

Abstract
The euryarchaeal transcriptional repressor NrpR regulates a variety of nitrogen assimilation genes by 2-oxoglutarate-reversible binding to conserved palindromic operators. The number and positioning of these operators varies among promoter regions of regulated genes, suggesting NrpR can bind in different patterns. Particularly intriguing is the contrast between the nif and glnK1 promoter regions of Methanococcus maripaludis, where two operators are present but with different configurations. Here we study NrpR binding and regulation at the glnK1 promoter, where the two operator sequences overlap and occur on opposite faces of the double helix. We find that both operators function in binding, with a dimer of NrpR binding simultaneously to each overlapping operator. We show in vivo that the first operator plays a primary role in regulation and the second operator plays an enhancing role. This is the first demonstration of overlapping operators functioning in Archaea.
Keywords: NrpR, Methanococcus maripaludis, glnK, transcription, regulation
Introduction
Most known transcriptional regulators in the Archaea belong to families that are better known in the Bacteria. This is the case even though the archaeal transcription machinery resembles a simplified version of the eukaryal RNA polymerase II system (Geiduschek & Ouhammouch, 2005). However, a few regulators that have been studied are primarily archaeal. NrpR is a prime example—it is widely distributed in the euryarchaeota but is known in only a few genera of Bacteria (Lie et al., 2007). Discovered and characterized in Methanococcus maripaludis, all NrpR homologs appear to function similarly as transcriptional repressors of nitrogen assimilation genes. In M. maripaludis, NrpR represses transcription of genes for nitrogen fixation (nif, (Cohen-Kupiec et al., 1997)), ammonia assimilation (glutamine synthetase, glnA, (Cohen-Kupiec et al., 1999, Lie & Leigh, 2003)), and (shown here) a PII nitrogen sensing protein and an ammonium channel protein.
We are characterizing the mechanism of NrpR function in M. maripaludis. Work to date has focused on the regulation of the nif operon, whose promoter region contains two inverted repeat operators with the sequence GGAAN6TTCC, spaced three helical turns apart (Fig. 1A). In vitro binding studies and in vivo mutagenesis showed that each operator binds a dimer of NrpR, and both operators together bind two NrpR dimers. Both operators are required for maximal repression, with the first operator playing the primary role (Cohen-Kupiec et al., 1997, Lie et al., 2005, Lie & Leigh, 2002). The second operator, whose spacing from the first operator is critical, enhances binding affinity by adding a cooperative component, probably via tetramerization of two NrpR dimers. Binding is reversed by 2-oxoglutarate (2OG), which thus acts as the inducer ligand of NrpR. 2OG, the metabolic precursor of ammonia assimilation, is evidently a widespread indicator of nitrogen starvation in the Bacteria and the Euryarchaeota (Leigh & Dodsworth, 2007). In M. maripaludis 2OG varies in intracellular concentration in the range of 0.08 to 0.8 mM, under nitrogen excess and nitrogen deficient conditions respectively (Dodsworth et al., 2005).
Fig. 1. nif and glnK1 promoter regions.
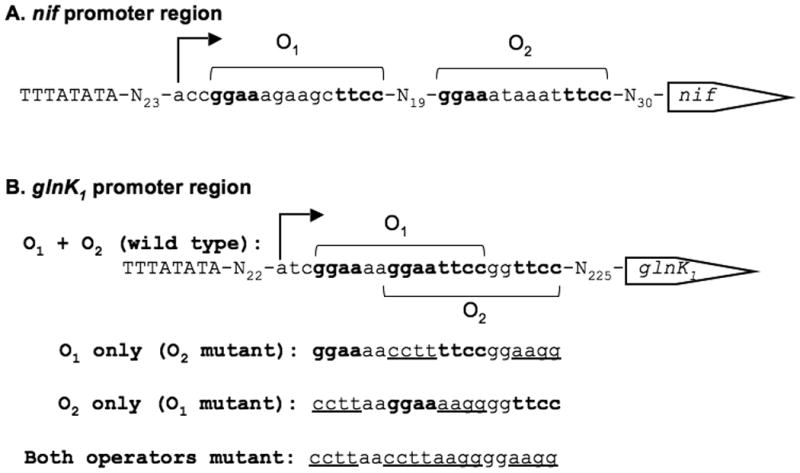
A. nif promoter region.
B. glnK1 promoter region. TATA boxes (caps), transcription start sites (bent arrows), and operators (bold) are shown. Mutant glnK1 operators are underlined.
Besides the nif promoter, the sequence GGAAN6TTCC is found in the promoter regions of five additional nitrogen-regulated genes in M. maripaludis: glnA, genes for two molybdate transporters, a gene for an alanine transporter, and an operon containing the genes glnK1 and amtB1 (Xia et al., 2009). All of these genes presumably function in nitrogen assimilation: besides the previously stated functions of Nif proteins and GlnA, molybdate transporters would help provide molybdenum as a component of the cofactor of nitrogenase, and alanine can serve as a nitrogen source. glnK1 encodes a nitrogen sensor protein of the PII family (Leigh & Dodsworth, 2007), and amtB1 encodes an ammonium channel protein. The GGAAN6TTCC sequence is completely conserved, suggesting a rigorous requirement for binding of NrpR. We refer to these sequences as nitrogen operators. Here we report on the roles of the nitrogen operators of the glnK1-amtB1 operon in NrpR binding and regulation. Like the nif operon, two operators are present, but with radically different configurations, suggesting two different patterns in which two NrpR dimers can bind to an operator pair.
Results
glnK1 is expressed in an operon and regulated by the nitrogen source
The M. maripaludis genome sequence (Hendrickson et al., 2004) revealed the presence of a gene cluster encoding three closely-related PII proteins and two ammonium channel proteins (Fig. 2). Two of the PII protein genes were designated glnK1 and glnK2 due to their presence immediately before the ammonium channel proteins amtB1 and amtB2, while the third PII protein gene was designated glnB. The sequences of the PII proteins are highly conserved and designation as glnK is usually made based on proximity to an amtB gene (Thomas et al., 2000).
Fig. 2. The glnK gene cluster.
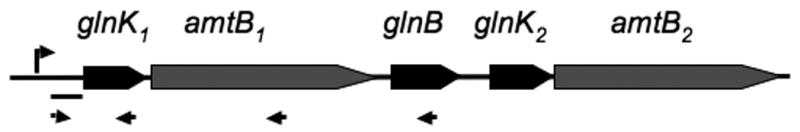
The location of the transcription start site is indicated by a bent arrow. The probe used for Northern blots is indicated by a short horizontal line. Primers used for RT-PCR are shown as arrows.
The coding regions for glnK1 and amtB1 are closely contiguous, suggesting that these two genes may constitute an operon. We characterized the transcript by primer extension, Northern, and RT-PCR analyses. Primer extension analysis (Fig. S1 in supporting information) showed a distinct 5′ end of RNA 248 nucleotides upstream of the putative start codon of glnK1. (This 5′ untranslated region is unusually long and its role in expression, if any, will be the subject of a future study.) A consensus TATA box (TTTATATA) is located 23 bp upstream of this transcription start site (Fig. 1B). For Northern analysis, high degrees of similarity between glnK1, glnB, and glnK2 and between amtB1 and amtB2 precluded specific probes in the coding regions. Consequently we used a probe for the 5′ untranslated region of glnK1. Two separate Northern experiments were conducted (Fig. S2), and both showed a band of 2.1 to 2.3 kb. Smaller RNAs were also detected and could be degradation or termination products. A transcript extending from the transcription start site to the end of amtB1 would be 1.8 kb and a transcript extending to the end of glnB would be 2.3 kb. We used RT-PCR to further determine the extent of the transcript (Fig. S3). RT-PCR products were detected that extended from the 5′ untranslated region into the coding regions of glnK1 and amtB1 but not glnB, indicating that the operon includes only glnK1 and amtB1.
nif and glnA expression varies with the nitrogen source (Lie et al., 2005, Lie & Leigh, 2002, Lie & Leigh, 2003, Cohen-Kupiec et al., 1997, Cohen-Kupiec et al., 1999). Both genes are repressed during growth with ammonia, expressed at intermediate levels during growth with alanine, and derepressed during growth with N2. The Northern analysis of the glnK1 transcript indicated a similar pattern of regulation (Fig. S2). One of the two Northern analyses also showed more mRNA fragmentation in preparations from N2-grown cells than cells grown with the other two nitrogen sources. This effect may not be specific to the glnK1 transcript, since we commonly observe fragmentation in mRNA preparations from N2-grown cells.
The glnK1 promoter region contains two overlapping nitrogen operators that bind NrpR
The promoter region of glnK1 contains two potential NrpR binding sites, O1 and O2, each with complete agreement to the consensus nitrogen operator sequence, GGAAN6TTCC (Fig. 1B). Unlike the nif promoter region, where two operators are centered 33 bp apart, in the glnK1 promoter region the two putative operators are centered six bp apart, overlap, and are on opposite faces of the DNA helix. Because the juxtaposition of the two putative operators is so different, we wanted to determine if both of them could bind NrpR. Electrophoretic gel mobility shift assays (EMSA) were conducted to assess the binding of NrpR to DNA containing both operators, only one operator (the other operator altered), or neither operator. The mutant operators (Fig. 1B) were designed as inverted sequences of the original operators, eliminating the binding sites but maintaining the palindromic nature the sequences. Each operator alone bound NrpR to produce a single shifted band. Both operators together produced two shifted bands; a faster migrating band dominated at low NrpR concentrations and corresponded to the shift that occurred when only one operator was present, while a more slowly migrating band dominated at high NrpR concentrations (Fig. 3A). With both operators altered, no shift occurred. This gel-shift pattern was similar to the nif promoter where a single operator bound one NrpR dimer and two operators bound two NrpR dimers (Lie et al., 2005). These results suggest that despite their overlapping nature, both glnK1 operators can simultaneously bind a dimer of NrpR. A binding curve for the wild type glnK1 promoter DNA (Fig. 3B, C) indicated a binding dissociation constant (Kd) of 0.3 nM for both operators together, similar to nif (Lie et al., 2005). Furthermore, when both operators were present, each bound a dimer of NrpR with high affinity, since the band with the greater shift dominated at NrpR concentrations as low as 0.4 nM. Binding curves for DNA containing mutant operators indicated Kd values of 1.7 nM for O1 and 3.6 nM for O2. However, these values must be interpreted with caution since the alteration of each operator necessarily changed nucleotides flanking the eight conserved base pairs of the other operator.
Fig. 3. Electrophoretic mobility shift analysis (EMSA) of NrpR binding to operator DNA.
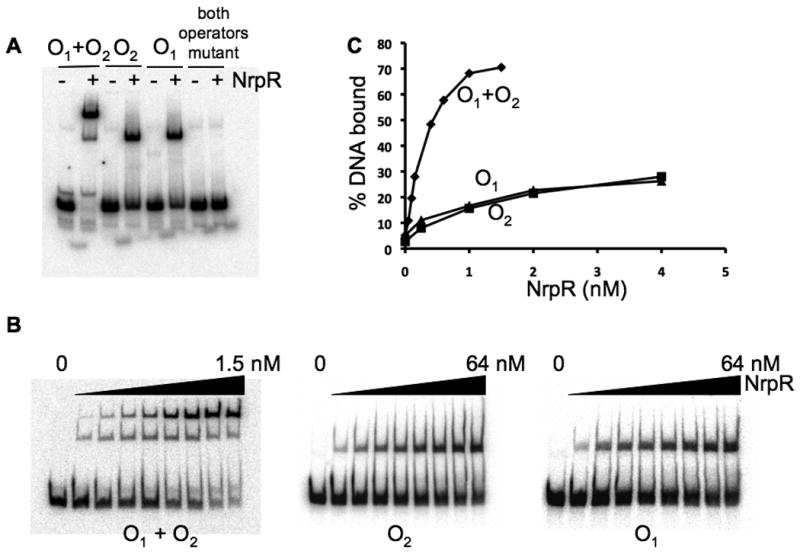
A. DNA probes containing operators shown in Fig. 1: wild type (O1+O2), O2 only (O1 mutant), O1 only (O2 mutant), and both operators mutant. No (-) or 120 nM (+) NrpR was mixed with the DNA.
B. EMSA at different NrpR concentrations: O1+O2, 0, 0.05, 0.1, 0.15, 0.2, 0.4, 0.6, 1.0, and 1.5 nM; O2 and O1, 0, 0.25, 1, 2, 4, 8, 16, 32, and 64 nM.
C. Plot of data from B.
DNAse I footprinting analysis was used to determine the region of DNA that is protected by binding of NrpR to the glnK1 operators. Footprinting was performed with DNA containing only the first operator and DNA containing both operators. With DNA containing only O1 (Fig. 4C and D), the footprint began 4 nucleotides 5′ of the inverted repeat on each strand and extended 11 and 7 nucleotides 3′ of the inverted repeat (coding and non-coding strand respectively). Hence, the footprint covered the inverted repeat and had a somewhat longer extension to the 3′ on each stand. With both operators present (Fig. 4A and B), the footprint conformed to that expected if two NrpR dimers bind, one to each inverted repeat. Hence, the upstream end of the footprint was similar: 4 nucleotides 5′ of the inverted repeat on the coding strand and 7 nucleotides 3′ on the non-coding strand. The downstream end was extended by the addition of the second inverted repeat: 8 nucleotides 3′ of the new inverted repeat on the coding strand and 3 nucleotides 5′ on the non-coding strand. With only O1, hypersensitive sites were present a few nucleotides 3′ of the inverted repeat on each strand. Hypersensitive sites were not detected when both operators were present.
Fig. 4. DNAse I footprinting analysis of NrpR binding to operator DNA.
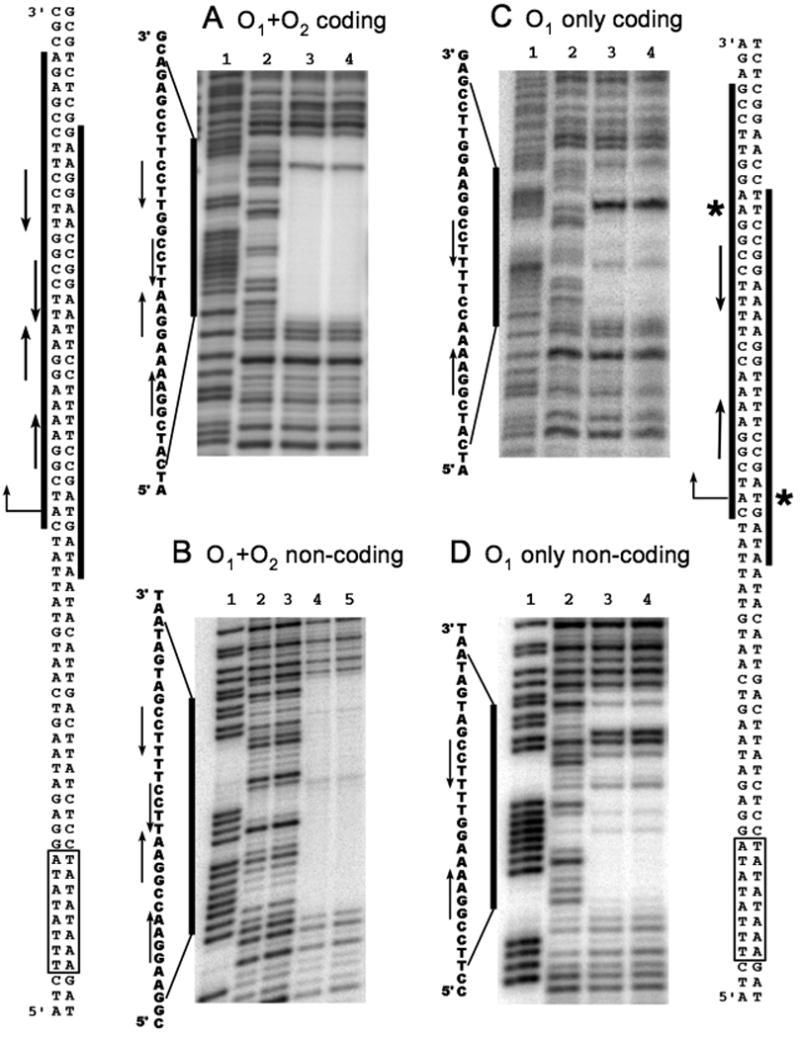
Footprints are delineated by bold lines and indicated with respect to the sequence. Double stranded DNA sequences on left and right represent O1+O2 and O1 only, respectively. Boxed sequences, TATA box; bent arrow, transcription start site; inverted arrow pairs, operator(s); asterisks, hypersensitive sites.
A, C and D. Lane 1, G+A ladder; lane 2, no NrpR; lane 3, 200 nM NrpR; lane 4, 400 nM NrpR. Units of DNAse I, 0.005
B. Lane 1, G+A ladder; lanes 2 and 3, no NrpR, lanes 4 and 5, 400 nM NrpR.; Units of DNAse I: lanes 2 and 4, 0.001; lanes 3 and 5, 0.002.
The difference in the juxtaposition of the two operators in the glnK1 promoter region compared to the nif promoter region suggests that the configuration in which two NrpR molecules bind would be different. Additional evidence that this is the case comes from a comparative gel shift experiment. Using segments of DNA of similar length, gel shifts at varying NrpR concentrations were used to obtain bands where one or two NrpR dimers were bound, similar to the experiment in Fig. 3B above. With nif and glnK1 operator regions, the migration was similar when one NrpR dimer was bound. However, with two NrpR dimers bound there was a greater shift with the glnK1 operators than with the nif operators (Fig. 5). This suggests that the NrpR-DNA complex with two dimers bound is less compact for glnK1 than for nif.
Fig. 5. EMSA of NrpR binding to nif and glnK1 operators.
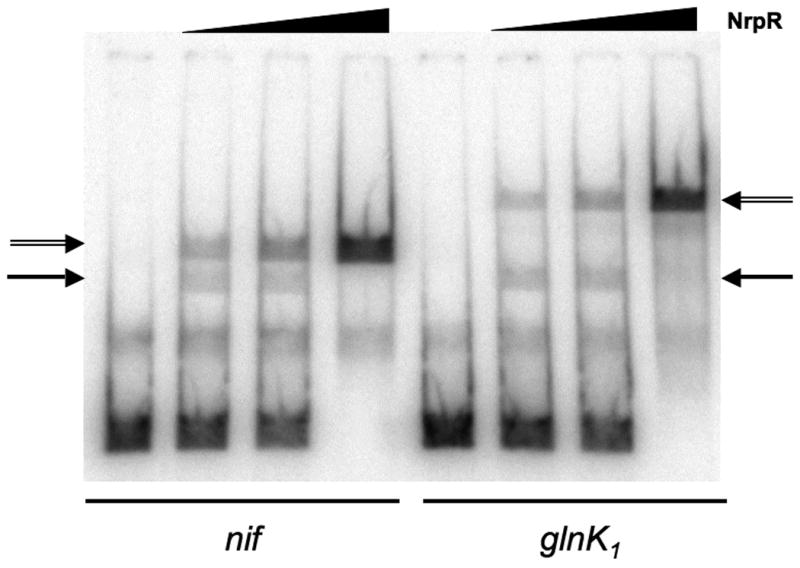
DNA probes of equal length contained nif O1 + O2 or glnK1 O1 + O2. Single and double arrows indicate shifts corresponding to NrpR binding to one or two operators respectively. NrpR concentrations were 0, 0.05, 0.1, and 2.0 nM.
NrpR binding to the operators in the nif promoter region is inhibited by 2OG (Lie et al., 2005). Binding curves for the wild type glnK1 operator region (Fig. S4) enabled us to calculate Kd values with 0.1 mM and 0.4 mM 2OG of 1.4 nM and 6.7 nM respectively, compared to 0.3 nM in the absence of 2OG. Hence, in the case of both the nif and the glnK1 promoter regions, 2OG indicates the nitrogen state of the cell and is the inducer ligand that controls NrpR binding to the operators.
The glnK1 operators both mediate nitrogen repression in vivo
To test the roles of the glnK1 operators in regulation of the glnK1 operon, lacZ reporter fusions were constructed. DNA with promoter regions containing wild type and mutant operators identical to those used for the gel shift analyses were used to make reporter constructs, by fusing the lacZ coding region to the glnK1 start codon. These constructs were incorporated into the M. maripaludis genome in single copy using a negative selection strategy (Moore & Leigh, 2005). In addition, an nrpR deletion mutation was introduced into the strain containing the lacZ fusion to the wild type promoter region. β-galactosidase assays were conducted with cultures grown on N2, alanine, or ammonia (Fig. 6). With the wild type promoter region (both O1 and O2 present), expression was high during growth with N2, partially repressed during growth with alanine, and markedly repressed during growth with ammonia, in agreement with the results of Northern blots (above). By comparison, no repression occurred when both operators were altered by mutation; in fact, β-galactosidase activities were higher with ammonia or alanine than with N2. We do not know why growth with N2 might decrease expression; however, nitrogen fixing cultures have higher energy demands and grow more slowly than non-nitrogen fixing cultures, complicating their direct comparison. Repression also did not occur with the wild type promoter region when nrpR was deleted, confirming the role of NrpR. In this case too, β-galactosidase activities were higher with ammonia or alanine than with N2, though not as markedly. The results with the double operator mutant and the nrpR deletion mutant both support the role of NrpR binding to the operators. However, the quantitative difference between these two mutants was unexpected. A direct comparison is complicated by the observation that nrpR mutants grow more slowly than nrpR+ strains for unknown reasons (Lie & Leigh, 2003).
Fig. 6. In vivo regulation of wild type and mutant glnK1 promoter reporter fusions.
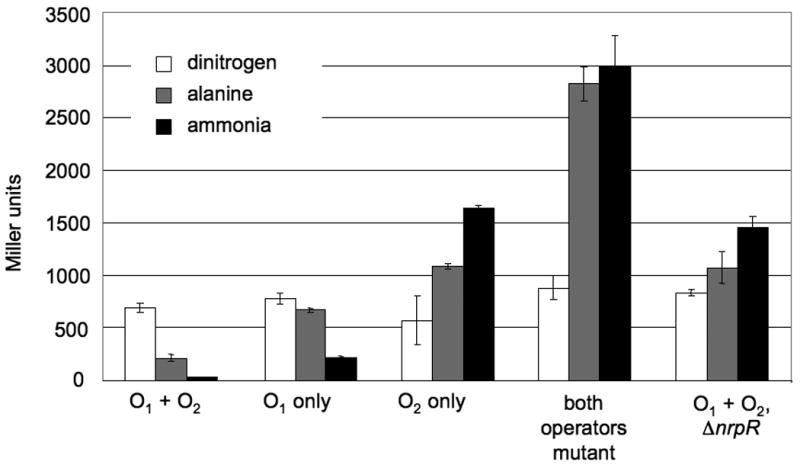
Strains containing lacZ fusions to promoters with wild type (O1 + O2) and mutant (O1 only, O2 only, or both operators mutant) operators were grown under three nitrogen conditions as indicated, and β-galactosidase assays performed as described in Experimental Procedures.
The lacZ fusion strains were also useful in determining the role of each operator individually. O1 alone mediated repression. This repression was not as marked as with both operators present, suggesting either that O2 enhances repression or that the mutations altering O2 lessened the effect of O1 due to the nucleotide changes flanking O1. O2 alone had little discernible effect, since the expression pattern with this mutant resembled that with both operators mutant.
Discussion
Taken together, the results of NrpR binding experiments and the measurements of gene expression support a model in which NrpR binding to both operators results in maximal repression. O1 appears to play a primary role in mediating repression, and due to its proximity to the transcription start site may block some step in transcription initiation such as recruitment of RNA polymerase. Repression in vivo was more rigorous when both operators were unaltered than when O2 was altered. This suggests that O2 enhances repression, with the caveat that mutations flanking O1 could have weakened it. NrpR binding studies bolster the idea that O2 plays an enhancing role. Gel shift and footprinting analyses strongly suggest that NrpR can bind to either operator, and that two NrpR dimers bind to both operators simultaneously. Thus, a greater gel shift occurred when both operators were intact than when only one operator was present, and the footprint with both operators was larger by the expected amount than when only O1 was present. Why should O2, which has a perfect 8-bp consensus sequence as does O1, play only an enhancing role in repression? One possibility is that the binding affinity of O2 for NrpR is lower, possibly due to differences in flanking sequences. Another explanation is the greater distance of O2 from the transcription start site, hampering its ability alone to block a step in transcription initiation. In either case, two mechanisms seem possible by which O2 could enhance repression. First, O2 could increase the affinity of NrpR binding to the operator region through cooperativity, either through a direct interaction of two NrpR dimers or by an effect on the conformation of the DNA, enhancing the binding of NrpR to O1. The presence of DNase hypersensitive sites on NrpR-bound O1 DNA suggests that effects on DNA conformation do occur. Second, NrpR binding to O2 could exacerbate the effect of NrpR binding to O1, by helping to block the same or a different step in transcription initiation. By any of these mechanisms, the enhancement of NrpR binding by O2 would presumably increase the sensitivity of repression to the nitrogen starvation signal 2OG. In addition, if binding of NrpR is cooperative, the effect of 2OG might also be cooperative. Further experiments are needed to distinguish among these possibilities.
To our knowledge, this is the first example in Archaea where two transcriptional regulator molecules bind to overlapping binding sites, although several examples are known in the Bacteria (Augustus et al., 2006, Lawson & Carey, 1993, Phillips & Stockley, 1994, Dahl et al., 1994, White et al., 1998). Structurally, NrpR appears to be unusually versatile in its ability to bind multiply to promoter regions in two different configurations, one where two operators overlap and one where two operators are separated. Models for binding to the nif and glnK1 promoter regions are shown in Fig. 7. NrpR binding to the nif promoter region is known to be cooperative (Lie et al., 2005), and tetramerization of two NrpR dimers involving faces distant from the DNA binding sites seems likely. In the case of the glnK1 promoter region, if cooperativity occurs, either a different dimer-dimer interaction involving faces of NrpR close to the DNA binding sites, or an effect mediated by a DNA conformational change as mentioned above, may occur. Structural studies are underway to test these models.
Fig. 7. Models for the binding of NrpR to the nif and glnK1 promoter regions.
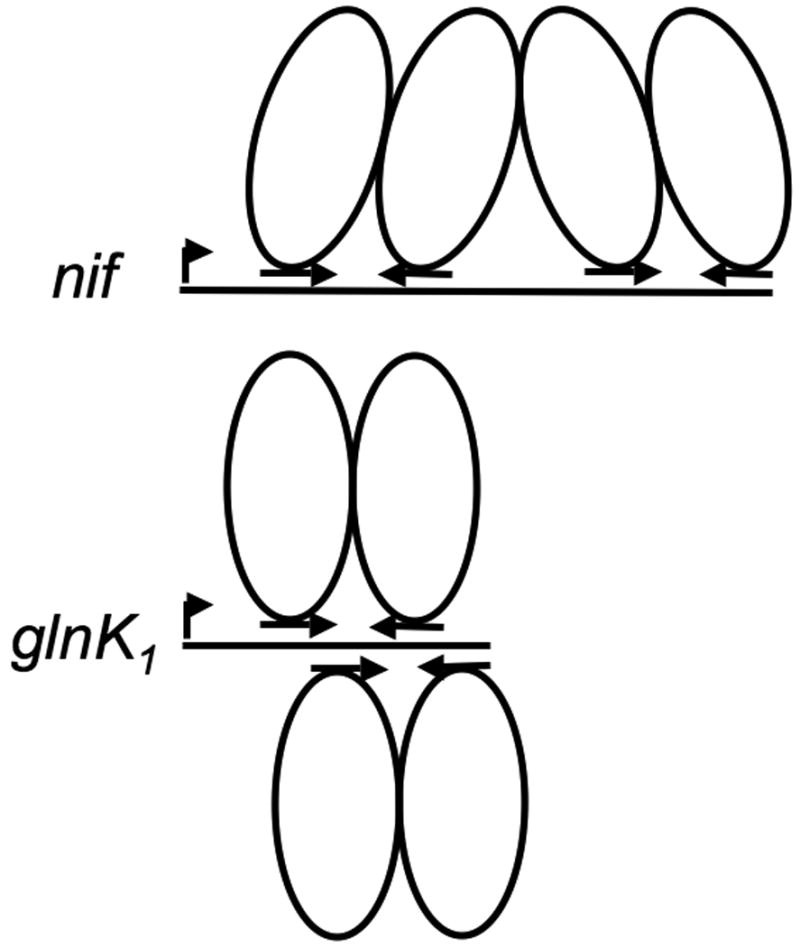
A dimer of dimers (tetramer) binds to the nif promoter region, which contains two operators centered three helical turns apart on the same face of the DNA helix. Two separate dimers bind on opposite sides of the helix in the glnK1 promoter region, which contains two overlapping operators spaced only 6 bp apart.
Experimental procedures
Cultivation of M. maripaludis
Cells were grown under strict anaerobic condition at 37° C as described (Balch et al., 1979). Nitrogen-free medium (Blank et al., 1995, Lie & Leigh, 2002) was supplemented with L-alanine (10 mM), NH4Cl (10 mM), or N2 (40% H2:20% CO2:40% N2 replaced 80% H2:20% CO2). Growth of M. maripaludis in a chemostat was done using a minimal defined medium as described (Xia et al., 2009, Haydock et al., 2004).
Plasmid construction, site directed mutagenesis, and strain construction
Plasmids are listed in Table S1. M. maripaludis DNA was extracted using the MasterPure™ Complete DNA and RNA Purification Kit (Epicentre Biotechnologies) according to the manufacturer's protocol. The wild type promoter region of glnK1 was PCR amplified from M. maripaludis S2 DNA with Herculase® polymerase (Stratagene) and primers PglnK1fw and PglnKrv. Digestion of the PCR product with SpeI and NsiI was followed by ligation into vector pWLG40 resulting in pWLG40K1. Similar plasmids with mutant operators were made as follows: PCR products were generated from M. maripaludis S2 DNA with Taq-polymerase, forward primer PglnK1fw, and either of three reverse primers, deltaO1rv, deltaO2rv or deltaO1/O2rv to produce mutations in O1, O2, and both operators respectively. The products served as “megaprimers” (Sarkar & Sommer, 1990) together with primer Pglnk1rv in a second set of PCR amplifications from M. maripaludis S2 DNA to generate products, which were cloned into pCR2.1®-TOPO, excised with SpeI and NsiI, and ligated into pWLG40+lacZ resulting in pWLG40glnK1O1, pWLG40glnK1O2, and pWLG40glnK1O1+O2. pCR2.1glnK1pro was constructed by amplifying the glnK1 promoter region from S2 genomic DNA with primers FpglnKFNotI and FpglnKRNotI and cloning into pCR2.1 TOPO® (Invitrogen).
A plasmid containing the wild type glnK1 promoter fused to lacZ was constructed as follows: pWLG40K1 DNA was used as template with primers PglnKAscIfw and LacZAscIrv to generate a product that was treated with AscI followed by ligation into pBLprt resulting in pBLPrtK1. Similar plasmids with mutant operators were constructed from pWLG40glnK1O1, pWLG40glnK1O2 and pWLG40glnK1O1+O2 DNA by PCR with primers PglnK1fw and PglnKrv, digestion with AscI, and insertion into pBLPrt to yield pBLPrtglnK1O1, pBLPrtglnK1O2 and pBLPrtglnK1O1+O2. Each plasmid contained the glnK1 promoter-lacZ construct in reverse orientation to upt. Constructs were confirmed by sequencing.
Strains are listed in Table S1. Strains Mm1127, Mm1213, Mm1128 and Mm1164 were made by transforming Mm900 with plasmids pBLPrtK1, pBLPrtglnK1O1, pBLPrtglnK1O2 and pBLPrtglnK1O1+O2. Transformation (Tumbula et al., 1994) and selection were carried out as described (Moore & Leigh, 2005), resulting in replacement of the upt site with each reporter construct. Constructs were confirmed by sequencing. Strain Mm1198 containing an in-frame deletion of nrpR was made by transforming Mm1127 with the plasmid pCRPrtNeoΔnrp and selecting for integration and vector removal as described (Moore & Leigh, 2005). The construct was confirmed by PCR analysis using the primers LfRepKO1 and RtRepKO1.
Electrophoretic mobility shift assays
Probes for EMSA were obtained by PCR from plasmid templates, gel purified, and end labeled with T4 polynucleotide kinase and 32P- γ-ATP. For the experiment in Fig. 3, probes containing the wild type operators or mutations in the first, second or both operators region were amplified from pWLG40K1, pWLG40glnK1O1, pWLG40glnK1O2, or pWLG40glnK1O1+O2 respectively with primers P7 and P8, P9 and P13, P10 and P12, or P11 and P14. For the experiment in Fig. 5, probes containing the nif or glnK1 operators were amplified from pMmp1.1 or pCR2.1glnK1pro respectively with primers Mmniffor1 and Mmnifrev, or FPglnKF and FPglnKrev2, resulting in products 161 and 154 bp in length. Conditions for gel-shift and image analysis were as described previously (Lie et al., 2005). 2OG and His-tagged NrpR (Lie et al., 2005) were added to the final concentrations indicated.
Footprinting
Forward (FpglnKF) and reverse (FpglnKrev2) primers for amplifying wildtype (with pCR2.1glnK1pro as a template) or O1 only (with pBLPrtglnK1O2) glnK1 promoter regions were labeled at the 5′ end using [γ–32P] ATP and T4 Polynucleotide kinase (NEB), ethanol precipitated, and washed before being used for PCR amplification. PCR products were purified (Qiagen) and resolved on a non-denaturing polyacrylamide gel (5%). Product (154 bp) was identified by UV shadowing, cut out, and incubated at 37°C in DNA elution buffer (10 mM TRIS pH 8, 1 mM EDTA, 1M LiCl, 0.1% SDS) overnight without agitation. The eluate was filtered through 4 mm PVDF syringe filters (National Scientific) with 0.2 μm pore size, ethanol precipitated, phenol-chloroform extracted, and ethanol precipitated again. The purity and concentration of product were determined spectrophotometrically. Products were then diluted to a final concentration of 200 nM in TE and stored at -80°C until needed. A G+A ladder of the PCR product was generated as described (Mukherjee & Sousa, 2003). Binding reactions contained DNA (2 nM), TRIS pH 7.5 (10 mM), glycerol (10%), KCl (150 mM), DTT (10 mM) and NrpR in a final volume of 50 μl, and were incubated at 37°C for 30 minutes. DNAse I co-factor solution (50 μl of 10 mM MgCl2 and 5 mM CaCl2) was then added followed by DNAse I (Fermentas), and incubation was continued for 2 min before addition of stop buffer (100 μl of 200 mM KCl, 1% SDS, 20 mM EDTA). Samples were then extracted with phenol-chloroform, ethanol precipitated, resuspended in TE, and frozen at -80°C until used. Samples were run on 8% non-denaturing polyacrylamide sequencing gels. Gels were dried, exposed to phosphor screens, and imaged with Storm® imaging software.
Reporter gene assays
Reporter strains were grown in triplicate with N2, alanine, or ammonia and β-glactosidase assays were carried out with 0.2 ml of each culture as described (Lie & Leigh, 2002). Samples were taken from cultures grown to OD660 between 0.3 and 0.7.
Primer extension analysis
RNA was extracted with the RNAeasy kit (Qiagen) with modifications as described previously (Lie & Leigh, 2002). The reaction contained 2 pmol of 32P-labeled primer Exglnk1a, 30 μg RNA from a nitrogen-fixing culture of M. maripaludis strain Mm900, and 0.5 μl of each deoxynucleoside triphosphate in 12 μl H2O. The reaction was heated to 70°C and slowly cooled to 42°C. The reverse transcription was carried out in 20 μl reaction buffer (Invitrogen Life Technologies) containing 10 mM dithiotreithol and 200 U of Superscript II Reverse Transcriptase, according to the supplier's instructions. After 30 min at 42°C nucleic acids were precipitated with isopropanol, resuspended in 6 μl H2O and 4 μl Sequenase stop solution (Sequenase Version 2 DNA Sequencing Kit, USB) and separated on a polyacrylamide sequencing gel next to a sequencing reaction performed with the same primer. The template for sequencing was generated by PCR amplification from a DNA bank of M. maripaludis using primers PglnK1fw and PglnKrv and ligating the product into pCR2.1®-TOPO. The sequencing reaction was carried out with the Sequenase Version 2 DNA Sequencing Kit, USB, according to the manufacturer's protocol.
Northern blots
RNA extractions and Northern analyses were done as described previously (Lie & Leigh, 2002). The probe (see Fig. 2) was amplified from plasmid pWLG40K1 with primers utrglnK1fw and utrglnK1rv. 10 (Fig. S2A) or 5 (Fig. S2B) μg of total RNA was loaded into each lane.
Reverse transcription analysis of the glnK1 transcript
PCR and RT-PCR were performed using the OneStep RT-PCR Kit (Qiagen) following the manufacturer's protocol. Conditions were: 50°C 30 min; 95°C 15 min; 30 cycles of 95°C 45 sec, 54°C 45 sec, and 72°C 3 min; and 72°C 10 min. For non-RT reactions, enzyme supplied in the kit was substituted with Platinum Taq DNA polymerase (Invitrogen). Primers were (forward) Glnkutr and (reverse) Glnk1rtr (Fig. S3 lanes 4, 5, and 8), Amtbrtr (lanes 3, 6, and 9), or Glnbrtr (lanes 2, 8, and 10). RNA was extracted from a nitrogen-fixing chemostat culture of Mm900 using the RNAeasy kit (Qiagen) with modifications as described previously ((Xia et al., 2006)). Genomic DNA was purified from M. maripaludis strain S2.
Supplementary Material
Acknowledgments
This work was supported by grant GM55255 from the National Institutes of Heath. We thank Mohamed Ouhammouch for his kind advice on footprinting procedures.
References
- Augustus AM, Reardon PN, Heller WT, Spicer LD. Structural basis for the differential regulation of DNA by the methionine repressor MetJ. J Biol Chem. 2006;281:34269–34276. doi: 10.1074/jbc.M605763200. [DOI] [PubMed] [Google Scholar]
- Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS. Methanogens: reevaluation of a unique biological group. Microbiol Rev. 1979;43:260–296. doi: 10.1128/mr.43.2.260-296.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CE, Kessler PS, Leigh JA. Genetics in methanogens: transposon insertion mutagenesis of a Methanococcus maripaludis nifH gene. J Bacteriol. 1995;177:5773–5777. doi: 10.1128/jb.177.20.5773-5777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kupiec R, Blank C, Leigh JA. Transcriptional regulation in Archaea: in vivo demonstration of a repressor binding site in a methanogen. Proc Natl Acad Sci U S A. 1997;94:1316–1320. doi: 10.1073/pnas.94.4.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kupiec R, Marx CJ, Leigh JA. Function and regulation of glnA in the methanogenic archaeon Methanococcus maripaludis. J Bacteriol. 1999;181:256–261. doi: 10.1128/jb.181.1.256-261.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl MK, Degenkolb J, Hillen W. Transcription of the xyl operon is controlled in Bacillus subtilis by tandem overlapping operators spaced by four base-pairs. J Mol Biol. 1994;243:413–424. doi: 10.1006/jmbi.1994.1669. [DOI] [PubMed] [Google Scholar]
- Dodsworth JA, Cady NC, Leigh JA. 2-Oxoglutarate and the PII homologues NifI1 and NifI2 regulate nitrogenase activity in cell extracts of Methanococcus maripaludis. Mol Microbiol. 2005;56:1527–1538. doi: 10.1111/j.1365-2958.2005.04621.x. [DOI] [PubMed] [Google Scholar]
- Gardner WL, Whitman WB. Expression vectors for Methanococcus maripaludis: overexpression of acetohydroxyacid synthase and beta-galactosidase. Genetics. 1999;152:1439–1447. doi: 10.1093/genetics/152.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiduschek EP, Ouhammouch M. Archaeal transcription and its regulators. Mol Microbiol. 2005;56:1397–1407. doi: 10.1111/j.1365-2958.2005.04627.x. [DOI] [PubMed] [Google Scholar]
- Haydock AK, Porat I, Whitman WB, Leigh JA. Continuous culture of Methanococcus maripaludis under defined nutrient conditions. FEMS Microbiol Lett. 2004;238:85–91. doi: 10.1016/j.femsle.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Hendrickson EL, Kaul R, Zhou Y, Bovee D, Chapman P, Chung J, Conway de Macario E, Dodsworth JA, Gillett W, Graham DE, Hackett M, Haydock AK, Kang A, Land ML, Levy R, Lie TJ, Major TA, Moore BC, Porat I, Palmeiri A, Rouse G, Saenphimmachak C, Soll D, VanDien S, Wang T, Whitman BW, Xia Q, Zhang Y, Larimer FW, Olson MV, Leigh JA. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J Bacteriol. 2004;186:6956–6969. doi: 10.1128/JB.186.20.6956-6969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CL, Carey J. Tandem binding in crystals of a trp repressor/operator half-site complex. Nature. 1993;366:178–182. doi: 10.1038/366178a0. [DOI] [PubMed] [Google Scholar]
- Leigh JA, Dodsworth JA. Nitrogen regulation in bacteria and archaea. Annu Rev Microbiol. 2007;61:349–377. doi: 10.1146/annurev.micro.61.080706.093409. [DOI] [PubMed] [Google Scholar]
- Lie TJ, Dodsworth JA, Nickle DC, Leigh JA. Diverse homologues of the archaeal repressor NrpR function similarly in nitrogen regulation. FEMS Microbiol Lett. 2007;271:281–288. doi: 10.1111/j.1574-6968.2007.00726.x. [DOI] [PubMed] [Google Scholar]
- Lie TJ, Leigh JA. Regulatory response of Methanococcus maripaludis to alanine, an intermediate nitrogen source. J Bacteriol. 2002;184:5301–5306. doi: 10.1128/JB.184.19.5301-5306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie TJ, Leigh JA. A novel repressor of nif and glnA expression in the methanogenic archaeon Methanococcus maripaludis. Mol Microbiol. 2003;47:235–246. doi: 10.1046/j.1365-2958.2003.03293.x. [DOI] [PubMed] [Google Scholar]
- Lie TJ, Wood GE, Leigh JA. Regulation of nif expression in Methanococcus maripaludis: roles of the euryarchaeal repressor NrpR, 2-oxoglutarate, and two operators. J Biol Chem. 2005;280:5236–5241. doi: 10.1074/jbc.M411778200. [DOI] [PubMed] [Google Scholar]
- Moore BC, Leigh JA. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. J Bacteriol. 2005;187:972–979. doi: 10.1128/JB.187.3.972-979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Sousa R. Use of Site-Specifically Tethered Chemical Nucleases to Study Macromolecular Reactions. Biol Proced Online. 2003;5:78–89. doi: 10.1251/bpo49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SE, Stockley PG. Similarity of met and trp repressors. Nature. 1994;368:106. doi: 10.1038/368106a0. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- Thomas G, Coutts G, Merrick M. The glnKamtB operon. A conserved gene pair in prokaryotes. Trends Genet. 2000;16:11–14. doi: 10.1016/s0168-9525(99)01887-9. [DOI] [PubMed] [Google Scholar]
- Tumbula DL, Makula RA, Whitman WB. Transformation of Methanococcus maripaludis and identification of a PstI-like restriction system. FEMS Microbiol Lett. 1994;121:309–314. [Google Scholar]
- White A, Ding X, vanderSpek JC, Murphy JR, Ringe D. Structure of the metal-ion-activated diphtheria toxin repressor/tox operator complex. Nature. 1998;394:502–506. doi: 10.1038/28893. [DOI] [PubMed] [Google Scholar]
- Whitman WB, Shieh J, Sohn S, Caras DS, Premachandran U. Isolation and characterization of 22 mesophilic methanococci. System Appl Microbiol. 1986;7:235–240. [Google Scholar]
- Xia Q, Hendrickson EL, Zhang Y, Wang T, Taub F, Moore BC, Porat I, Whitman WB, Hackett M, Leigh JA. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Mol Cell Proteomics. 2006;5:868–881. doi: 10.1074/mcp.M500369-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang T, Hendrickson EL, Lie TJ, Hackett M, Leigh JA. Quantitative proteomics of nutrient limitation in the hydrogenotrophic methanogen Methanococcus maripaludis. BMC Microbiol. 2009;9:149. doi: 10.1186/1471-2180-9-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.