 30 min). B, Representative eEPSCs from a stargazer mouse showing the failure to elevate RI following HFS. C, Line graph showing depression in the amplitude of wild-type eEPSCs at −60 mV 15 and 30 min following HFS (control: 294.2 ± 25.1 pA, n = 33; 15 min: 232.6 ± 26.6 pA, n = 33; 30 min: 252.4 ± 24.7 pA, n = 22). Average eEPSC amplitudes following HFS, presented as absolute values, were significantly depressed relative to control (15 min: p < 0.0001 relative to control; 30 min: p < 0.001 relative to control, paired Wilcoxon rank sum test). D, Stargazer eEPSCs at −60 mV showed a similar depression following HFS (control: 296.6 ± 36.0 pA, n = 16; 15 min: 223.6 ± 36.7 pA, n = 16; 30 min: 276.2 ± 36.9 pA, n = 10). Average eEPSC amplitudes following HFS, presented as absolute values, were significantly depressed relative to control (15 min: p < 0.0001 relative to control; 30 min: p < 0.005 relative to control, paired Wilcoxon rank sum test). E, Line graph demonstrating a significant change in the average RI of eEPSCs of wild-type PF-SC synapses 15 and 30 min following HFS (control: 0.44 ± 0.03 pA, n = 33; 15 min: 0.52 ± 0.03 pA, n = 33; 30 min: 0.51 ± 0.04 pA, n = 22). Average RI values of eEPSCs following stimulation were significantly enhanced relative to control (15 min: p < 0.001 relative to control; 30 min: p < 0.005 relative to control, paired Wilcoxon rank sum test). F, At stargazer PF-SCs, however, average RI values did not show any significant enhancement (control: 0.23 ± 0.03 pA, n = 16; 15 min: 0.23 ± 0.03 pA, n = 16, N.S.; 30 min: 0.22 ± 0.03 pA, n = 10, N.S., paired Wilcoxon rank sum test). Asterisks indicate significance: ***p < 0.0005, **p < 0.005, N.S. is not significant. Note: Average values with SEM are presented for each time point. However, in each condition, a minority of cells failed to reach the 30 min time point, so paired statistics between control and 30 min conditions were only performed on those cells for which three points could be measured.
In terms of plasticity in synaptic AMPAR rectification, however, wild-type and stargazer SCs had divergent phenotypes. In wild-type SCs, HFS of PFs elicited a modest but significant enhancement in RI measured ∼15 and 30 min following the stimulus. Control average RI was 0.44 ± 0.03 (n = 33); after 15 min the average RI had increased to 0.52 ± 0.03 (n = 33) (p < 0.001 relative to control), and after 30 min the average RI had remained elevated at 0.51 ± 0.03 (n = 22) (p < 0.005 relative to control) (Fig. 3A,E). In stargazer SCs however, HFS failed to elicit a significant enhancement in RI. Control average RI was 0.23 ± 0.03 (n = 16); after 15 min the average RI remained stable at 0.23 ± 0.03 (n = 16), and after 30 min the average RI was 0.22 ± 0.03 (n = 10) (Fig. 3B,F). If the enhanced rectification observed in the stargazer mouse is merely the result of reduced excitatory drive from granule cells, then HFS should still have elicited an activity-dependent change in rectification. Thus, the impairment in synaptic plasticity in stargazer SCs suggests that diminished basal excitatory input is unlikely to explain the strong inward rectification of synaptic I–Vs.
Overall, we found that stargazin is necessary for synaptic plasticity in SCs, manifested as an activity-dependent change in the rectification of synaptic I–Vs. In wild-type SCs, eEPSCs at −60 mV were depressed and RI was enhanced following HFS, generally consistent with previous descriptions of PF-SC plasticity (Liu and Cull-Candy, 2000; Gardner et al., 2005). A notable exception, however, is that we rarely observed potentiation of the eEPSC amplitude at +40 mV, making our results inconsistent with the selective insertion of GluA2-containing AMPARs. In our hands, the significant change in RI was accounted for by a preservation and/or slower decline in the eEPSC amplitude at +40 mV than at −60 mV. In the prevailing model of synaptic plasticity in cerebellar SCs, GluR2-containing AMPARs are located extrasynaptically and are poised to diffuse laterally into synapses following HFS, replacing GluR2-lacking AMPARs (Liu and Cull-Candy, 2000, 2005; Gardner et al., 2005; Cull-Candy et al., 2006). Perhaps the enhanced inward rectification of synaptic I–Vs and the absence of an activity-dependent change in rectification evident at stargazer PF-SC synapses can be explained by a diminished supply of extrasynaptic GluA2-containing AMPARs. We therefore examined the properties of extrasynaptic/somatic AMPARs in the stargazer mouse.
Extrasynaptic/somatic AMPARs are preserved in stargazer mice
Given the severe synaptic phenotype we observed in SCs from stargazer mice, we next asked whether the same is true of extrasynaptic/somatic AMPARs and whether it could explain our failure to induce plasticity in the rectification of PF-SC synapses. To address this question, we pulled nucleated outside-out patches from the somata of SCs (see Materials and Methods). We chose to use nucleated patches to sample a larger population of extrasynaptic/somatic AMPARs and hence acquire higher resolution traces of agonist-evoked currents than one could achieve with conventional outside-out patches. Nucleated patches were subjected to the application of 500 μm glutamate in the presence of 100 μm CTZ at a membrane potential of −60 mV. In contrast to synaptic recordings, we were surprised to find that nucleated patches from stargazer SCs did not exhibit any significant deficit in the amplitude of glutamate/CTZ-evoked currents compared with those from wild-type SCs. The average amplitude of glutamate/CTZ-evoked currents from wild-type nucleated patches was 394.9 ± 35.5 pA (n = 56), while that of stargazer mice was 330.3 ± 28.3 pA (n = 43) (Fig. 4A,B). These data indicate that the synaptic deficit in AMPAR-mediated transmission in stargazer mice is not secondary to a reduction in the pool of extrasynaptic receptors and that AMPARs are fully capable of being trafficked to the extrasynaptic/somatic membrane compartment in the absence of stargazin but are impaired in their ability to be targeted to synapses.
Figure 4.

Extrasynaptic/somatic AMPARs are preserved in stargazer mice and stargazin is the only type I TARP expressed in SCs. A, Typical traces of glutamate/CTZ-evoked currents recorded from nucleated patches pulled from wild-type (left) and stargazer (right) SCs at a holding potential of −60 mV. Glutamate (500 μm) application is represented by gray bars while CTZ (100 μm) is present in all solutions and is represented by white bars. B, Bar plot representing collected amplitude of glutamate/CTZ-evoked currents from nucleated patch recordings, presented as absolute values. No significance difference was found between wild-type (wt) (average amplitude = 394.9 ± 35.5 pA, n = 56) and stargazer (stg) (average amplitude = 330.3 ± 28.3 pA, n = 43, N.S., Wilcoxon rank sum test). C, Effect of bath application of CNQX (10 μm) and TCM (400 μm) in whole-cell recordings from SCs in cerebellar slices from wild-type and stargazer mice at a holding potential of −60 mV. Control ACSF contained picrotoxin, d-AP5, and TTX. Bath application of CNQX/TCM consistently elicited an inward current from wild-type SCs but failed to elicit any current from stargazer SCs. Error bars represent SEM. D, Bar plot of the amplitude of holding current changes elicited by CNQX, presented as absolute values (wt: 78.6 ± 6.3 pA, n = 7; stg: 1.0 ± 0.4 pA, n = 3; p < 0.05, Wilcoxon rank sum test). E, Representative traces of currents evoked by 500 μm kainate (black bars) and 500 μm glutamate (gray bars) in single nucleated patches from wild-type (left) and stargazer (right) SCs at a holding potential of −60 mV. CTZ is present in all solutions and is represented by white bars. F, Bar plots showing the average KA/Glu ratios from nucleated patch recordings. KA/Glu ratios from stargazer mice (average KA/Glu ratio: 0.67 ± 0.02, n = 12) exhibited a modest but significant decrease relative to wild type (average KA/Glu ratio = 0.78 ± 0.02, n = 6; p < 0.001, Wilcoxon rank sum test). Asterisks indicate significance: **p < 0.005, *p < 0.05, N.S. is not significant.
Might the trafficking of AMPARs to extrasynaptic/somatic sites require another TARP family member? To address this question we turned to two pharmacological tools that have been developed based on the ability of TARPs to modulate AMPAR agonist efficacy. Previous work has shown that CNQX, a commonly used AMPAR antagonist, behaves as a partial agonist only when AMPARs are associated with a TARP family member (Menuz et al., 2007). This behavior is manifested as inward whole-cell current elicited by bath application of CNQX in the presence of TCM, a blocker of AMPAR desensitization. This phenomenon has been shown in a variety of neuronal cell-types as well as in heterologous expression systems where GluA1 homomers are coexpressed with any of the TARP family members γ-2, γ-3, γ-4, or γ-8 (Menuz et al., 2007). Using these same methods, we found that bath application of 10 μm CNQX and 400 μm TCM at a holding potential of −60 mV elicited an inward current in wild-type SCs with an average amplitude of 78.6 ± 6.3 pA (n = 7) (Fig. 4C), consistent with results from other neurons. In stargazer SCs however, no inward current was evident (n = 3) (Fig. 4D), suggesting that stargazin fully accounts for the complement of canonical or type I TARPs (Kato et al., 2008) present in SCs at the whole-cell level.
Another informative metric for TARP association is the KA/Glu ratio. In the presence of type I TARPs, the efficacy of the AMPAR partial agonist kainate is greatly enhanced (Tomita et al., 2005; Turetsky et al., 2005). The KA/Glu ratio has since been shown to be a useful tool in determining not just the presence or absence of TARPs but for estimating AMPAR-TARP stoichiometry (Shi et al., 2009). Our prediction for the KA/Glu ratio experiments, based on our results from whole-cell application of CNQX, was that the KA/Glu ratio would be substantially reduced in the stargazer mouse, reflecting near complete loss of TARP-association. In CA1 pyramidal neurons where TARP γ-8 is the primary type I TARP, the KA/Glu ratio from wild-type mice was ∼0.55 whereas in γ-8−/− mice the ratio was ∼0.20, representing a reduction of >60% (Shi et al., 2009). To determine the KA/Glu ratio of extrasynaptic/somatic AMPARs, we pulled nucleated patches from SC somata and sequentially applied 500 μm kainate and 500 μm glutamate, all in the presence of 100 μm CTZ. Surprisingly, we found that the KA-Glu ratio in nucleated patches was only mildly disrupted in the stargazer mouse. The average KA-Glu ratio in wild-type SCs was 0.78 ± 0.02 (n = 6), whereas in stargazer mice it was 0.67 ± 0.02 (n = 12) (Fig. 4E,F). Despite being significantly different (p < 0.001), it represented only a modest ∼14% decline.
Are the remaining AMPARs in the stargazer mouse TARP-associated? Our measurement of whole-cell CNQX-evoked current in wild-type and stargazer SCs provided clear evidence that stargazin accounts for the entirety of type I TARPs associated with synaptic and extrasynaptic AMPARs in SCs. However, a paradox arises when comparing the binary nature of the CNQX result with the surprisingly subtle effects on KA/Glu ratio and mEPSC decay kinetics (Fig. 1). Although both measures indicate that TARP association is perturbed, it was not consistent with the complete absence of TARPs. The gating mechanisms governing the TARP-dependent enhancement of agonist efficacy have been hypothesized to share a common structural mechanism in the modulation of the ligand-binding domains (Milstein and Nicoll, 2008). The current data suggest, at least in SCs, that these effects are dissociable. A plausible explanation for these data is that another protein, possibly a noncanonical or type II TARP (Kato et al., 2008), is expressed in SCs and shares the property of type 1 TARPs in significantly enhancing kainate efficacy but is incapable of converting CNQX into a partial agonist or effectively trafficking AMPARs to synapses (see Discussion).
Extrasynaptic/somatic AMPARs in stargazer mice exhibit changes in rectification index
To define the subunit composition of extrasynaptic/somatic AMPARs in nucleated patches from stargazer mice, we applied a voltage ramp from −100 to +100 mV over 100 ms during the application of 500 μm glutamate and 100 μm CTZ. I–V curves were acquired for each nucleated patch following subtraction of the average control trace from the average glutamate/CTZ application trace (Fig. 5A). RI was calculated using slope ratios from point measurements at −60, 0, and +40 mV on the subtracted current trace (see Materials and Methods) so that RI values could be directly compared with synaptic RI values. We found that wild-type extrasynaptic I–Vs were moderately inwardly rectifying with an average RI value of 0.38 ± 0.01 (n = 51), whereas extrasynaptic I–Vs in stargazer SCs exhibited a significantly lower average RI value of 0.24 ± 0.02 (n = 38; p < 0.0005 relative to wild type) (Fig. 5C and D). Similar to synaptic AMPARs, the subunit composition of extrasynaptic AMPARs is moderately rectifying, consistent with a mix of GluA2-lacking and GluA2-containing AMPARs. Also, like synaptic AMPARs, extrasynaptic AMPARs in the stargazer mouse are significantly more rectifying. These data reflect either a specific impairment in the ability of stargazer SCs to traffic GluA2-containing AMPARs to the cell surface and to synapses, thus explaining our failure to induce synaptic plasticity at PF-SC synapses in stargazer mice or a TARP-dependent change in the affinity of the AMPAR pore for intracellular spermine (Soto et al., 2007).
Altogether, we found that SCs exhibit a relatively homogeneous distribution of AMPARs with mixed subunit composition between synaptic and extrasynaptic compartments. This is a striking departure from previous reports, which proposed a dramatic compartmentalization based on subunit composition (Liu and Cull-Candy, 2000, 2002; Gardner et al., 2005). These studies showed small amplitude (∼0.5–10 pA), agonist-evoked currents from somatic outside-out patches, the I–Vs of which varied from linear to strongly outwardly rectifying. In addition, this compartmentalization is a component of the prevailing model of synaptic plasticity in SCs (Liu and Cull-Candy, 2000; Cull-Candy et al., 2006). Formally, one possible explanation for the discrepancy between our data and previous reports is that we are sampling synaptic AMPARs in addition to somatic AMPARs. This is unlikely for several reasons. First, conventional outside-out patches in our hands were similarly inwardly rectifying (Fig. 5B), with an average RI of 0.31 ± 0.07 (n = 5). In addition, we did not find a single nucleated patch (out of 51) from wild-type SCs, which exhibited an I–V curve that approached linearity (Fig. 5C). Finally, electron microscopic analysis indicates that there are few if any glutamatergic synapses on the somata of mouse cerebellar SCs (Lemkey-Johnston and Larramendi, 1968).
Defining AMPAR subunit composition using use-dependent block by PhTx-433
Recent work has shown that GluA2-lacking AMPARs associated with stargazin exhibit a reduced affinity for intracellular spermine (Soto et al., 2007) suggesting that AMPAR channel properties are profoundly influenced by the presence or absence of TARPs and that the use of RI may not be an unambiguous metric for GluA2-content. As an alternative method for determining the GluA2-context of SC AMPARs, we turned to the use of polyamine toxins, which have been shown to selectively block GluA2-lacking AMPARs in a use-dependent manner (Blaschke et al., 1993; Brackley et al., 1993; Herlitze et al., 1993; Washburn and Dingledine, 1996). We chose to use PhTx-433, a polyamine toxin commonly used to block synaptic GluA2-lacking AMPARs in brain slice preparations (Tóth and McBain, 1998; Terashima et al., 2004; Ho et al., 2007). Early characterization of the mechanism of action of PhTx-433 in Xenopus oocytes showed that kainate-evoked currents through GluA3 homomers were selectively blocked by PhTx-433 with relatively low affinity (IC50 ∼1 μm) (Washburn and Dingledine, 1996). The minimal concentration required for complete block of GluA2-lacking AMPARs without affecting heteromeric GluA2-containing AMPARs was found to be ∼10 μm PhTx-433. Kainate-evoked currents through GluA1 homomers were similarly blocked by 10 μm PhTx-433 (IC50 ∼3 μm) (Brackley et al., 1993). We therefore began by using 10 μm PhTx-433 as a way of quantifying GluA2-content of AMPAR-mediated current in nucleated patches from wild-type SCs. For this purpose we obtained both an RI value from voltage ramps and extent of block by 10 μm PhTx-433 in single nucleated patches. Our prediction was that if 10 μm PhTx-433 was selective for block of GluA2-lacking AMPARs, there would be a component of moderately rectifying glutamate/CTZ-evoked current that would be PhTx insensitive and would roughly correlate with RI. RI values from a subset of wild-type SC nucleated patches were moderately rectifying with an average RI of 0.33 ± 0.01 (n = 7), suggesting that some component of the current arises from GluA2-containing AMPARs (Fig. 6A,B). We were therefore puzzled to find that in every case the current was rapidly and completely blocked by 10 μm PhTx-433 (average remaining current = 1.6 ± 0.6%; n = 7) (Fig. 6A,B). These data led us to consider one of two possibilities; either AMPARs in SCs are indeed entirely GluA2 lacking and the I–V gave us a spurious measure of GluA2-content, or that 10 μm PhTx-433 was blocking nonselectively and is, in fact, a poor measure of GluA2 content.
Figure 6.

Calibrating the selectivity of use-dependent block of GluA2-lacking AMPARs by PhTx-433 using GluA2−/− mice. A, Typical example of a nucleated patch from a wild-type SC that was subjected to both a voltage ramp (left) and block by 10 μm PhTx-433 (right). The RI value was 0.36, yet the current was rapidly and completely blocked by 10 μm PhTx-433. B, Bar graph showing RI values of wild-type SC nucleated patches subjected to block by 10 μm PhTx-433 (average RI = 0.33 ± 0.01, n = 7) (left). Bar graph showing remaining current (%) in 10 μm PhTx-433 in the same patches (average remaining current = 1.6 ± 0.6%, n = 7) (right). Open circles represent values from individual cells. C, Representative examples of glutamate/CTZ-evoked currents in nucleated patches from GluR2−/− SCs blocked by varying concentrations of PhTx-433 (1, 3, and 100 nm). Note rapid and complete block using 100 nm PhTx-433. D, Representative example of an outside-out patch recording from a hippocampal CA1 pyramidal neuron where glutamate/CTZ-evoked current was untouched by 100 nm PhTx-433. E, Normalized I–V curves from nucleated patches pulled from GluR2−/− SCs. Individual curves are shown in gray and the average curve is in black (left). Bar graph shows average RI of GluR2−/− I–V curves (average RI = 0.11 ± 0.01, n = 19) (right). F, Bar graph representing the percentage glutamate/CTZ-evoked current remaining following block by progressively increasing concentrations of PhTx-433: 1 nm (percentage remaining = 41.9 ± 8.5%, n = 4); 3 nm (percentage remaining = 20.1 ± 2.7%, n = 10); 10 nm (percentage remaining = 4.8 ± 0.3%, n = 4); 30 nm (percentage remaining = 2.2 ± 0.2%, n = 3); and 100 nm (percentage remaining = 0%, n = 2).
To resolve this issue, we turned to GluA2 knock-out mice (GluA2−/−) (Jia et al., 1996) where, by definition, SCs would express a pure population of GluA2-lacking AMPARs. If 10 μm PhTx-433 is blocking heteromeric GluA2-containing AMPARs nonselectively, we could use SC nucleated patches to calibrate the concentration of PhTx-433 required to completely and selectively block GluA2-lacking AMPARs. As expected, the AMPAR-mediated currents in GluA2−/− SCs showed strong inward rectification (average RI = 0.11 ± 0.01; n = 19) (Fig. 6E). Remarkably, we found PhTx-433 to be a far more potent blocker of GluA2-lacking AMPARs than previously described. Even 1 nm PhTx-433 was capable of blocking, on average, almost 60% of the current (Fig. 6C,F). We found that the minimal concentration of PhTx-433 required to completely block GluA2-lacking receptors was 100 nm (Fig. 6C,F). This concentration had no effect on currents generated by GluA2-containing receptors from wild-type hippocampal pyramidal neurons (Fig. 6D,F), consistent with previous work (Lu et al., 2009).
Stargazer mice do not exhibit a specific defect in GluA2 trafficking
Armed with this information, we set out to precisely quantify GluA2 content by directly comparing PhTx-433 sensitivity with RI in single nucleated patches from wild-type and stargazer SCs (Fig. 7A,B). To our surprise, we found that although RI was significantly lower in SCs from stargazer mice as expected (wild type, average RI = 0.40 ± 0.02, n = 19; stargazer, average RI = 0.27 ± 0.02, n = 15; p < 0.0001) (Fig. 7C), there was no difference in PhTx-433 sensitivity (wild type, average remaining current = 21.0 ± 2.4%, n = 19; stargazer, average remaining current = 21.2 ± 3.3%, n = 15; P = N.S.) (Fig. 7D). GluA2 content in both wild-type and stargazer SCs, as precisely measured by block with 100 nm PhTx-433 block, were ∼20%. These results suggest that the dramatic changes in rectification we observed in AMPARs from stargazer SCs are unlikely to be explained by a difference in subunit composition and therefore are not the result of a specific defect in the trafficking of GluA2-containing AMPARs.
Figure 7.
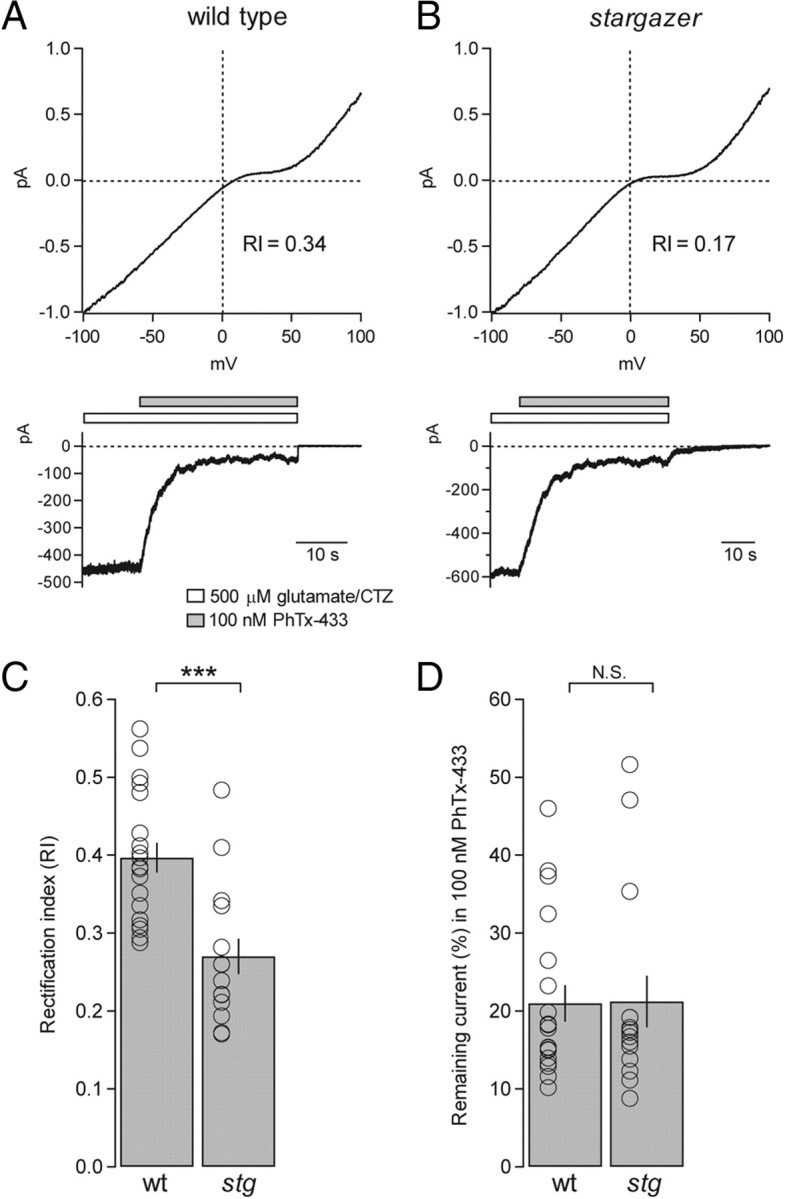
Using PhTx-433 to quantify GluA2-content in nucleated patches from wild-type and stargazer SCs. A, Representative example of a nucleated patch from a wild-type SC that was subjected to both a voltage ramp (top) and block by 100 nm PhTx-433 (bottom). In this case, the RI value was 0.34 and the current that remained unblocked by 100 nm PhTx-433 was 10.1%. B, In a representative example of a nucleated patch from a stargazer SC, the RI value was 0.17 and the remaining current was 12.2%. C, Bar graph comparing RI values from wild-type and stargazer SC nucleated patches, also subjected to block by 100 nm PhTx-433. Consistent with data from Figure 5, the average RI of wild-type (wt) SCs was significantly higher than that of stargazer (stg) SCs (wt: average RI = 0.40 ± 0.02, n = 19; stg: average RI = 0.27 ± 0.02, n = 15; p < 0.0001, Wilcoxon rank sum test). D, Bar graph showing that despite the significant difference in RI between wild-type and stargazer RIs, the current that remained unblocked in 100 nm PhTx-433 was not significantly different (wt: average remaining current = 21.0 ± 2.4%, n = 19; stg: average remaining current = 21.2 ± 3.3%, n = 15; N.S., Wilcoxon rank sum test). Open circles represent values from individual cells. Asterisks indicate significance, ***p < 0.0005, N.S. is not significant.
To probe the GluA2 content of synaptic AMPARs using PhTx-433, we again turned to the GluA2−/− mouse. We began by comparing the ability of 100 nm PhTx-433 to block synaptic GluA2-lacking AMPARs by first determining synaptic RI and then bath applying PhTx (Fig. 8A). We found that although 100 nm PhTx-433 rapidly and completely blocked a pure population of GluA2-lacking AMPARs when applied to nucleated patches, it was ineffective at blocking synaptic GluA2-lacking AMPARs (average remaining current = 90.4 ± 9.3%; n = 3) (Fig. 8C). Subsequently we turned to 10 μm PhTx-433, which, despite blocking rapidly but nonselectively when applied to patches, was relatively effective at blocking synaptic GluA2-lacking AMPARs (average remaining current = 28.2 ± 7.3%; n = 4) (Fig. 8B,C) and has been shown to have no effect on GluA2-containing synaptic AMPAR from wild-type hippocampal CA1 pyramidal neurons (Adesnik and Nicoll, 2007). The dramatic differences in the efficacy of PhTx-433 block between the two preparations can be explained by the mode of application, the mechanism of channel activation, and accessibility of the toxin to the channel. The rate and extent of block of synaptic AMPARs by a use-dependent polyamine blocker like PhTx-433 will be a function of the rate of stimulation (Mainen et al., 1998; Zaitsev et al., 2011) and all the variables that determine EPSC time course, which include features of presynaptic release, glutamate concentration in the cleft, and AMPAR deactivation and desensitization kinetics (Jonas, 2000). Nevertheless, we next attempted to define GluA2-content of wild-type and stargazer PF-SC synapses by directly comparing synaptic RI values with the extent of block by 10 μm PhTx-433 (Fig. 8D,E). Consistent with our results from nucleated patches, we found that wild-type and stargazer synaptic AMPARs did not exhibit a significant difference in PhTx-433 sensitivity (wild type: average remaining current = 65.7 ± 4.8%, n = 10; stargazer: average remaining current = 57.8 ± 3.8%, n = 7; N.S.) (Fig. 8G) despite a significant difference in RI (wild type: average RI = 0.45 ± 0.05, n = 10; stargazer: average RI = 0.24 ± 0.03, n = 7; p < 0.05) (Fig. 8F). These results reinforce the notion that the rectification change we observed in synaptic AMPARs from stargazer SCs is unlikely to be the result of a difference in subunit composition and more likely to be explained by a difference in the pore properties of AMPARs conferred by the presence of stargazin.
Figure 8.
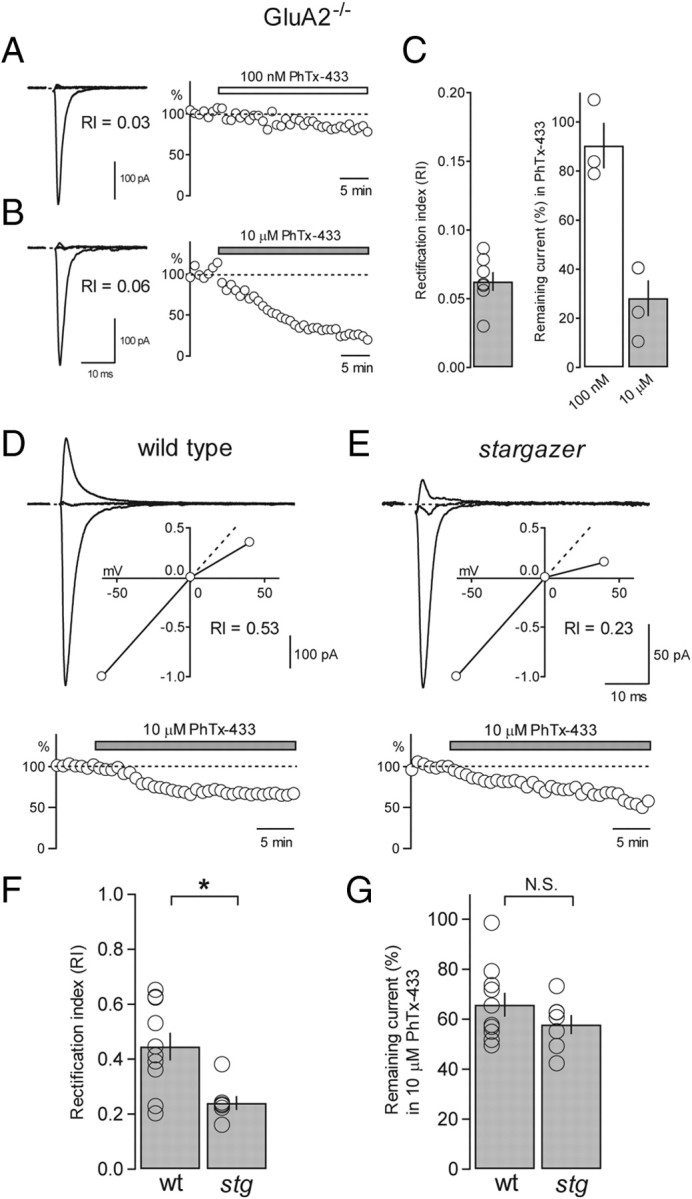
Using PhTx-433 to quantify GluA2 content of synaptic AMPARs from wild-type and stargazer SCs. A, Overlay of synaptic eEPSCs from a GluA2−/− SC (−60, 0, and +40 mV; RI = 0.03) with stimulus artifacts blanked (left). Time course of block of the same cell subjected to bath application of 100 nm PhTx-433 (remaining current = 83.6%) at a holding potential of −60 mV (right). B, Synaptic eEPSCs from another GluA2−/− SC (RI = 0.06) (left). The same cell subjected to bath application of 10 μm PhTx-433 (remaining current = 22.2%) (right). C, Bar graph showing average RI of synaptic I–V curves from GluR2−/− SCs (average RI = 0.06 ± 0.01, n = 7) (left). Bar graph comparing the average remaining current (%) following bath application of 100 nm PhTx (white bar; average remaining current = 90.4 ± 9.3%, n = 3), and 10 μm PhTx (gray bar; average remaining current = 28.2 ± 7.3%, n = 4) (right). D, Overlay of eEPSCs from a wild-type SC with an inset showing the normalized I–V (RI = 0.53) (top). Time course of block of the same cell subjected to bath application of 10 μm PhTx-433 (remaining current = 65.2%) (bottom). E, eEPSCs from a stargazer SC with an inset showing the normalized I–V (RI = 0.23) (top). Time course of block of the same cell subjected to bath application of 10 μm PhTx-433 (remaining current = 55.0%) (bottom). F, Bar graph comparing collected RI values from wild-type (wt) and stargazer (stg) PF-SC synapses, also subjected to bath application of 10 μm PhTx-433 (wt: average RI = 0.45 ± 0.05, n = 10; stg: average RI = 0.24 ± 0.03, n = 7; p < 0.05, Wilcoxon rank sum test). G, Bar graph showing that the current that remained unblocked in 10 μm PhTx-433 was not significantly different between conditions (wt: average remaining current = 65.7 ± 4.8%, n = 10; stg: average remaining current = 57.8 ± 3.8%, n = 7; N.S., Wilcoxon rank sum test). Open circles represent values from individual cells. Asterisks indicate significance, *p < 0.05. N.S., not significant.
Discussion
In stargazer mutant mice we showed that despite the severe loss of synaptic AMPARs, extrasynaptic AMPARs are largely preserved, suggesting that stargazin is essential for the synaptic localization of AMPARs but not for surface delivery, thus implicating stargazin in compartment-specific AMPAR trafficking. We also found that activity-dependent synaptic plasticity in AMPAR rectification is impaired in stargazer mice. Although, both synaptic and extrasynaptic AMPAR I–Vs in stargazer SCs are more strongly inwardly rectifying than in wild-type SCs, using PhTx-433 to quantify GluA2-content of both extrasynaptic and synaptic AMPARs allowed us to determine that GluA2-content is in fact unchanged in the stargazer mouse. These data suggest that the changes in RI seen in stargazer mice are unlikely to be the result of a specific defect in GluA2 trafficking but are more likely attributable to a TARP-dependent change in AMPAR gating.
The role of TARP family members in the regulation of synaptic and extrasynaptic AMPARs
Our result indicating that stargazin is essential for the synaptic localization of AMPARs in SCs is consistent with the notion that the expression of one or more TARP family members is crucial for AMPAR function in the CNS (Nicoll et al., 2006; Coombs and Cull-Candy, 2009; Kato et al., 2010). In stargazer cerebellar granule cells, where stargazin is likely the only type I TARP family member expressed, both synaptic and extrasynaptic AMPARs are virtually absent (Hashimoto et al., 1999; Chen et al., 2000). In sharp contrast, stargazer cerebellar Golgi cells do not exhibit any significant defect in AMPAR-mediated transmission. Severe deficits only emerged when TARP γ-3 was also knocked out, suggesting that stargazin and γ-3 are capable of compensating for the loss of the other, despite the presence of remaining TARP γ-7 (Menuz et al., 2008). Purkinje cells, which express both stargazin and γ-7 at high levels (Fukaya et al., 2005; Lein et al., 2007), exhibit reductions in AMPAR-mediated synaptic transmission, but no change in agonist-evoked currents from somatic patches, in stargazer mice (Menuz and Nicoll, 2008). Furthermore, transgenic mice lacking both stargazin and γ-7 exhibit profound deficits in AMPAR transmission (Yamazaki et al., 2010). Similar compensatory effects by TARP subtypes have been described in other brain regions (Menuz et al., 2009). Together, these data suggest that when stargazin is the only type I TARP family member expressed in a given cell type, both surface and synaptic AMPAR trafficking are severely impaired. But when stargazin is expressed along with a type II TARP, the latter may be capable of trafficking receptors to extrasynaptic sites but is much less effective at carrying out synaptic targeting in the absence of stargazin. In fact, one possible explanation for our paradoxical measurements of CNQX-evoked current, kainate efficacy (Fig. 4),and mEPSC decay kinetics (Fig. 1) in stargazer SCs may be that a type II TARP like γ-7 is at play in SCs and is capable of enhancing kainate efficacy but fails to facilitate CNQX current. At the moment, there is no clear evidence that γ-7 can mediate CNQX-evoked current through AMPARs, but γ-7 is capable of boosting kainate efficacy and slowing deactivation and desensitization kinetics (Kato et al., 2007), making it a reasonable candidate as a remaining TARP in stargazer SCs. Indeed, γ-7 displays low to moderate expression in SCs (Fukaya et al., 2005; Lein et al., 2007).
An informative comparison can also be made with hippocampal CA1 pyramidal neurons, where stargazin and TARPs γ-3, γ-4, γ7,and γ-8 are expressed (Fukaya et al., 2005; Lein et al., 2007). Although γ-8 is the predominant type I TARP in pyramidal neurons (Rouach et al., 2005), as stargazin is in SCs, their loss affects each cell type differently. Whereas γ-8−/− pyramidal neurons only showed a modest reduction in synaptic AMPARs, they exhibited a near complete loss of extrasynaptic AMPARs (Rouach et al., 2005). Stargazer SCs, however, exhibit the opposite phenotype: near complete loss of synaptic AMPARs without any significant effect on extrasynaptic receptors. Given this remarkable compartmentalization, it is tempting to speculate that distinct TARP subtype-dependent pathways regulate AMPAR trafficking to different intracellular compartments. The cytoplasmic C-tails of AMPARs (Ziff, 2007) and/or newly characterized AMPAR auxiliary subunits (Tomita, 2010; Díaz, et al., 2010; Guzman and Jonas, 2010) may also have a role to play in TARP-independent AMPAR trafficking.
Refining the use of PhTx-433 block as a metric for GluA2-content
Neuronal GluA2-content is generally determined by one of two methods: (1) measurements of RI from I–V relationships; and (2) use-dependent block by extracellular polyamine-based compounds (Strømgaard and Mellor, 2004; Strømgaard et al., 2005). AMPAR RI has been used as a measure of GluA2 content but is subject to several confounding variables. First, stargazin association has been shown to substantially change the rectification of heterologously expressed GluA2-lacking AMPARs owing to TARP-dependent changes in spermine affinity (Soto et al., 2007). Second, given the diversity of TARP subtype-dependent effects on mEPSC kinetics (Milstein et al., 2007, Cho et al., 2007) and other gating characteristics (Suzuki et al., 2008; Kott et al., 2007, 2009), it is conceivable that different TARP subtypes will have differential effects on RI, independent of GluA2 content. Third, intracellular polyamine metabolism has been shown to be dynamically regulated, resulting in changes in rectification that may not reflect GluA2-content (Aizenman et al., 2002; Shin et al., 2005, 2007). PhTx-433 and related compounds are frequently used as a relative measure of GluA2 content in native systems. Previous characterizations of the dose–response relationship of PhTx-433 showed that the minimal concentration required for complete and selective block of homomeric GluA2-lacking AMPARs is 10 μm (Brackley et al., 1993; Washburn and Dingledine, 1996). We found, however, that 100 nm PhTx-433 provided a much better estimate of GluA2 content. A likely explanation for this discrepancy is that previous characterizations examined the PhTx sensitivity of AMPAR-mediated currents activated by the partial agonist kainate (Washburn and Dingledine, 1996; Brackley et al., 1993), whereas we used the full agonist glutamate. Indeed, recent work from our laboratory shows that PhTx-433 block is actually a steep function of agonist efficacy and TARP association, although TARP association does not affect block of GluA2-lacking AMPARs in the presence of a full agonist (Jackson et al., unpublished data). In the past, the enhanced rectification of both synaptic and extrasynaptic I–Vs that we observed in stargazer SCs would have been unequivocal evidence of a change in subunit composition. However, recent work (Soto et al., 2007) casts doubt on a linear relationship between RI and GluA2-content. The present data demonstrate that I–V rectification, as well as block by commonly used concentrations of polyamine-based toxins, can each be misleading metrics for GluA2 content. Our use of a calibrated concentration of PhTx in nucleated patches allowed us to show, in a native system, that voltage-dependent block by intracellular polyamines can be dissociable from use-dependent block by external polyamine toxins.
Plasticity at PF-SC synapses
The prevailing model of plasticity at PF-SC synapses involves an activity-dependent change in the GluA2 content of synaptic AMPARs based on observed changes in rectification (Liu and Cull-Candy, 2000; Cull-Candy et al., 2006). However, our data and those of others (Soto et al., 2007) show that stargazin association itself can directly modulate AMPAR rectification independently of GluA2 content. Formally, this leaves several distinct possibilities that could explain the AMPAR plasticity in rectification: (1) subunit-selective AMPAR trafficking by stargazin in which GluA2-lacking AMPARs are selectively removed and GluA2-containing AMPARs are retained at synapses following HFS; (2) nonselective AMPAR trafficking by stargazin in which stargazin anchors AMPARs at synapses, regardless of their subunit composition, while HFS acts to destabilize AMPARs not associated with stargazin.
We found little evidence for subunit-selective trafficking by stargazin despite an apparent change in rectification. Based on our data, we favor a simpler model in which stargazin traffics AMPARs in a largely nonselective manner and anchors them at synapses. HFS may then trigger the destabilization and removal of a subpopulation of AMPARs that are not associated with stargazin. This process would leave more strongly anchored AMPARs that are less rectifying. In the stargazer mouse, synaptic AMPARs would be removed from synapses following stimulation without a change in rectification. The AMPARs that are not associated with stargazin may either be TARP-less or associated with a remaining type II TARP like γ-7, which has an atypical PDZ-binding domain and has been shown to bind only weakly to membrane-associated guanylate kinase proteins such as PSD-95 (Kato et al., 2007), likely leaving them poorly anchored at synapses. This process may also involve other protein–protein interactions (Liu and Cull-Candy, 2005; Gardner et al., 2005), although our data are inconsistent with the synaptic delivery of GluA2-containing AMPARs from extrasynaptic pools.
Related forms of plasticity have been characterized in various brain regions that manifest as changes in synaptic AMPAR rectification. Aside from variations of postsynaptic plasticity at PF-SC synapses (Sun and Liu, 2007; Kelly et al., 2009; Lu et al., 2009), qualitatively similar forms of plasticity have been shown in the ventral tegmental area (Bellone and Lüscher, 2005, 2006) and at mossy fiber-CA3 pyramidal cell synapses in the hippocampus (Ho et al., 2007). In addition, developmental changes in AMPAR rectification have been reported throughout the CNS (Kumar et al., 2002; Shin et al., 2005, 2007; Ho et al., 2007). It is of considerable interest to determine whether these changes observed during development and plasticity involve TARP subtype-selective streams for trafficking GluA2-lacking and GluA2-containing AMPARs and/or that TARP-dependent modulation of spermine affinity contribute to the observed changes in rectification.
Footnotes
A.C.J. is supported by Ruth L. Kirschstein National Research Service Award F32MH081430 from the National Institute of Mental Health (NIMH). R.A.N. is supported by grants from the NIMH. We are grateful to A. Tzingounis, K. Menuz, M. Howard, A. Milstein, A. Priel, A. Chesler, W. Lu, and G. Soler-Llavina and for helpful discussions and comments on the manuscript, all members of the Nicoll Laboratory for helpful discussions, and K. Bjorgan and M. Cerpas for technical support.
References
- Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman CD, Muñoz-Elias G, Cline HT. Visually driven modulation of glutamatergic synaptic transmission is mediated by the regulation of intracellular polyamines. Neuron. 2002;34:623–634. doi: 10.1016/s0896-6273(02)00674-8. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci. 2005;21:1280–1288. doi: 10.1111/j.1460-9568.2005.03979.x. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit-specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proc Natl Acad Sci U S A. 1993;90:6528–6532. doi: 10.1073/pnas.90.14.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochet P, Audinat E, Lambolez B, Crépel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Brackley PT, Bell DR, Choi SK, Nakanishi K, Usherwood PN. Selective antagonism of native and cloned kainate and NMDA receptors by polyamine-containing toxins. J Pharmacol Exp Ther. 1993;266:1573–1580. [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci. 2005;8:1310–1318. doi: 10.1038/nn1534. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Cho CH, St-Gelais F, Zhang W, Tomita S, Howe JR. Two families of TARP isoforms that have distinct effects on the kinetic properties of AMPA receptors and synaptic currents. Neuron. 2007;55:890–904. doi: 10.1016/j.neuron.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs ID, Cull-Candy SG. Transmembrane AMPA receptor regulatory proteins and AMPA receptor function in the cerebellum. Neuroscience. 2009;162:656–665. doi: 10.1016/j.neuroscience.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Díaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32:261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Yamazaki M, Sakimura K, Watanabe M. Spatial diversity in gene expression for VDCC-gamma subunit family in developing and adult mouse brains. Neurosci Res. 2005;53:376–383. doi: 10.1016/j.neures.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman SJ, Jonas P. Beyond TARPs: the growing list of auxiliary AMPAR subunits. Neuron. 2010;66:8–10. doi: 10.1016/j.neuron.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Fukaya M, Qiao X, Sakimura K, Watanabe M, Kano M. Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J Neurosci. 1999;19:6027–6036. doi: 10.1523/JNEUROSCI.19-14-06027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Raditsch M, Ruppersberg JP, Jahn W, Monyer H, Schoepfer R, Witzemann V. Argiotoxin detects molecular differences in AMPA receptor channels. Neuron. 1993;10:1131–1140. doi: 10.1016/0896-6273(93)90061-u. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channel depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Jonas P. The time course of signaling at central glutamatergic synapses. News Physiol Sci. 2000;15:83–89. doi: 10.1152/physiologyonline.2000.15.2.83. [DOI] [PubMed] [Google Scholar]
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Zhou W, Milstein AD, Knierman MD, Siuda ER, Dotzlaf JE, Yu H, Hale JE, Nisenbaum ES, Nicoll RA, Bredt DS. New transmembrane AMPA receptor regulatory protein isoform, γ-7, differentially regulates AMPA receptors. J Neurosci. 2007;27:4969–4977. doi: 10.1523/JNEUROSCI.5561-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Siuda ER, Nisenbaum ES, Bredt DS. AMPA receptor subunit-specific regulation by a distinct family of type II TARPs. Neuron. 2008;59:986–996. doi: 10.1016/j.neuron.2008.07.034. [DOI] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES, Bredt DS. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat Neurosci. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- Koh DS, Burnashev N, Jonas P. Block of native Ca(2+)-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J Physiol. 1995;486:305–312. doi: 10.1113/jphysiol.1995.sp020813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott S, Werner M, Körber C, Hollmann M. Electrophysiological properties of AMPA receptors are differentially modulated depending on the associated member of the TARP family. J Neurosci. 2007;27:3780–3789. doi: 10.1523/JNEUROSCI.4185-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott S, Sager C, Tapken D, Werner M, Hollmann M. Comparative analysis of the pharmacology of GluR1 in complex with transmembrane AMPA receptor regulatory proteins gamma2, gamma3, gamma4, and gamma8. Neuroscience. 2009;158:78–88. doi: 10.1016/j.neuroscience.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Leitch B, Shevtsova O, Kerr JR. Selective reduction in synaptic proteins involved in vesicle docking and signalling at synapses in the ataxic mutant mouse stargazer. J Comp Neurol. 2009;512:52–73. doi: 10.1002/cne.21890. [DOI] [PubMed] [Google Scholar]
- Lemkey-Johnston N, Larramendi LM. Types and distribution of synapses upon basket and stellate cells of the mouse cerebellum: an electron microscopic study. J Comp Neurol. 1968;134:73–112. doi: 10.1002/cne.901340106. [DOI] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, 2nd, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J Neurosci. 2002;22:3881–3889. doi: 10.1523/JNEUROSCI.22-10-03881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat Neurosci. 2005;8:768–775. doi: 10.1038/nn1468. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca22+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabó G, Zukin RS, Liu SJ. A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat Neurosci. 2010;13:223–231. doi: 10.1038/nn.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Jia Z, Roder J, Malinow R. Use-dependent AMPA receptor block in mice lacking GluR2 suggests postsynaptic site for LTP expression. Nat Neurosci. 1998;1:579–586. doi: 10.1038/2812. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Dingledine R. Heterogeneity of synaptic glutamate receptors on CA3 stratum radiatum interneurones of rat hippocampus. J Physiol. 1993;462:373–392. doi: 10.1113/jphysiol.1993.sp019560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Nicoll RA. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J Neurosci. 2008;28:10599–10603. doi: 10.1523/JNEUROSCI.2732-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Stroud RM, Nicoll RA, Hays FA. TARP auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science. 2007;318:815–817. doi: 10.1126/science.1146317. [DOI] [PubMed] [Google Scholar]
- Menuz K, O'Brien JL, Karmizadegan S, Bredt DS, Nicoll RA. TARP redundancy is critical for maintaining AMPA receptor function. J Neurosci. 2008;28:8740–8746. doi: 10.1523/JNEUROSCI.1319-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Kerchner GA, O'Brien JL, Nicoll RA. Critical role for TARPs in early development despite broad functional redundancy. Neuropharmacology. 2009;56:22–29. doi: 10.1016/j.neuropharm.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Zhou W, Karimzadegan S, Bredt DS, Nicoll RA. TARP subtypes differentially and dose-dependently control synaptic AMPA receptor gating. Neuron. 2007;55:905–918. doi: 10.1016/j.neuron.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- Rancillac A, Crépel F. Synapses between parallel fibres and stellate cells express long-term changes in synaptic efficacy in rat cerebellum. J Physiol. 2004;554:707–720. doi: 10.1113/jphysiol.2003.055871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CA, Leitch B. Phenotype of cerebellar glutamatergic neurons is altered in stargazer mutant mice lacking brain-derived neurotrophic factor mRNA expression. J Comp Neurol. 2005;481:145–159. doi: 10.1002/cne.20386. [DOI] [PubMed] [Google Scholar]
- Rouach N, Byrd K, Petralia RS, Elias GM, Adesnik H, Tomita S, Karimzadegan S, Kealey C, Bredt DS, Nicoll RA. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Sager C, Tapken D, Kott S, Hollmann M. Functional modulation of AMPA receptors by transmembrane AMPA receptor regulatory proteins. Neuroscience. 2009;158:45–54. doi: 10.1016/j.neuroscience.2007.12.046. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lu W, Milstein AD, Nicoll RA. The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron. 2009;62:633–640. doi: 10.1016/j.neuron.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Shen F, Huguenard JR. Polyamines modulate AMPA receptor dependent synaptic responses in immature layer V pyramidal neurons. J Neurophysiol. 2005;93:2634–2643. doi: 10.1152/jn.01054.2004. [DOI] [PubMed] [Google Scholar]
- Shin J, Shen F, Huguenard J. PKC and polyamine modulation of GluR2-deficient AMPA receptors in immature neocortical pyramidal neurons of the rat. J Physiol. 2007;581:679–691. doi: 10.1113/jphysiol.2007.130963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat Neurosci. 2006;9:798–806. doi: 10.1038/nn1698. [DOI] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Kelly L, Farrant M, Cull-Candy SG. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat Neurosci. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Renzi M, Zonouzi M, Farrant M, Cull-Candy SG. Selective regulation of long-form calcium-permeable AMPA receptors by an atypical TARP, gamma-5. Nat Neurosci. 2009;12:277–285. doi: 10.1038/nn.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strømgaard K, Mellor I. AMPA receptor ligands: synthetic and pharmacological studies of polyamines and polyamine toxins. Med Res Rev. 2004;24:589–620. doi: 10.1002/med.20004. [DOI] [PubMed] [Google Scholar]
- Strømgaard K, Jensen LS, Vogensen SB. Polyamine toxins: development of selective ligands for ionotropic receptors. Toxicon. 2005;45:249–254. doi: 10.1016/j.toxicon.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu SJ. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E, Kessler M, Arai AC. The fast kinetics of AMPA GluR3 receptors is selectively modulated by the TARPs gamma 4 and gamma 8. Mol Cell Neurosci. 2008;38:117–123. doi: 10.1016/j.mcn.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24:5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S. Regulation of ionotropic glutamate receptors by their auxiliary subunits. Physiology. 2010;25:41–49. doi: 10.1152/physiol.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- Tóth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Turetsky D, Garringer E, Patneau DK. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci. 2005;25:7438–7448. doi: 10.1523/JNEUROSCI.1108-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Dingledine R. Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther. 1996;278:669–678. [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000;20:4414–4422. doi: 10.1523/JNEUROSCI.20-12-04414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Fukaya M, Hashimoto K, Yamasaki M, Tsujita M, Itakura M, Abe M, Natsume R, Takahashi M, Kano M, Sakimura K, Watanabe M. TARPs gamma-2 and gamma-7 are essential for AMPA receptor expression in the cerebellum. Eur J Neurosci. 2010;31:2204–2220. doi: 10.1111/j.1460-9568.2010.07254.x. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Kim KK, Fedorova IM, Dorofeeva NA, Magazanik LG, Tikhonov DB. Specific mechanism of use-dependent channel block of calcium-permeable AMPA receptors provides activity-dependent inhibition of glutamatergic neurotransmission. J Physiol. 2011 doi: 10.1113/jphysiol.2011.204362. Advance online publication. Retrieved March 3, 2011. PMID: 21282285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziff EB. TARPs and the AMPA receptor trafficking paradox. Neuron. 2007;53:627–633. doi: 10.1016/j.neuron.2007.02.006. [DOI] [PubMed] [Google Scholar]