

Abstract

It is reported that stable glycosyl sulfonium salts can be generated via direct anomeric S-methylation of ethylthio glycosides. Mechanistically, this pathway represents the first step in the activation of thioglycosides for glycosidation, however, it can further allow for the synthesis and isolation of quasi-stable sulfonium ions, representing a new approach for studying these key intermediates.
Existing as the most abundant class of organic compounds, carbohydrates are involved in a myriad of life-sustaining and life-threatening processes.1 While Nature flawlessly and repeatedly executes the glycosylation reaction to yield complex poly- and oligosaccharides,2 chemical installation of the glycosidic linkage remains cumbersome, even with the aid of modern technologies.3-8 In the past three decades, much effort has been dedicated to refining glycosylation reaction conditions.9 However, enhancements resulting from this effort are still not sufficient to control the outcome of many glycosylations. Thioglycosides serve as a prime example of this, even though they have been one of the most studied and applied classes of glycosyl donors.10 Amongst the various methodologies developed for thioglycoside activation, the alkylation pathway is commonly accessed utilizing the methylating reagent, MeOTf.11 It is assumed that the reaction begins with the formation of a glycosyl sulfonium ion (Scheme 1), which is quickly converted into other reaction intermediates, such as an oxacarbenium ion, glycosyl triflate (or a combination thereof), eventually leading to a glycoside. While the latter stages of glycosylation have been extensively studied,12-14 the first activation step has never been proven, nor has the postulated sulfonium ion intermediate of such glycosylation been isolated.
Scheme 1.
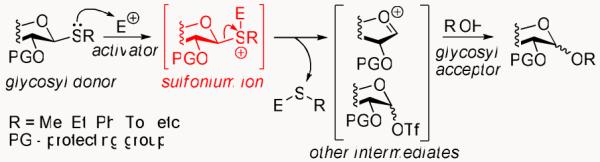
Glycosidation of thioglycosides
Since the pioneering studies by Schuerch et al.15 and Sun et al.,16 there has been an increased interest in the detailed investigation of anomeric sulfonium ions. Synthetic approaches to the formation of identifiable anomeric sulfonium ions, however, are indirect and often lack stereoselectivity, or the products are contaminated with other species (Scheme 2a).17,18 The generation of bicyclic sulfonium ions, wherein the anomeric sulfur is tethered elsewhere to the sugar ring, has proven to be more stereoselective (Scheme 2b), but their preparation requires multiple synthetic steps.19-22 Furthermore, unlike other classes of cyclic sulfonium salts (such as those used as enzyme inhibitors),23 these anomeric reaction intermediates have been found to be relatively labile, existing mainly in situ,24,25 and at low temperatures. Herein, we report a simple and direct method to generate anomerically pure sulfonium salts, such as 2a, that can be isolated, characterized, and stored (Scheme 2c).
Scheme 2.
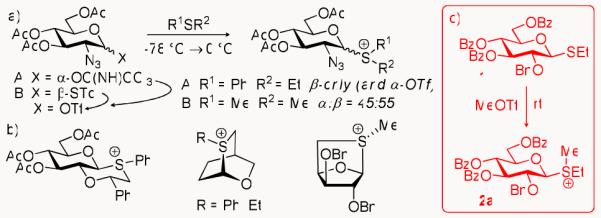
Glycosyl sulfonium ions: (a) obtained via anomeric triflate;17,18 (b) bicyclic sulfonium ions;19-21 (c) obtained via direct methylation of the leaving group (this work).
While expanding our investigations of the O-2/O-5 cooperative effect26 to thioglycosides,27,28 we noted that superdisarmed glycosyl donor 1 provided consistently lower yields in glycosylation in comparison to that of per-acylated glycosyl donor 3.11 Thus, the glycosylations between standard glycosyl acceptor 429,30 and glycosyl donors 1 and 3 (in the presence of MeOTf) were both quenched at 4 h. At this time, it was determined that the reaction between 3 and 4 was complete. However, the progress of the reaction between 1 and 4 was less clear, as there was a significant amount of acceptor 4 remaining, but none of glycosyl donor 1, and so it too was quenched for further investigation. Subsequently, upon analysis of the two glycosylation reactions, it was found that disaccharide 5 (derived from glycosyl donor 1), was only formed in 53% yield, whereas disaccharide 631 (from 3) was obtained in 84% yield (Scheme 3).
Scheme 3.
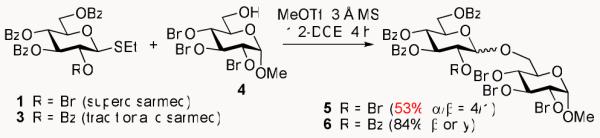
Unexpectedly low yield of disaccharide 5.
In further studying thioglycoside 1 in glycosylation, it was noticed that concomitant with the formation of disaccharide 5, an unusually polar species also formed at the baseline of the TLC plate (ethyl acetate – toluene, 1/9, v/v; for comparison Rf (1) = 0.55). In addition, when investigated in a more polar TLC system (methanol-CH2Cl2, 1/9, v/v), the unknown compound was visualized as an elongated, yet well-defined, spot with an Rf spanning 0.40-0.55. In addition, when subjected to aqueous work-up, it decomposed into the corresponding hemiacetal 7 (shown in Scheme 4). Being significantly more polar than all other reaction components (including 7), and finding it susceptible to hydrolysis, it was hypothesized that this unknown “baseline species” corresponded to anomeric glycosyl sulfonium salt 2a (shown in Schemes 2 and 4), which was formed upon methylation of the thioethyl leaving group. Upon repeating the reaction between 1 and 4, it was found that the reaction required an additional 2 h (6 h total) in order for the baseline spot (2a) to completely disappear/react. Resultantly, disaccharide 5 was isolated in a significantly improved yield of 87%.
Scheme 4.
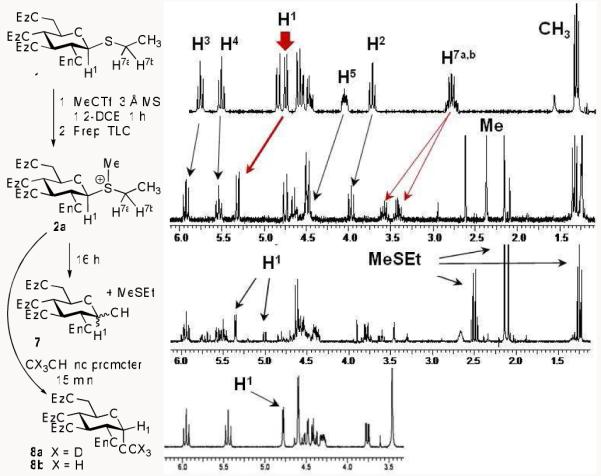
Formation and hydrolysis of salt 2a monitored by 300 MHz 1H NMR in CDCl3.
We next reacted thioglycoside 1 with MeOTf in the absence of the glycosyl acceptor, which resulted in the near exclusive formation of the anticipated sulfonium salt 2a (in about 1 h). The reaction mixture was then concentrated and the residue was purified by preparative TLC (acetone/CH2Cl2, 3.5/6.5, v/v). 1H NMR and mass spectral analyses of the isolated product were consistent with those expected for ethylmethylsulfonium salt 2a. In comparing the 1H NMR spectra of 1 vs. 2a recorded at 300 MHz in CDCl3 (depicted in Scheme 4), a downfield shift on a number of signals was noted. Most significantly, was that of the anomeric H1 signal ( Δδ = 0.59 ppm; while retaining its β-configuration: J1,2 = 9.9 Hz) and the H7a,b signal, which corresponds to the methylene protons of the leaving group ( Δδ = 0.75 ppm). The appearance of a new singlet at 2.44 ppm was consistent with the newly acquired methylthio group. Several other downfield shifts were also noticed, including those of the H2, H3 and H5 protons. The mass spectrum of 2a exhibited an ion peak at m/z 641.2219 (calculated for C37H37O8S+, 641.2209).
A follow-up 1H NMR spectrum recorded after 16 h revealed that salt 2a had hydrolyzed completely, and the resulting mixture consisted of α/ β–hemiacetal 7, and liberated ethylmethylsulfide (Scheme 4). On a side note, our initial attempt to record the spectrum in CD3OD, gave rise to a follow up spectrum (recorded after 16 h) showing the exclusive formation of methyl(D3) α-glucoside 8a. Likewise, if compound 2a was handled in the presence of methanol used as a co-solvent for the preparative TLC, the corresponding α-methyl glucoside 8b was isolated as the sole product (spectrum shown in Scheme 4). It is noteworthy, that the isolated 2a yielded a similar glycosidation stereoselectivity to that obtained in reactions wherein 2a was generated and allowed to react with glycosyl acceptor in situ.
We attribute the unusual stability of 2a, to the electronic consequences resulting from the “superdisarming” (2-O-“non-participating alkyl”-3,4,6-tri-O-“electron withdrawing acyl”) protecting group motif.26,28 This protecting group combination renders leaving group departure energetically unfavorable, as the resulting carbocation intermediate is incapable of achieving adequate stabilization.32 Although this low-reactivity donor was initially developed to improve stereocontrol in the glycosylation reaction, it was subsequently found to be invaluable in the chemoselective introduction of a trans-cis or cis-cis oligosaccharide pattern, which was not directly accessible by the traditional armed-disarmed technique.33 At present, it is this superdisarmed approach that has also allowed us, for the first time, to detect, trap and even isolate the key intermediates formed during the glycosidation of thioglycosides.
In our attempt to isolate other sulfonium salts, per-benzoylated (disarmed) thioglycoside 3 and its per-benzylated (armed) counterpart, were each treated with MeOTf (3 equiv.) in the presence of molecular sieves, in 1,2-dichloroethane at rt. While the armed thioglycoside did not yield a sulfonium salt, the less reactive disarmed glycosyl donor 3 showed nominal signs of sulfonium salt formation. However, all efforts made to isolate this per-benzoylated sulfonium salt were unsuccessful, as were attempts to detect this species using low temperature NMR monitoring.31 Other superdisarmed glycosyl donors equipped with sulfur-based leaving groups including S-phenyl, S-tolyl, and S-benzoxazolyl were also investigated for their potential ability to form sulfonium ions. Although all glycosyl donors underwent glycosylation in the presence of methyl triflate, no salt formation was observed. These results made us believe that these intermediate sulfonium salts are significantly more reactive than ethylthio glycoside-derived salt 2a.
Next, we decided to investigate the role that the (often overlooked) counter-anion could be playing. To accomplish this task, we chose to generate a variety of “methylating promoters” in situ. Methyl iodide, which alone is too weak a promoter to activate S-ethyl glycosides, was chosen as the source of methyl cation (Me+). On the other hand, a series of commercially available silver salts (AgX, X = BF4, PF6, ClO4, OTs, OMs, or NO3) were chosen as the source of counteranion, because these reagents alone also do not promote thioglycoside glycosidations. Exploiting the known affinity of silver compounds to readily undergo anion exchange with alkyl halides (such as MeI), we were then able to generate a series of new “methylating promoters” in situ.34 Using these reagents, a range of sulfonium salts (each containing a different counter-anion) could be generated in the absence of the glycosyl acceptor as follows. Thioglycoside 1 was stirred for 30 min with excess MeI (9 equiv.), followed by the addition of the desired silver salt (Table 1) to generate the corresponding promoter. Accordingly, as the various sulfonium salts began to form, the precipitation of yellow AgI was noticed among the reactions between MeI and AgBF4, AgPF6 and AgClO4, yielding sulfonium salts 2b-d (Entries 1-3). However, it should be noted that in the reactions between MeI and AgOTs, AgOMs or AgNO3, little-to-no AgI precipitate was observed, even after 16 h (Table 1, entries 4-6). It therefore followed that in these cases no sulfonium salt was formed, as the anion exchange did not occur. At this point, sulfonium salts 2b-d were purified by preparative layer chromatography and subsequent NMR spectra were recorded.
Table 1.
Formation of β-sulfonium salts 2b-d using in situ generated methylating promoters.
 | ||||
|---|---|---|---|---|
| entry | AgX | promoter34 | time | salt |
| 1 | AgBF4 | MeBF4 | 0.5 ha | 2b |
| 2 | AgPF6 | MePF6 | 0.5 ha | 2c |
| 3 | AgClO4 | MeClO4 | 0.5 ha | 2d |
| 4 | AgOTs | MeOTs | 16 h | none |
| 5 | AgOMs | MeOMs | 16 h | none |
| 6 | AgNO3 | MeNO3 | 16 h | none |
–time at which significant amount of AgI formation was detected
Interestingly, unlike the solitary H-1 signal seen at 5.31 ppm in the spectrum of 2a (Scheme 4), the 1H NMR spectra of sulfonium salts 2b-d recorded at 300 MHz in CDCl3 revealed the presence of two new downfield H-1 signals. As exemplified in the reaction between 1 and MeI/AgClO4, the NMR spectrum of 2d showed the new H-1 signals to be at 5.30 ppm and 5.17 ppm (varies slightly for each counter-anion), and to each have a coupling constant consistent with that of a β-glycoside (9.7 Hz and 9.8 Hz, respectively). Additionally, these H-1 shifts could each be linked (via integration) to a different set of S-ethyl protons, and to a new singlet indicative of an acquired methyl group (see the SI). Furthermore, while there was splitting seen for the H-1 protons and the leaving group protons, the rest of the signals remained overlapping. This led us to believe that these were diastereomeric β-sulfonium salts (2da and 2db, Figure 1), as this occurrence has been documented previously.17
Figure 1.
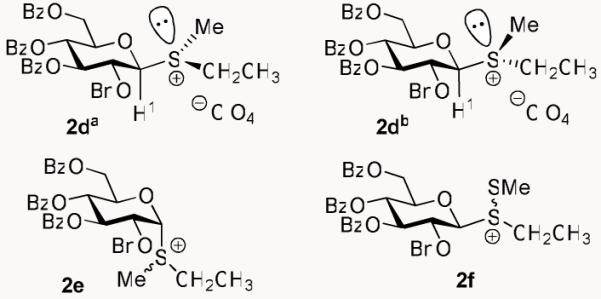
Structures of sulfonium salts 2da, 2db, 2e and 2f.
As an extension of our findings with β-sulfonium salt 2a, we also attempted to synthesize its α-epimer (2e, Figure 1). Immediately, it became apparent that the reactivity of 2e is much greater than that observed with its β-counterpart 2a, and only small amounts of 2e were detected. Interestingly, traceable 2e could only be generated when utilizing the in situ generated promoters MeI/AgBF4 and MeI/AgPF6, and no salt was observed with MeOTf. The crude 1H NMR spectra of 2e revealed the presence of two new α-anomeric signals at around 6.21 and 6.31 ppm (see the SI). However, the spectrum indicated the presence of large amounts of the starting material and by-products. Reinforcing these findings, are the similar results found by both Yoshida and Boons, wherein β-sulfonium species were found to be more stable than their α-counterparts.17,18
In order to further investigate the ability of glycosyl donor 1 to form a “stable” cationic species, we screened other common thioglycoside promoters that could potentially form a detectable β-sulfonium salt. As a result, it was found that when 1 was treated with dimethyl(methylthio)sulfonium triflate (DMTST),35,36 it too gave rise to the baseline spot on TLC, indicative of a polar sulfonium species. When attempts were made to isolate this proposed thiomethylated salt (2f, Figure 1), this species was found to be less stable than its methylated analog 2a. The 1H NMR of purified compound 2f recorded at 300 MHz in CDCl3 contained a significant amount of hemiacetal 7, but when a crude NMR of 2f was acquired, a new H-1 peak could easily be identified at 6.45 ppm (see the SI). Interestingly, unlike the H-1 signal seen in the NMR spectrum of methylated salt 2a, the H-1 signal of donor 2f was much more deshielded and displayed a significantly smaller coupling constant (J1,2 = 4.5 Hz). Upon first glance, it seemed as though the thiomethylated leaving group had anomerized. Soon after however, other peculiarities were noticed, such as the unusually small coupling constants of H-3 (J2,3 = J3,4 = 5.3 Hz) and the 0.88 ppm downfield shift of H-2. This led us to believe that the pyranose ring in 2f was no longer residing in a 4C1 chair conformation,37 but may have undergone a conformational change, as the NMR data was more indicative of a half-chair conformation.38 Of further interest, was that upon treatment of 2f with large access of p-toluenethiol the corresponding α-tolyl thioglycoside was obtained stereoselectively.
In conclusion, we believe that further investigation of quasi-stable reaction intermediates and expansion to studying the means by which these intermediates convert into glycoside products, can contribute to understanding the reaction mechanisms of glycosylation. For instance, while Boons et al. found that the glycosidation of sulfonium salts results in excellent SN2-like stereoselectivity,17,19,39,40 several research groups have conversely encountered poor or unanticipated anomeric selectivities when dealing with these key intermediates. Yoshida et al. found that both the α- and β-sulfonium species fail to undergo the anticipated inversion.18 Likewise, Woerpel et al. found an intramolecular glycosyl sulfonium species which also failed to yield an inverted product, giving instead, a stereoselectivity arising from the predominance of the open cation (SN1) pathway over a concerted (SN2) displacement.20,41 Thus, due to such experimental inconsistencies, we anticipate that the simple approach to glycosyl sulfonium ions described herein, will aid in the investigation of traceable reaction intermediates in glycosylation. This discovery may also offer a reliable system for studying the controversial reaction mechanism by which anomeric sulfonium ions are displaced by nucleophiles. It is our belief, that a carefully controlled reaction pathway will ultimately lead to the development of a highly stereocontrolled glycosylation. This study may ultimately impact areas outside of the glycosciences, as sulfonium salts of similar structural composition have been found to be valuable substrates42,43 and reagents44 in synthetic chemistry, and represent valuable targets for computational studies.45
Supplementary Material
Acknowledgment
This work was supported by awards from the NIGMS (GM077170) and NSF (CHE-0547566). Dr. Winter and Mr. Kramer (UM – St. Louis) are thanked for HRMS determinations.
Footnotes
Supporting Information Available: Experimental procedures, extended experimental data, 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Stanley P, Hart GW, Etzler ME. Essentials of Glycobiology. Second ed. CSH Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- (2).Hehre EJ. Carbohydr. Res. 2001;331:347–368. doi: 10.1016/s0008-6215(01)00042-8. [DOI] [PubMed] [Google Scholar]
- (3).Crich D, Banerjee A, Yao Q. J. Am. Chem. Soc. 2004;126:14930–14934. doi: 10.1021/ja047194t. [DOI] [PubMed] [Google Scholar]
- (4).Polat T, Wong CH. J. Am. Chem. Soc. 2007;129:12795–12800. doi: 10.1021/ja073098r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee CC, Chang KL, Hung SC. Nature. 2007;446:896–899. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- (6).Seeberger PH, Werz DB. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- (7).Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem. Commun. 2009:1834–1836. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- (9).Demchenko AV. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim, Germany: 2008. pp. 1–27. [Google Scholar]
- (10).Zhong W, Boons G-J. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim, Germany: 2008. pp. 261–303. [Google Scholar]
- (11).Lonn H. J. Carbohydr. Chem. 1987;6:301–306. [Google Scholar]
- (12).Whitfield DM. Adv. Carbohydr. Chem. Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- (13).Mydock LK, Demchenko AV. Org. Biomol. Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- (14).Crich D. Acc. Chem. Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- (15).West AC, Schuerch C. J. Am. Chem. Soc. 1973;95:1333–1335. [Google Scholar]
- (16).Sun LH, Li P, Zhao K. Tetrahedron Lett. 1994;35:7147–7150. [Google Scholar]
- (17).Park J, Kawatkar S, Kim JH, Boons GJ. Org. Lett. 2007;9:1959–1962. doi: 10.1021/ol070513b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nokami T, Shibuya A, Manabe S, Ito Y, Yoshida J. Chem. Eur. J. 2009;15:2252–2255. doi: 10.1002/chem.200802293. [DOI] [PubMed] [Google Scholar]
- (19).Kim JH, Yang H, Park J, Boons GJ. J. Am. Chem. Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- (20).Beaver MG, Billings SB, Woerpel KA. J. Am. Chem. Soc. 2008;130:2082–2086. doi: 10.1021/ja0767783. [DOI] [PubMed] [Google Scholar]
- (21).Stalford SA, Kilner CA, Leach AG, Turnbull WB. Org. Biomol. Chem. 2009;7:4842–4852. doi: 10.1039/b914417j. [DOI] [PubMed] [Google Scholar]
- (22).Fascione MA, Adshead SJ, Stalford SA, Kilner CA, Leach AG, Turnbull WB. Chem. Commun. 2009:5841–5843. doi: 10.1039/b913308a. [DOI] [PubMed] [Google Scholar]
- (23).Jayakanthan K, Mohan S, Pinto BM. J. Am. Chem. Soc. 2009;131:5621–5626. doi: 10.1021/ja900867q. [DOI] [PubMed] [Google Scholar]
- (24).Boltje TJ, Kim J-H, Park J, Boons G-J. Org. Lett. 2011;13:284–287. doi: 10.1021/ol1027267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Concomitantly with this study Yoshida et al discovered glycosyl sulfonium ion that can be stored at −80 °C for 24 h. Nokami T, Nozaki Y, Saigusa Y, Shibuya A, Manabe S, Ito Y, Yoshida J. Org. Lett. 2011;13:1544–1547. doi: 10.1021/ol200242u.
- (26).Kamat MN, Demchenko AV. Org. Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- (27).Premathilake HD, Mydock LK, Demchenko AV. J. Org. Chem. 2010;75:1095–1100. doi: 10.1021/jo9021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kamkhachorn T, Parameswar AR, Demchenko AV. Org. Lett. 2010;12:3078–3081. doi: 10.1021/ol101089u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kuester JM, Dyong I. Justus Liebigs Ann. Chem. 1975:2179–2189. [Google Scholar]
- (30).Liptak A, Jodal I, Nanasi P. Carbohydr. Res. 1975;44:1–11. [Google Scholar]
- (31).Garcia BA, Gin DY. J. Am. Chem. Soc. 2000;122:4269–4279. [Google Scholar]
- (32).Note: While per-acylated glycosyl donors (such as 3) also suffer from an electron deficiency, the positive charge acquired upon leaving group departure can be better stabilized through acyloxonium ion formation. In fact, although a faint baseline spot could be also detected during the glycosidation of 3, our attempts to isolate this species have been unsuccessful.
- (33).Fraser-Reid B, Udodong UE, Wu ZF, Ottosson H, Merritt JR, Rao CS, Roberts C, Madsen R. Synlett. 1992:927–942. and references therein.
- (34).Note: It should be noted that assuming the independent existence of such new MeX species is not entirely correct, as it is more likely that the methylation of the leaving group would occur concomitantly with counter-anion exchange through a more complex transition state. Herein, however, it is referred to as such for the purpose of simplification. To verify that no reaction took place prior to the generation of the active promoter in situ, two glycosylations were attempted in the presence of MeI and separately in the presence of the silver salt (AgX), wherein no reactions were observed.
- (35).Ravenscroft M, Roberts RMG, Tillett JG. J. Chem. Soc. Perkin Trans. 2. 1982:1569–1972. [Google Scholar]
- (36).Fugedi P, Garegg PJ. Carbohydr. Res. 1986;149:c9–c12. [Google Scholar]
- (37).Lemieux RU, Hendriks KB, Stick RV, James K. J. Am. Chem. Soc. 1975;97:4056–4062. and references therein.
- (38).Pedersen CM, Nordstrom LU, Bols M. J. Am. Chem. Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- (39).Kim JH, Yang H, Boons GJ. Angew. Chem. Int. Ed. 2005;44:947–949. doi: 10.1002/anie.200461745. [DOI] [PubMed] [Google Scholar]
- (40).Kim JH, Yang H, Khot V, Whitfield D, Boons GJ. Eur. J. Org. Chem. 2006:5007–5028. [Google Scholar]
- (41).Billings SB, Woerpel KA. J. Org. Chem. 2006;71:5171–5178. doi: 10.1021/jo060077r. [DOI] [PubMed] [Google Scholar]
- (42).Kim S, Lee BS, Park JH. Bull. Korean Chem. Soc. 1993;14:654–655. [Google Scholar]
- (43).Xu Y, Fletcher M, Dolbier WR., Jr. J. Org. Chem. 2000;65:3460–3465. doi: 10.1021/jo000015f. [DOI] [PubMed] [Google Scholar]
- (44).Kaczmarczyk G, Jończyk A. Synlett. 1997:921–922. [Google Scholar]
- (45).Buckley N, Oppenheimer NJ. J. Org. Chem. 1996;61:8048–8062. doi: 10.1021/jo960748t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.