

Abstract
Autophagy is a protective mechanism that renders cells viable in stressful conditions. Emerging evidence suggests that this cellular process is also a tumor suppressor pathway. Previous studies showed that cyclin-dependent kinase inhibitors (CDKI) induce autophagy. Whether retinoblastoma protein (RB), a key tumor suppressor and downstream target of CDKIs, induces autophagy is not clear. Here, we show that RB triggers autophagy and that the RB activators p16INK4a and p27/kip1 induce autophagy in an RB-dependent manner. RB binding to E2 transcription factor (E2F) is required for autophagy induction and E2F1 antagonizes RB-induced autophagy, leading to apoptosis. Downregulation of E2F1 in cells results in high levels of autophagy. Our findings indicate that RB induces autophagy by repressing E2F1 activity. We speculate that this newly discovered aspect of RB function is relevant to cancer development and therapy.
Introduction
Autophagy is a cellular process that engulfs organelles and cytoplasmic contents to digest and recycle these materials to sustain cellular metabolism (1). In addition to providing a basic catabolic function, autophagy is also used by the cell to cope with stressful conditions to improve survival (2). Emerging evidence suggests that autophagy is a tumor suppressor pathway (2, 3), although the detailed mechanisms governing this pathway are largely unknown. Indeed, several tumor suppressor proteins, including phosphatase and tensin homologue (PTEN) and p53 (2), induce autophagy. Conversely, multiple oncogenes, including Bcl-2 and the AKT/target of rapamycin (TOR) pathway, inhibit autophagy (2). Of particular importance, the inactivation of several key autophagy regulators, such as Beclin 1 and ATG4, results in spontaneous tumors (4) or increases the formation of tumors by carcinogens in mice (5).
Retinoblastoma protein (RB) is considered the prototype of tumor suppressor proteins, and as such, inactivation of RB is found in an ample array of cancers (6). In tumors with wild-type RB gene, RB is frequently inactivated by the abnormal expression of activators, such as the p16INK4a (p16) protein, and repressors, including cyclin D1 and cyclin-dependent kinase-4 (6). Interestingly, loss of RB in cancer cells leads to increased sensitivity to therapy (7–9) and activation of RB through transfer of p16 increases chemoresistance (10). The mechanisms underlying this RB-mediated resistance are not completely understood. Recently, autophagy has been connected to resistance to cancer therapy (2). On the basis of previous studies in which cyclin-dependent kinase inhibitors (CDKI), activators of RB, were shown to induce autophagy (11, 12), we hypothesized that RB, a key tumor suppressor and downstream target of CDKIs, induces autophagy.
Moreover, the physical interaction between RB and E2 transcription factor 1 (E2F1) is perhaps the best-understood mechanism of how RB functions. These two proteins play opposite roles in the regulation of the cell cycle and apoptosis. RB promotes growth arrest, whereas E2F1 positively regulates the transition from the G1 to the S phase (13); RB protects from apoptosis (7), whereas E2F1 induces apoptosis (14). Thus, we asked whether RB and E2F1 also play antagonistic roles in the regulation of autophagy.
In this study, we examined the role of RB/E2F1 in the regulation of autophagy. Our data indicate that RB induces autophagy by repressing E2F1 activity, and E2F1 antagonizes RB activity to prevent autophagy.
Materials and Methods
Cell culture
Human osteosarcoma (U-2 OS), sarcoma osteogenic (Saos-2), hepatoma (Hep3B), and glioblastoma-astrocytoma (U-87 MG) cells (American Type Culture Collection) were cultured in DMEM/F-12 supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories, Inc.), 100 μg/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). The 293 cell line (Qbiogene, Inc.) was cultured in DMEM supplemented with 10% FBS and antibiotics. Brain tumor (MDNSC23) stem cells were established from acute cell dissociation of surgical specimens of human glioblastoma multiforme and maintained in DMEM/F-12 supplemented with B27 (Invitrogen), epidermal growth factor, and basic fibroblast growth factor (20 ng/mL each; Sigma-Aldrich) according to procedures described elsewhere (15).
Reagents and antibodies
Acridine orange was obtained from Sigma-Aldrich. Rabbit polyclonal p16 (N-20), rabbit polyclonal E2F1 (C-20), and goat polyclonal actin (I-19) antibodies were obtained from Santa Cruz Biotechnology. Rabbit polyclonal Bcl-2, light chain 3B (LC3B), and Beclin 1 antibodies were obtained from Cell Signaling Technology. Mouse monoclonal RB was obtained from Abcam, and monoclonal anti–α-tubulin (B-5-1-2) was obtained from Sigma-Aldrich.
Adenoviruses
To construct recombinant adenoviruses expressing RB mutants, we made the Δ22 deletion (amino acids 738–775; ref. 16) and R661W mutation (17) required for E2F binding. The site-directed mutagenesis for the deletion and mutation was conducted with the use of the QuikChange II XL site-directed mutagenesis kit (Stratagene) in pXCJL-RB, a shuttle vector that includes the expression cassette for wild-type RB driven by human cytomegalovirus (CMV) promoter (18). We confirmed the mutations in RB cDNA and the fidelity of the mutagenesis reaction by sequencing the whole RB cDNA region in the resultant pXCJL-RBΔ22 and pXCJL-RB R661W plasmids. We then cotransfected the pXCJL-RBΔ22 or pXCJL-RB R661W plasmid with pBHG10 (Microbix Biosystems, Inc.) into 293 cells to generate an AdCMV-RBΔ22 or AdCMV-RB R661W adenovirus. The recombinant adenoviruses were prepared as previously described (19). Briefly, the viruses were amplified in 293 cells, purified by three successive bandings on cesium, and stored at −80°C. The viral titer was assayed by the tissue culture infection dose 50 (TCID50) and determined as plaque-forming units (pfu) per milliliter, according to the validated method developed by Quantum Biotechnology. Other recombinant adenoviruses used in the study included AdCMV-RB (18), AdRSV-p16 (18), AdCMV, AdCMV-green fluorescent protein (GFP), AdCMV-E2F1 (20), AdMH4p27 (11), and AdCMV-β-gal (all prepared by the Keck Institutional Vector Core at M.D. Anderson Cancer Center).
Immunoprecipitation and immunoblotting
The cells were suspended and lysed in ice-cold PBS plus 1% NP40 and a protease inhibitor cocktail (Sigma-Aldrich). For each sample, 2 μg of anti-E2F1 antibody and 40 μL of protein A-agarose beads (Upstate Biotechnology) were added to 500 μg of cell lysate proteins. After 2 hours of immunoprecipitation at 4°C, the beads were pelleted and washed. The precipitated proteins or cell lysate proteins were dissolved in 1× SDS loading buffer, separated by SDS-PAGE, and probed with antibodies. Finally, the protein bands were visualized with the ECL Western blot detection system (Amersham Pharmacia Biotech).
Subcellular localization of light chain-3 fusion proteins
Saos-2 and U-87 MG cells were transfected with enhanced green fluorescent protein-light chain 3 (EGFP-LC3; a kind gift from Dr. S. Kondo, The University of Texas M.D. Anderson Cancer Center) or mRFP-EGFP-LC3 (Addgene; ref. 21) fusion protein–expressing plasmid by FuGENE 6 (Roche Molecular Biochemicals). The cells were then screened with G418 (800 μg/mL for Saos-2, 700 μg/mL for U-87 MG). Positive clones were selected and maintained in culture medium plus G418. The cells expressing EGFP-LC3 were first fixed in 4% paraformaldehyde in PBS for 30 minutes at 4°C. Saos-2-mRFP-EGFP-LC3 cells were observed alive. Images were taken and processed on a Zeiss Axiovert Zoom fluorescence microscope (Carl Zeiss, Inc.) equipped with an AxioCam MRM camera and a 40×/1.3 oil EC Plan-NEOFLUAR objective, using Immersol (n = 1.518) at room temperature. The acquisition software was AxioVision Release 4.7.1 (Carl Zeiss). Images were processed by Photoshop (Adobe) with the use of proportional adjustments. We counted 150 to 200 cells from three independent experiments for quantitative analysis.
Electron microscopy
Samples were fixed with a solution containing 3% glutaral-dehyde plus 2% paraformaldehyde in 0.1 mol/L cacodylate buffer (pH 7.3) for 1 hour. After fixation, the samples were washed and treated with 0.1% Millipore-filtered cacodylate-buffered tannic acid (Sigma-Aldrich), postfixed for 1 hour with 1% buffered osmium tetroxide (Sigma-Aldrich), and then stained with 1% Millipore-filtered uranyl acetate (Sigma-Aldrich). The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and then embedded in Spurr’s low-viscosity medium. The samples were polymerized in an oven for 2 days at 70°C. Ultrathin sections were cut in a Leica Ultracut microtome (Leica), stained with uranyl acetate and lead citrate in a Leica EM stainer, and examined by using a JEM-1010 transmission electron microscope (JEOL USA, Inc.) at an accelerating voltage of 80 kV. Digital images were obtained with the use of an imaging system from Advanced Microscopy Techniques.
RNA interference
The siRNA for Beclin 1 depletion was obtained from Santa Cruz Biotechnology. We purchased custom and control siRNAs from Dharmacon, Inc. The E2F1 siRNA (siE2F1) sequence was as follows: 5′-GGCCCGAUCGAUGUUUUC-CUU-3′ (22). The cells were mock or siRNA transfected with INTERFERin (Polyplus-transfection, Inc.) according to the manufacturer’s protocol. To knock down RB expression in U-87 MG cells, we transfected the cells with shGFP- or shRB-expressing plasmids (23) by FuGENE 6. The cells were screened, and stable cell clones were maintained in culture medium plus 0.5 μg/mL puromycin.
Autophagy PCR array
Total RNA was extracted from cells with the RNeasy Mini Kit (Qiagen, Inc.). The RNA samples were then treated with TURBO DNA-free kit to remove any DNA contamination (Ambion) and sent to SABiosciences for analysis with the Autophagy PCR Array.
Luciferase assay
Cells were seeded in 24-well plates (3 × 104 cells per well) and infected with adenovirus vectors expressing GFP, RB, or its mutants (100 pfu/cell). Twenty-four hours later, we cotransfected the cells with 250 ng of the E2F1 reporter plasmid (24) and 1 ng of pRL-CMV (Promega Life Science) as a control for transfection efficiency with FuGENE 6 (Roche Molecular Biochemicals). Twenty-four hours after transfection, the cells were lysed, and luciferase activity was assayed with a luminometer.
Quantification of apoptotic cells with Annexin V-FITC staining
Apoptotic cells were stained with Annexin V-FITC using an Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer’s instructions. Stained cells were then analyzed by flow cytometry with a FACScan cytometer and CellQuest software (both from Becton Dickinson).
Statistical analyses
We performed a two-tailed Student’s t test to determine the statistical significance of the experiments. P < 0.05 was considered statistically significant. Data were given as mean ± SD.
Results
RB induces autophagic responses in RB-deficient cells
To test our hypothesis that RB induces autophagy, we first transduced RB into the following RB-defective human cancer cells: sarcoma osteogenic cells (Saos-2), hepatoma cells (Hep3B), and brain tumor stem cells (MDNSC23; ref. 15). The exogenous RB protein induced autophagy in all three cell lines (Fig. 1; Supplementary Fig. S1). Specifically, we examined whether the transfer of RB induced the conversion of the microtubule-associated protein LC3-I to LC3-II, a biochemical marker of autophagy that is correlated with the formation of autophagosomes. Immunoblotting analyses revealed an accumulation of LC3-II in the cells treated with RB, but not in the mock-treated or GFP-treated cells (Fig. 1A). Consistently, RB also triggered the formation of autophagosomes/autolysosomes visualized by punctate EGFP-LC3 in Saos-2 cells that constitutively express the fusion protein (Fig. 1B). In more than 80% of the RB-treated cells, EGFP-LC3 was not soluble and presented green fluorescent dots, whereas less than 20% of the control cells displayed the same pattern (Fig. 1B). Ultimately, these data were confirmed by an ultrastructural study of the cells with a transmission electron microscope, which revealed accumulation of membrane-rimmed vacuoles with characteristics of autophagosomes/autolysosomes in Saos-2 (Fig. 1C) and Hep3B (Supplementary Fig. S1) cells after RB expression.
Figure 1.

RB induces autophagy responses in RB-deficient cells. A, immunoblot analysis of LC3 and RB. Cells were infected with AdCMV-RB (100 pfu/cell). After 72 h, cell lysates were collected. Note the accumulation of LC3-II in RB-expressing cells but not in mock-treated or GFP-expressing cells. Actin was used as a loading control. AdCMV-GFP was used as a control for viral infection and nonspecific protein expression. B, green fluorescence punctation in Saos-2 cells constitutively expressing EGFP-LC3. The cells were infected with AdCMV-RB (100 pfu/cell). Seventy-two hours later, the cells were fixed and then examined by fluorescence microscopy. Left, representative images of the cells with the indicated treatments. Right, quantification of the cells with EGFP-LC3 dots (columns, mean; bars, SD). *, P = 0.003; **, P > 0.05. AdCMV (Vector) was used as a control for viral infection. Bar, 20 μm. C, representative transmission electron microscopy images of autophagosome/autolysosome accumulation in Saos-2 cells. The cells were infected with AdCMV-RB (100 pfu/cell) for 72 h. AdCMV-GFP was used as a control for viral infection and nonspecific protein expression. Membrane-bordered vacuoles contain cytoplasmic components (arrows) in the cytoplasm of RB-expressing cells (bottom left) but not in mock-treated or GFP-expressing cells (top). A close-up of the vacuoles revealed double membrane in the autophagosome with darker content or single membrane in autolysosome with lighter content (bottom, arrowheads). Bar, 500 nm. D, immunoblot of LC3, Beclin 1, and RB. The cells were first transfected with the indicated siRNA (10 nmol/L). After 24 h, the cells were infected with AdCMV-RB (100 pfu/cell) for 72 h. Actin was used as loading control. CsiRNA, control siRNA; siBeclin 1, siRNA against Beclin 1.
We then challenged the autophagy mechanism by down-modulating Beclin 1, a key activator of the pathway. As expected, silencing Beclin 1 in Saos-2 cells with siRNA decreased the RB-mediated processing of the formation of autophagosomes/autolysosomes and membrane-bound LC3-II (Fig. 1D; Supplementary Fig. S2). In agreement with these results was our observation that RB expression in Saos-2 cells, in a PCR-based array, stimulated the expression of several autophagy-related genes (Supplementary Fig. S3), such as ATG4A and ATG7. However, Bcl-2, which inhibits the autophagy process and whose gene is a well-studied transcriptional target of E2F1 (25, 26), was down-modulated (Supplementary Fig. S3). Overall, our data indicate that RB restoration in RB-deficient cells triggers autophagy.
RB promotes autophagosome formation and maturation
The accumulation of autophagosomes and LC3-II in the cells could be due to either the increased formation or the decreased turnover of vesicles. To determine how RB affects the autophagy flux, we took advantage of the double-tagged mRFP-EGFP-LC3 construct (ptfLC3; ref. 21), which is detected as yellow fluorescence (green merged with red) in nonacidic structures (autophagosomes and amphisomes) and as red only in autolysosomes due to the quenching of EGFP in these acidic structures. As shown in Fig. 2, more than 70% of the RB-transduced cells displayed tremendous red fluorescence punctation, whereas only less than 10% of the mock- or AdCMV-treated cells showed this phenotype. However, when the cells were treated with bafilomycin A1 (which inhibits the fusion of autophagosomes with lysosomes) to stop the turnover of autophagosomes, different from the cells transduced with RB, we found an extraordinary amount of punctate yellow fluorescence in the cytoplasm (Fig. 2A). These data indicate that RB increases the transport of mRFP-EGFP-LC3 to acidic lysosomes (i.e., formation of autolysosomes). Moreover, electron microscopic examination of the cells revealed that bafilomycin A1 caused the accumulation of vacuoles in RB-expressing cells that filled most of the space in the cytoplasm, whereas the vacuoles in mock or GFP-expressing cells occupied less than half of the space in the cytoplasm (Supplementary Fig. S4), indicating that RB stimulated a higher rate of autophagosome formation. Taken together with the previous observations, we conclude that RB mediates the initiation and maturation of autophagosomes instead of inhibiting the fusion of autophagosomes and lysosomes.
Figure 2.

RB mediates autophagosome maturation in Saos-2 cells. Saos-2 cells constitutively expressing mRFP-EGFP-LC3 fusion protein were infected with AdCMV-RB at 100 pfu/cell in the absence or presence of bafilomycin A1 (BA1). Three days later, live cells were observed by fluorescence microscopy. A, representative images of the cells with the indicated treatments. Bar, 20 μm. B, quantification of the cells with red fluorescent dots (columns, mean; bars, SD). *, P = 0.008. AdCMV (Vector) was used as a control for viral infection.
CDKIs cause autophagy in an RB-dependent manner
Because RB activity is positively regulated by CDKIs, such as p16 and p27/kip1 (p27), we asked whether activation of the RB pathway by CDKIs would result in autophagy in these cells. To this end, we transferred p16 to glioblastoma-astrocytoma (U-87 MG) and human osteosarcoma (U-2 OS) cell lines that express wild-type RB. The ectopic p16 protein triggered the lipidation of LC3, resulting in an increase of LC3-II in both cell lines (Fig. 3A). Accordingly, more than 60% of the U-87-EGFP-LC3 cells showed a marked punctate pattern of green fluorescence after transduction of p16, whereas less than 20% of the control cells showed a similar pattern (Fig. 3B). Moreover, when we transferred p16 to U-87-shRB cells, in which RB expression had been knocked down by shRNA against RB (Fig. 3C), we observed that down-modulation of RB prevented the conversion of LC3-I to LC3-II, indicating that it rendered U-87 MG cells resistant to p16-mediated autophagy (Fig. 3C).
Figure 3.

CDKIs trigger autophagy in an RB-dependent manner. A, immunoblot analysis of LC3 and p16. The cells were infected with AdRSV-p16 (300 pfu/cell) for 72 h. AdCMV-GFP was used as a control for viral infection and nonspecific protein expression. Actin was used as a loading control. B, green fluorescence punctation in U-87-EGFP-LC3. The cells were infected with AdRSV-p16 (300 pfu/cell); 72 h later, the cells were fixed and then examined by fluorescence microscopy. Top, representative images of the cells with the indicated treatments. Bottom, quantification of the cells with EGFP-LC3 dots (columns, mean; bars, SD). *, P = 0.004. Bar, 20 μm. C, top, immunoblot analysis of RB in U-87 MG cells transfected with shGFP or shRB-expressing plasmid. α-Tubulin was used as a loading control. Bottom, immunoblot analysis of LC3 and p16. The cells were infected with AdRSV-p16 (300 pfu/cell) for 72 h. AdCMV-β-gal was used as a control for viral infection and nonspecific protein expression. Actin was used as a loading control. D, immunoblot analysis of LC3. Cells were infected with AdMH4p27 at the indicated doses (pfu/cell). Seventy-two hours later, cell lysates were collected for immunoblot analysis. AdCMV-β-gal (100 pfu/cell) was added as a control for nonspecific protein expression. Actin was used as a loading control.
In addition to p16, the best-documented CDKI that is involved in the positive regulation of autophagy is p27 (11, 12). Unlike p16, p27 has other targets in addition to RB (27–29). When we transferred p27 to U-2 OS and Saos-2 cells, U-2 OS cells were much more sensitive to p27 in a dose-dependent manner, resulting in a conversion of LC3-I to LC3-II that was not observed in Saos-2 cells (Fig. 3D), suggesting that RB might be one of the factors involved in p27-mediated autophagy.
Collectively, these data indicate that RB expression is required for p16-mediated autophagy and that RB could be one of the downstream targets involved in p27-induced autophagy. These data therefore reinforce the role of the RB pathway in autophagy regulation.
Binding to E2F is required for RB to trigger autophagy
The best-known mechanistic model of RB indicates the physical binding and repression of E2F transcription factors, including E2F1 (30). Thus, we speculated whether RB mutants that are deficient for E2F binding would lose the capability to induce autophagy. We selected two RB mutants naturally found in cancer cells that are deficient in binding E2F. Coimmunoprecipitation assays showed that both RB mutants Δ22 (16) and R661W (17) displayed impaired ability in binding E2F1 (Fig. 4A). Accordingly, the two RB mutants were deficient in blocking the transactivation of the E2F1 responsible element, as quantified by luciferase assays (Fig. 4B). Then, we examined autophagy occurrence in Saos-2 cells transduced with the two mutants. We found that the expression of Δ22 or R661W resulted in EGFP-LC3 punctation in less than 20% of the cells, whereas wild-type RB induced green fluorescence dots in more than 80% of the cells (Fig. 4C). Consistently, neither Δ22 nor R661W induced significant LC3-I to LC3-II conversion, according to immunoblot analysis (Fig. 4D). Therefore, RB mutants with impaired binding to E2F are deficient for autophagy induction, indicating that RB binding to E2F is required for RB-induced autophagy.
Figure 4.

RB mutants deficient for E2F binding are unable to induce autophagy in Saos-2 Cells. A, immunoprecipitation analysis of RB and its mutants interacting with E2F1. The cells were infected with AdCMV-RB (100 pfu/cell) and AdCMV-E2F1 (5 pfu/cell), and cell lysates were collected for immunoprecipitation analysis after 48 h. B, effect of RB and RB mutants on E2F1 activity. The cells were infected with the indicated vectors (100 pfu/cell). After 24 h, the cells were transfected with a luciferase-reporting plasmid with an E2F1 promoter. Twenty-four hours later, the luciferase activity of the samples was determined (columns, mean; bars, SD). *, P = 0.0007; #, P = 0.0004; $, P = 0.003; **, P = 0.4. C, green fluorescence punctation in EGFP-LC3–expressing Saos-2 cells. The cells were infected with adenovirus vectors expressing RB and its mutants (100 pfu/cell) for 72 h. Left, representative images of the cells with the indicated treatments. Right, quantification of the cells with EGFP-LC3 dots (columns, mean; bars, SD). *, P = 0.0002; **, P > 0.05. Bar, 20 μm. D, immunoblot analysis of RB and LC3 in Saos-2 cells infected with adenoviruses expressing RB or its mutants. The cells were infected with the indicated vectors for 72 h. The numbers on the top of the protein bands represent the doses of the viruses for infection (pfu/cell). Actin was used as a loading control.
E2F1 antagonizes RB-mediated autophagy
RB and E2F1 exert contrary functions in the regulation of the cell cycle and apoptosis (7, 13). We previously reported that expression of ectopic E2F1 overrides the cell cycle arrest induced by RB (20). In this study, we were interested in ascertaining whether the expression of E2F1 would be sufficient to antagonize RB-mediated autophagy. Thus, we transferred RB and then E2F1 into Saos-2 cells 24 hours later. Examination of the cells 48 hours after the transfer of E2F1 revealed that E2F1 was able to partially override RB function and prevent the formation of the green-dot phenotype and the conversion of LC3-I to LC3-II (Fig. 5A and B). These studies showed a progressive decrease in the percentage of cells with punctate green fluorescence: from almost 100% in cells treated with RB, to 40% in cells treated with both RB and E2F1, to less than 20% in cells treated with E2F1 alone (Fig. 5A). Consistently, Bcl-2 protein, an autophagy inhibitor, was downregulated by RB but was upregulated by E2F1 (Fig. 5B).
Figure 5.

RB/E2F1 interaction regulates autophagy and apoptosis. A, green fluorescence punctation in EGFP-LC3–expressing Saos-2 cells. The cells were first infected with AdCMV-RB (100 pfu/cell). After 24 h, the cells were infected with AdCMV-E2F1 (50 pfu/cell) for another 48 h. Top, representative images of the cells with the indicated treatments. Bar, 20 μm. Bottom, quantification of the cells with EGFP-LC3 punctation (columns, mean; bars, SD). *, P = 0.03. B, immunoblot analysis of LC3, E2F1, and RB in Saos-2 cells. The cells were first infected with AdCMV-RB (100 pfu/cell). After 24 h, the cells were infected with AdCMV-E2F1 (50 pfu/cell). Proteins from the cell lysates were collected for analysis after 48 h. C, cells were treated as described in B and were stained with Annexin V and analyzed by flow cytometry. AdCMV-β-gal was used as a control for viral infection and nonspecific protein expression. Columns, mean from at least three independent experiments; bars, SD. *, P = 0.007; #, P = 0.001.
Because expression of E2F1 induces apoptosis (20), we asked whether the cells that were transduced by E2F1 were undergoing cell death through apoptosis. Optical microscopy showed that although RB-transduced cells displayed a flat morphology and remained attached to the culture dish, some of the cells transduced with both RB and E2F1 became rounded, birefringent, and detached from the dish (data not shown), strongly suggesting decreased viability of the culture. To determine whether the cause of the cell death was apoptosis, we used flow cytometry to examine the ability of the cells to bind to Annexin V, a reagent to detect the translocation of the membrane phospholipid phosphatidylserine from the inner to the outer leaflet of the plasma membrane—one of the earliest indications of apoptosis. Although the percentage of autophagic cells was less than 10% in E2F1-treated cells (Fig. 5A), the treatment caused about 40% of the cells to undergo apoptosis (Fig. 5C). Thus, we conclude that E2F1 antagonizes RB-mediated autophagy and leads to apoptosis. These results suggest that RB and E2F1 proteins play opposite roles in the control of both autophagy and apoptosis.
Downregulation of E2F1 results in autophagy
Because RB needs to bind to E2F to induce autophagy, and E2F1 antagonizes RB-mediated autophagy, we hypothesized that cells lacking E2F1 expression would be more susceptible to spontaneous autophagy and that the levels of autophagy in these cells would be significantly higher than the levels in control cells. To test this hypothesis, we used siRNA to down-modulate E2F1 expression in U-87 MG cells. Silencing the E2F1 gene resulted in the formation of autophagic vacuoles, as documented by a dramatic lipidation of LC3 that caused EGFP-LC3 punctation in more than 60% of the cells (Fig. 6A and B). In contrast, less than 10% of the control cells showed a dotted pattern (Fig. 6B). Consistently, immunoblotting analysis revealed that E2F1 siRNA downregulated E2F1 protein level and caused the conversion of LC3-I to LC3-II (Fig. 6C). These results suggest that down-modulation of E2F1 is sufficient to trigger autophagy and that E2F1 plays an active role in suppressing autophagy. Thus, the increased level of autophagy in E2F1–down-modulated cells indirectly points to the inhibition of E2F1 as the mechanism for RB-mediated autophagy.
Figure 6.

Downregulation of E2F1 results in autophagy. A, green fluorescence punctation in U-87 MG cells constitutively expressing EGFP-LC3. The cells were transfected with siRNA (20 nmol/L) for 48 h. Representative images of the cells with the indicated treatments. B, quantification of the cells with dotted EGFP-LC3 (columns, mean; bars, SD). *, P = 0.002; **, P = 0.3 (Student’s t test). Bar, 20 μm. C, immunoblot analysis of LC3 and E2F1 in U-87 MG cells treated as described in A. α-Tubulin was used as a loading control.
Discussion
The RB/E2F1 pathway, which is crucial in regulating cell growth and apoptosis, is disrupted in virtually all human cancers (31). Here, we report that RB positively regulates autophagy by repressing E2F1 activity. Our results indicate that, instead of preventing autophagosome turnover in the cells, RB activity, or lack of E2F1 activity, promotes autophagy flux, from the formation of autophagosomes to their fusion with lysosomes. Our data reveal a previously unknown function of the RB/E2F1 pathway that might contribute to its role in cancer suppression and resistance to cancer therapy.
A balanced RB/E2F1 pathway is required to maintain cellular homeostasis. We found that excessive RB activity or lack of E2F1 causes autophagy, and RB/E2F interaction is required for RB-mediated autophagy. Moreover, excessive E2F1 activity antagonizes RB-induced autophagy, leading to apoptosis. Therefore, in addition to the opposite roles of RB and E2F1 in regulating the cell cycle and apoptosis (30), our data suggest a model in which the interplay between RB and E2F1 also regulates autophagy (Fig. 7). We speculate that adequate RB/E2F1 activity ensures a basal level of autophagy that is critical to maintaining normal cellular and molecular processes in the cell. Given the negative effects of autophagy on tumorigenesis (2), autophagy could be part of the mechanism through which RB acts as a tumor suppressor. In this regard, the involvement of PTEN, p53, and RB, the three most frequently mutated genes in human cancers, in regulating autophagy suggests a strong mechanistic link between stress-sensing pathways, autophagy, and tumor suppression. Tumor suppressors, such as RB, may induce autophagy as a housekeeping mechanism to recycle damaged macromolecules and organelles to ensure the stability of the genome (32). In fact, it has been reported that the basal levels of proteolysis or autophagic degradation in cancer cells were lower than those in their normal counterparts (33). In this regard, cells that were transformed by SV40, which can inactivate RB and p53, had decreased levels of proteolysis compared with nontransformed or nontreated cells (34). Lately, autophagy was indicated as an effector mechanism of senescence (35). Given the critical role that RB plays during cellular senescence (36) and in the response to cancer therapeutics such as CDK inhibitors (37), there might be a link between RB-mediated autophagy and senescence and its consequential tumor suppression.
Figure 7.
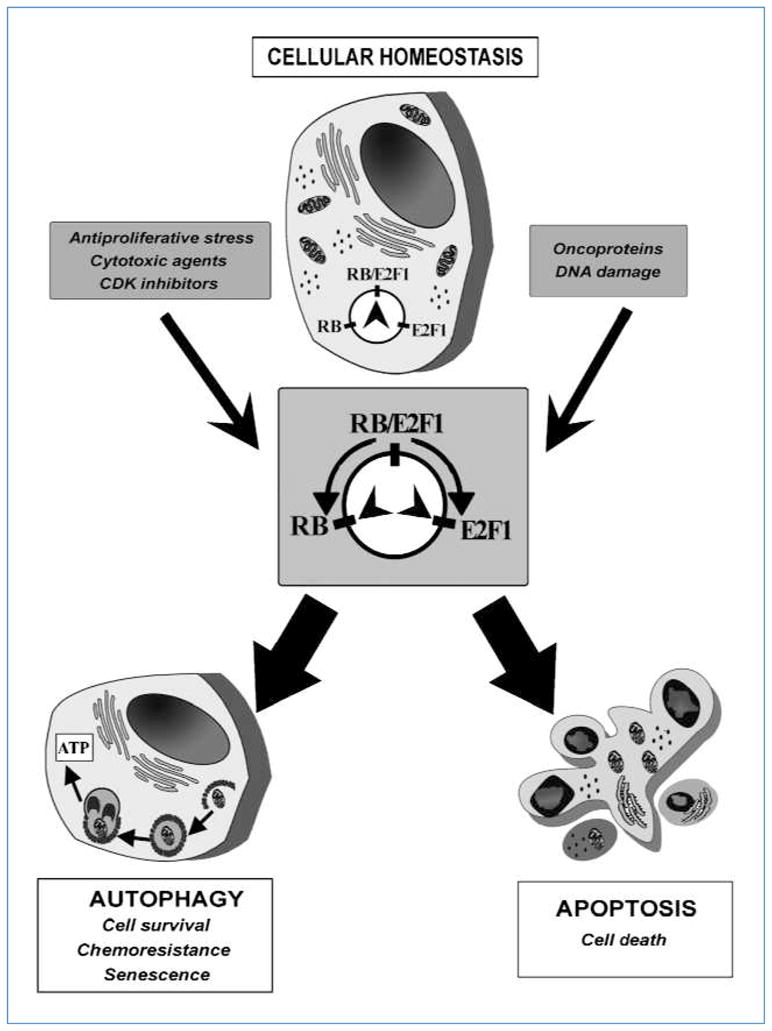
Rheostat model for the interplay of RB and E2F1 to regulate autophagy and apoptosis. Balanced RB and E2F1 activity ensures homeostasis in the cell. Strong stimuli may result in either excessive RB activity to induce autophagy or immoderate E2F1 function to cause apoptosis.
Aside from its housekeeping function, autophagy can also act as a protective mechanism to prolong cell survival under stressful conditions (38). In certain scenarios, autophagy constitutes a stress adaptation that suppresses apoptosis to avoid cell death (39). In addition to its central role in inhibiting cell proliferation, RB also plays a crucial role in inhibiting apoptosis (7). Studies in RB-mutant mice show massive apoptosis in neurons (40). Interestingly, Atg7 deficiency in the central nervous system also causes massive neuronal loss in the cerebral and cerebellar cortices (41), suggesting a cross-talk between autophagy and apoptosis (39). Thus, on top of autophagy/apoptosis regulation by RB/E2F1 interplay (Fig. 7), RB-mediated autophagy might inhibit apoptosis as well. Moreover, during its growth, the tumor is usually poorly vascularized in the center area, and in this environment, autophagy has a more prominent role in sustaining cell viability in cancer cells (42). Previous work has shown that most sporadic cancers inactivate RB by phosphorylation, rather than by mutations in the RB gene itself (7). Because phosphor-ylation is reversible, altering this posttranslational modification pathway of RB not only allows cancer cells to grow aggressively but also gives these cells the option of reactivating RB for protection against apoptotic stimuli to exploit the survival advantage conferred by RB under stress (7). For instance, glucose-regulated stress, a potent inducer of autophagy (12), causes stabilization of p27 (12) and decreased expression of cyclin D1 (43), resulting in hypophosphorylation of RB, which is the active form of the protein (6). Our data show that CDKIs, such as p27 and p16, induce autophagy in a RB-dependent manner (Fig. 3), indicating the involvement of RB-mediated autophagy in the stress response. Thus, it is of clinical relevance to determine whether RB activity in cancer cells expressing wild-type RB is partially restored under these conditions to induce autophagy to prevent apoptotic cell death. Furthermore, results from experimental anticancer therapies suggest that autophagy activation represents a cellular attempt to survive the stress induced by cytotoxic agents (2). In fact, it has been reported that restoration of the RB protein in RB-deficient cells results in resistance to DNA-damaging agents including cisplatin, etoposide, 5-fluorouracil, and doxorubicin (44, 45). In addition, expression of p16 is associated with chemoresistance in glioma and bladder cancer cells (10, 46). Overall, our study suggests that RB-induced autophagy provides a possible new mechanistic link between RB, cancer cell survival, and resistance to cancer therapy.
Although the role of RB in modulating autophagy seems to be well defined here, the role of E2F1 could be more complex. In fact, E2F1 has a dual role as a tumor suppressor and an oncogene and, as such, can positively regulate both cell proliferation and cell death (47). Thus, E2F1 may exert a contextual role in autophagy as well. A weak E2F1 transactivation of autophagy-related genes was observed in a drug-induced E2F1-expressing system (48). However, our data show that overactivated E2F1 blocked RB-induced autophagy and led to apoptosis (Fig. 5). One of the possible molecular targets of E2F1 in the suppression of autophagy is Bcl-2, which blocks autophagy by binding Beclin 1 and prevents activation of the PI3Kc3 complex (25). Our previously published results revealed that Bcl-2 is a transcriptional target of E2F1 (26). Here, we show that repression of E2F1 by RB leads to down-modulation of Bcl-2 (Supplementary Fig. S3; Fig. 5), contributing to the triggering of autophagy. The mammalian TOR (mTOR) pathway is a key negative regulator of autophagy (49). We examined the phosphorylation status of p70S6K, a substrate of mTOR (50). We did not observe any changes in the total p70S6K level and its phosphorylation level after introducing RB into Saos-2 and MDNSC23 cells or p16 into U-87 MG and U-2 OS cells (data not shown). We assume that mTOR does not play a role under this circumstance. Thus, RB-induced autophagy is more relevant to E2F1-mediated Bcl-2 expression than affecting the mTOR pathway. However, it is now clear that E2F1 transactivates more than 2,000 genes and that the role of E2F1 in regulating transcription is highly dependent on context (47). Therefore, it is overly simplistic to identify one or two players to completely explain the delicate balance between RB and E2F1 in the regulation of autophagy. Besides, further study will be required to determine whether RB/E2F1 modulation of autophagy is independent of its role in cell cycle regulation.
On the other hand, RB interacts with more than 200 proteins (6), one of which is the hypoxia-inducible factor, which is a potent inducer of autophagy under hypoxic conditions (51). In these extreme circumstances, RB protein seems to negatively modulate hypoxia-inducible factor–induced autophagy by attenuating BNIP3 induction (52). However, in our system, transfer of RB to cancer cells under normoxia resulted in the upregulation of BNIP3 (Supplementary Fig. S3), suggesting a marked difference between the regulation of autophagy under normoxic and hypoxic conditions.
In summary, our data unequivocally show that the RB/E2F1 pathway plays a role in the regulation of autophagy. Moreover, the existence of such a rheostat-like module to regulate the delicate balance between autophagy and apoptosis (Fig. 7) provides a new interpretation for the role of RB in tumor suppression during development, cell survival under stressful conditions, and cancer cell resistance to therapy.
Supplementary Material
Acknowledgments
We thank Dr. Jinsong Liu (Department of Pathology, M.D. Anderson) for the shRB plasmid, Dr. Seiji Kondo for EGFP-LC3 plasmid (Brain Tumor Center, M.D. Anderson), Tamara K. Locke and Joseph Munch (Department of Scientific Publications, M.D. Anderson) for editorial assistance, and Kenneth Dunner, Jr. for electron microscopy analysis.
Grant Support
National Cancer Institute (NIH/P50CA127001 to J. Fueyo), The Marcus Foundation (to J. Fueyo), The University of Texas M.D. Anderson Cancer Center (Institutional Research Grant to J. Fueyo), and the High Resolution Electron Microscopy Facility (Core Grant CA-16672 to M.D. Anderson Cancer Center).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–4. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 4.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–83. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 6.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 7.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 8.Stengel KR, Dean JL, Seeley SL, Mayhew CN, Knudsen ES. RB status governs differential sensitivity to cytotoxic and molecularly targeted therapeutic agents. Cell Cycle. 2008;7:1095–103. doi: 10.4161/cc.7.8.5737. [DOI] [PubMed] [Google Scholar]
- 9.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–24. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grim J, D’Amico A, Frizelle S, Zhou J, Kratzke RA, Curiel DT. Adenovirus-mediated delivery of p16 to p16-deficient human bladder cancer cells confers chemoresistance to cisplatin and paclitaxel. Clin Cancer Res. 1997;3:2415–23. [PubMed] [Google Scholar]
- 11.Komata T, Kanzawa T, Takeuchi H, et al. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br J Cancer. 2003;88:1277–80. doi: 10.1038/sj.bjc.6600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Shao SH, Xu ZX, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 13.Hallstrom TC, Nevins JR. Balancing the decision of cell proliferation and cell fate. Cell Cycle. 2009;8:532–5. doi: 10.4161/cc.8.4.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polager S, Ginsberg D. E2F—at the crossroads of life and death. Trends Cell Biol. 2008;18:528–35. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Jiang H, Gomez-Manzano C, Aoki H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–4. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 16.Templeton DJ, Park SH, Lanier L, Weinberg RA. Nonfunctional mutants of the retinoblastoma protein are characterized by defects in phosphorylation, viral oncoprotein association, and nuclear tethering. Proc Natl Acad Sci U S A. 1991;88:3033–7. doi: 10.1073/pnas.88.8.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–54. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- 18.Fueyo J, Gomez-Manzano C, Puduvalli VK, et al. Adenovirus-mediated p16 transfer to glioma cells induces G1 arrest and protects from paclitaxel and topotecan: implications for therapy. Int J Oncol. 1998;12:665–9. doi: 10.3892/ijo.12.3.665. [DOI] [PubMed] [Google Scholar]
- 19.Jones N, Shenk T. Isolation of deletion and substitution mutants of adenovirus type 5. Cell. 1978;13:181–8. doi: 10.1016/0092-8674(78)90148-4. [DOI] [PubMed] [Google Scholar]
- 20.Fueyo J, Gomez-Manzano C, Yung WK, et al. Overexpression of E2F-1 in glioma triggers apoptosis and suppresses tumor growth in vitro and in vivo. Nat Med. 1998;4:685–90. doi: 10.1038/nm0698-685. [DOI] [PubMed] [Google Scholar]
- 21.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 22.Rogoff HA, Pickering MT, Frame FM, et al. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24:2968–77. doi: 10.1128/MCB.24.7.2968-2977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang G, Rosen DG, Colacino JA, Mercado-Uribe I, Liu J. Disruption of the retinoblastoma pathway by small interfering RNA and ectopic expression of the catalytic subunit of telomerase lead to immortalization of human ovarian surface epithelial cells. Oncogene. 2007;26:1492–8. doi: 10.1038/sj.onc.1209905. [DOI] [PubMed] [Google Scholar]
- 24.Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994;8:1514–25. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- 25.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Manzano C, Mitlianga P, Fueyo J, et al. Transfer of E2F-1 to human glioma cells results in transcriptional up-regulation of Bcl-2. Cancer Res. 2001;61:6693–7. [PubMed] [Google Scholar]
- 27.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 28.Baldassarre G, Belletti B, Nicoloso MS, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 29.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–76. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol. 2008;9:713–24. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- 31.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 32.Karantza-Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kisen GO, Tessitore L, Costelli P, et al. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 1993;14:2501–5. doi: 10.1093/carcin/14.12.2501. [DOI] [PubMed] [Google Scholar]
- 34.Gronostajski RM, Pardee AB. Protein degradation in 3T3 cells and tumorigenic transformed 3T3 cells. J Cell Physiol. 1984;119:127–32. doi: 10.1002/jcp.1041190120. [DOI] [PubMed] [Google Scholar]
- 35.Young AR, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas DM, Yang HS, Alexander K, Hinds PW. Role of the retino-blastoma protein in differentiation and senescence. Cancer Biol Ther. 2003;2:124–30. [PubMed] [Google Scholar]
- 37.Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 16:1094–9. doi: 10.1158/1078-0432.CCR-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 40.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 41.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 42.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomida A, Suzuki H, Kim HD, Tsuruo T. Glucose-regulated stresses cause decreased expression of cyclin D1 and hypophosphorylation of retinoblastoma protein in human cancer cells. Oncogene. 1996;13:2699–705. [PubMed] [Google Scholar]
- 44.Mayhew CN, Perkin LM, Zhang X, Sage J, Jacks T, Knudsen ES. Discrete signaling pathways participate in RB-dependent responses to chemotherapeutic agents. Oncogene. 2004;23:4107–20. doi: 10.1038/sj.onc.1207503. [DOI] [PubMed] [Google Scholar]
- 45.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–96. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 46.Weller M, Rieger J, Grimmel C, et al. Predicting chemoresistance in human malignant glioma cells: the role of molecular genetic analyses. Int J Cancer. 1998;79:640–4. doi: 10.1002/(sici)1097-0215(19981218)79:6<640::aid-ijc15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 47.Johnson DG, Degregori J. Putting the oncogenic and tumor suppressive activities of E2F into context. Curr Mol Med. 2006;6:731–8. doi: 10.2174/1566524010606070731. [DOI] [PubMed] [Google Scholar]
- 48.Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4. doi: 10.1038/onc.2008.117. [DOI] [PubMed] [Google Scholar]
- 49.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–6. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.