

Abstract
The Ca2+/Mg2+ sites (III and IV) located in the C-terminal domain of cardiac troponin C (cTnC) have been generally considered to play a purely structural role in keeping the cTnC bound to the thin filament. However, several lines of evidence, including the discovery of cardiomyopathy-associated mutations in the C-domain, have raised the possibility that these sites may have a more complex role in contractile regulation. To explore this possibility, the ATPase activity of rat cardiac myofibrils was assayed under conditions in which no Ca2+ was bound to the N-terminal regulatory Ca2+ -binding site (site II). Myosin-S1 was treated with N-ethylmaleimide to create strong-binding myosin heads (NEM-S1), which could activate the cardiac thin filament in the absence of Ca2+. NEM-S1 activation was assayed at pCa 8.0-6.5 and in the presence of either 1mM or 30 μM free Mg2+. ATPase activity was maximal when sites III and IV were occupied by Mg2+ and it steadily declined as Ca2+ displaced Mg2+. The data suggest that in the absence of Ca2+ at site II strong-binding myosin crossbridges cause the opening of more active sites on the thin filament if the C-domain is occupied by Mg2+ rather than Ca2+. This finding could be relevant to the contraction-relaxation kinetics of cardiac muscle. As Ca2+ dissociates from site II of cTnC during the early relaxing phase of the cardiac cycle, residual Ca2+ bound at sites III and IV might facilitate the switching off of the thin filament and the detachment of crossbridges from actin.
Keywords: troponin, thin filament, myosin crossbridges, magnesium, NEM-S1
Introduction
Cardiac troponin C (cTnC) has three functional Ca2+-binding sites, namely, site II in the N-terminal domain and sites III and IV in the C-terminal domain. Site II is specific for Ca2+, but with a lower Ca2+ affinity (K∼105 M-1) than sites III and IV, and binding to this site activates crossbridge cycling and force development. Sites III and IV can bind either Ca2+ or Mg2+ and have been generally considered to play a purely structural role in keeping cTnC bound to the thin filament [1]. However, suggestions have been made that sites III and IV might have a more complex role in contractile regulation [2; 3]. This idea is reinforced by the recent discovery of cardiomyopathy-associated mutations [4; 5] in the C-domain of cardiac troponin C. Evaluation of the possible regulatory role of the C-domain of cTnC must take into account the more complex cation-protein interactions at this site, as compared to the N-terminal domain. .
The free intracellular [Mg2+] in intact cardiac myocytes has been found to be in the range of 0.5-0.7 mM [6]. The Mg2+ binding constant to sites III and IV of cTnC is ∼5×l03 M-1 [7] and the binding constant for Ca2+ at these sites is ∼3×l08 M-1. Thus in the relaxed state, with pCa > 7.0, sites III and IV would be primarily occupied by Mg2+ (cTnC·2Mg2+). During activation the stronger binding Ca2+ ions would displace Mg2+ as well as bind to vacant site II (cTnC·3Ca2+). A regulatory role for the C-domain of cTnC has generally been discounted on the grounds that, in contrast to the rapid Ca2+ binding to site II, the Ca2+ -Mg2+ exchange at sites III and IV is very slow relative to the kinetics of contraction and relaxation [7]. Thus in a single twitch very little Ca2+ would bind to the C-domain, even as site II is occupied. However with the repetitive activity characteristic of cardiac muscle the Ca2+/Mg2+ binding ratio should change as a function of heart rate. It has been suggested that the C-domain of cTnC may have a role in modulating contractile activity in accordance with activity pattern [2]. To explore this possibility we have assayed thin filament activation of rat cardiac myofibrils under conditions in which no Ca2+ was bound to the N-terminal regulatory site (site II). Myofibrils were reacted with myosin S1 treated with N-ethylmaleimide (NEM-S1) to create strong binding myosin heads that can activate the thin filament in the absence of Ca2+. We have found that NEM-S1 induced significantly higher activation of myofibrillar ATPase when sites III and IV of cTnC were occupied by Mg2+ rather than Ca2+, suggesting a role for the C-domain of cTnC in modulation of crossbridge dependent thin filament activation.
Materials and Methods
Myofibrils were prepared from rat ventricle essentially as described by Solaro, et al [8], with minor modifications. They were stored at -20°C in a solution of 50% glycerol, 40mM KCl, 10 mM MOPS, pH 7.0, 0.5 mM MgCl2, and 0.25mM dithiothreitol. Rabbit skeletal myosin S-1 was prepared according to Weeds and Taylor [9]. The reaction of S1 with N-ethylmaleimide and the separation of the purified NEM-S1 was carried out according to Swartz and Moss [10]. However, instead of dialysis, repeated cycles of dilution and ultrafiltration with Amicon Ultra 10 kDa filters were used for purification and concentration of the final product. Myofibril protein concentration was determined with BCA reagent (Pierce).
For assay of myofibrillar ATPase activity, aliquots (50 μl) containing 0.2 mg/ml myofibril protein, ± 1 μM NEM-S1, 100 mM KCl, 20 mM MOPS (pH 7.0), 1 mM EGTA, 1 mM NTA, and required concentrations of MgCl2 and CaCl2 were incubated at 30°C in Eppendorf centrifuge tubes. ATPase activity was assayed with free Mg2+ set at 1 mM or 30 μM. The program MAXCHELATOR [11] was used to calculate the concentrations of free Mg2+ and Ca2+. The reaction was initiated with the addition of 1 mM ATP and it was terminated after 10 min by the addition of 50 μl of 10% trichloroacetic acid. Following centrifugation at 14000 RPM for 5 min in an Eppendorf microcentrifuge, 50 μl of supernatant was transferred to a glass tube, and inorganic phosphate was determined with malachite green reagent as described by Lanzetta, et al [12], with minor modifications. All measurements were carried out in a pCa range in which myofibrillar ATPase activity is at the lowest. Since the NEM-S1 has some residual ATPase activity, each assay involved a measurement of NEM-S1 alone, myofibrils alone, and myofibrils + NEM-S1, so that each measurement could be corrected for the contribution of the NEM-S1.
Results
Thin filament activation was brought about by addition of strong-binding NEM-S1 and ATPase activity was assayed with the free Mg2+ set at either 1 mM or 30 μM. Based on binding and kinetic studies [7] it is assumed that in the isolated myofibril exposed to 1 mM free Mg2+ sites III and IV will be occupied exclusively by Mg2+ at very low Ca2+ levels (pCa 8.0). As the Ca2+ concentration is elevated in the intermediate range (pCa 7.5-6.5) progressively more Mg2+ will be displaced by Ca2+. On the other hand, if the free Mg2+ is 30 μM these sites should be largely empty at pCa 8.0 and occupied solely by Ca2+ as the Ca2+ concentration is elevated. Variation in free [Ca2+] was restricted to a range in which little or no Ca2+ would be bound at the regulatory site II.
As shown in Fig 1, the addition of 1 μM NEM-S1 to a myofibril suspension in the presence of 1mM free Mg2+ at pCa 8.0 produced an ∼3.5–fold increase in myofibrillar ATPase activity. Further additions of Ca2+ up to pCa 6.5 produced no substantial change in baseline ATPase activity but the effect of NEM-S1 was attenuated with the increase in [Ca2+], until at pCa 6.5 the degree of activation was ∼2-fold. The stability of the baseline ATPase activity, obtained in the absence of NEM-S1, indicates that no Ca2+ binding to the regulatory site II in the N-domain of cTnC occurs in the pCa range studied and provides strong support for the assumption that Ca2+ binding at the C-domain is responsible for the observed results. With 30 μM free Mg2+ there was only a slight increase in baseline ATPase activity over the pCa range 8.0-6.5 (Fig. 2). Addition of NEM-S1 caused an ∼2-fold increase in ATPase activity at all pCa values. In Fig. 3 the percent NEM-S1 activation is plotted as a function of pCa at 1 mM Mg2+ and 30 μM Mg2+. The plots clearly differ at pCa 8.0 but converge at pCa 6.5.
Figure 1.

The effect of NEM-S1 on myofibrillar ATPase activity at varying free Ca2+ in the presence of 1 mM free Mg2+. Assay mixtures contained: 0.2 mg/ml protein, ± 1 μM NEM-S1, 100 mM KCl, 20 mM MOPS (pH 7.0), 1 mM EGTA, 1 mM NTA, 1mM ATP, and added MgCl2 and CaCl2 to set the required free metal ion concentrations. Temperature, 30°C; incubation time 10 min. Each point is mean±SEM of 3-12 measurements. The maximum Ca2+-activated ATPase activity was 159±15 nmol Pi/mg/min
Figure 2.
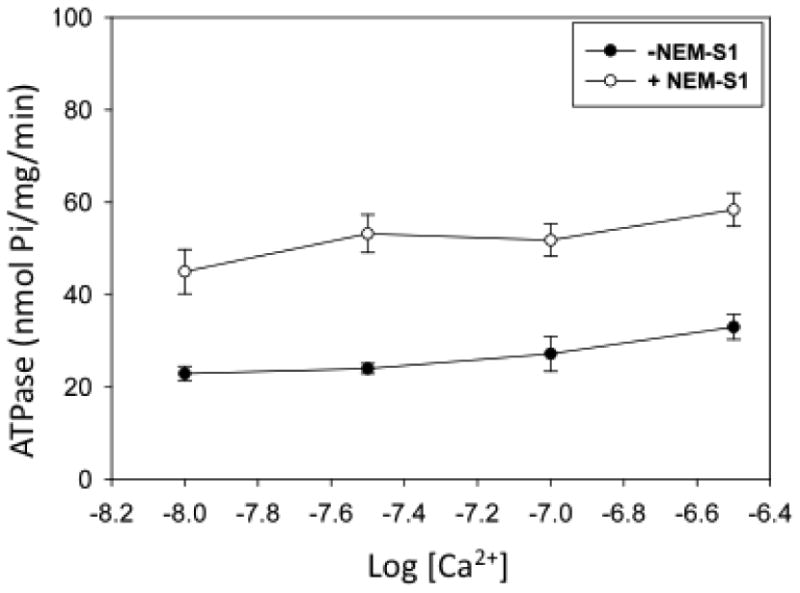
The effect of NEM-S1 on myofibrillar ATPase activity in the presence of 30 μM free Mg2+. Conditions were as in Figure 1, except for differences in added total Mg2+ and Ca2+. Each point is mean±SEM of 4-14 measurements.
Figure 3.
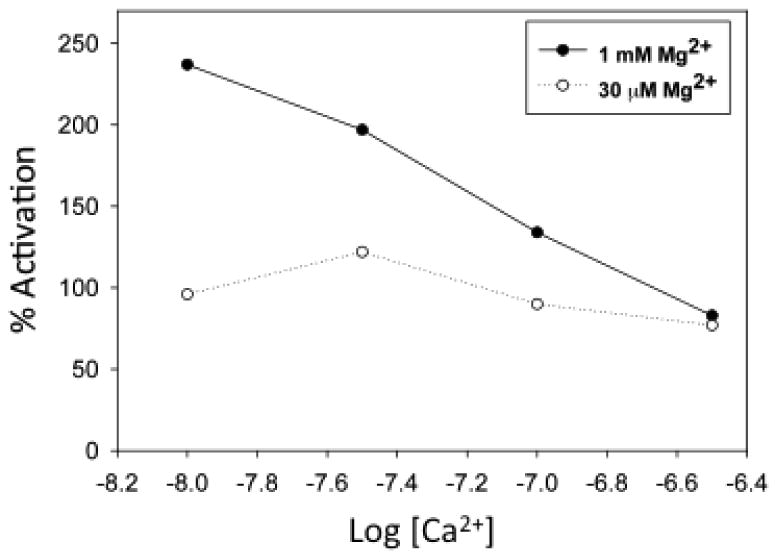
Effect of magnesium binding to site III and IV of cTnC on the relative activation of myofibrillar ATPase by NEM-S1. The % activation was calculated from data in figures 1 and 2 using the equation:
%Activation =(ANEM-S1/A − 1)× 100,
where A and ANEM-S1 are the ATPase activities in the absence and presence of NEM-S1, respectively at each free [Ca2+].
Discussion
Although suggestions have been made that the C-domain of cTnC might have a modulatory role in contractile regulation [2; 3] experimental data in support of this assertion are sparse. Such a role is suggested by studies showing that the structure of the C-domain and the strength of the interaction with cardiac troponin I (cTnI) depends on whether sites III and IV are occupied by Ca2+ or Mg2+ [2; 3]. The discovery of mutations in the C-domain which can cause either hypertrophic cardiomyopathy (D145E) or dilated cardiomyopathy (G159D) also suggest a more complex function for this region [4]. Another recent development relates to the effects of modifiers of Ca2+ sensitivity which act through the C-domain. Thus EMD 57033 sensitizes skinned cardiac fibers to Ca2+ [13], whereas the green tea polyphenol epigallocatechin 3-gallate reduces Ca2+ sensitivity [14]. Both bind to the C-domain [15; 16] but how they produce opposite effects remains to be determined.
By using NEM-S1 to activate the thin filament we could directly compare apo-cTnC, cTnC·2Mg2+, and cTnC·2Ca2+ with respect to any modulating effect on crossbridge-mediated thin filament activation. As Ca2+ is added in the range below that needed to activate ATPase activity (pCa 8.0 – 6.5) it is likely that there will be a variable population of mixed species (cTnC·Mg2+·Ca2+). However, based on known binding constants [7], it seems reasonable to assume that at pCa 8.0 and 1 mM Mg2+ the dominant species will be cTnC·2Mg2+ and at pCa 6.5, with either 1 mM or 30 μM Mg2+, the dominant species will be cTnC·2Ca2+. At 30 μM Mg2+ and pCa 8.0, the C-domain sites should be largely empty. Maximum ATPase activation by NEM-S1 occurred when Mg2+ was the only cation bound at sites III and IV. The level of activation declined as Mg2+ was displaced by Ca2+.
Our result can be explained in terms of the 3-state model of thin filament activation in which strong binding crossbridges can displace tropomyosin from its blocking position on F-actin [17; 18]. By employing NEM-S1 the Ca2+-dependent transition from the blocked state to the closed state is bypassed. It is less clear why the presence of Mg2+ at the C-domain of TnC would promote this effect. The high-resolution structure of the core domain of troponin suggests that the effect might involve the inhibitory segment of TnI. This segment binds to F-actin in the blocked state and to the N-terminal α-helix of site III of TnC in the activated state of the filament [19]. Since NEM-S1 induced thin filament activation requires the inhibitory segment of TnI to dissociate from its binding site on actin, the implication is that the interaction of that segment with TnC is stronger when sites III and IV are occupied by Mg2+ rather than Ca2+. Evidence for the Ca2+/Mg2+ dependent differences in the interaction between the C-domain of TnC and TnI have been presented based on NMR [3] and microcalorimetric studies [2]. Stereospecific metal-ligand interaction requirements and structural constraints may provide the basis for the difference in the Mg2+ vs. Ca2+ bound structure of TnC [20].
It is of interest to compare these results with data obtained in skinned skeletal and cardiac muscle fibers where NEM-S1 was added in the pCa range in which Ca2+ alone produced either no activation or just slight activation of force [21]. In these studies steady state force and the rate constant for force redevelopment following quick release (Ktr) were measured as a function of pCa. There is ample evidence that Ktr reflects the level of thin filament activation [22; 23]. NEM-S1 increased the steady-state force over the entire low Ca2+ range. It also increased Ktr but, unlike steady state force, this effect was diminished as [Ca2+] was increased. That is, Ktr was maximal when only Mg2+ was bound to TnC and apparently decreased as Ca2+ displaced Mg2+ from the C-domain. In fact, with only the cTnC·Mg2+ complex present (1 mM Mg2+, pCa 9.0) the Ktr value for skinned cardiac muscle was the same as that recorded at full Ca2+ saturation (pCa 4.5) when only the cTnC·3Ca2+ was present.
These data call attention to the possible differences between cTnC·3Ca2+, cTnC·2Ca2+, and cTnC·2Mg2+ with respect to the influence of each on myosin-mediated thin filament activation. In the latter two complexes the N-terminal regulatory site is unoccupied and, based on the data shown above, it would appear that the cTnC·2Mg2+ complex is more effective than cTnC·2Ca2+ at promoting myosin-mediated activation of the thin filament. With respect to physiological relevance, it is important to know which are the predominant cTnC complexes formed under a given set of conditions. Unfortunately, direct measurement in the intact muscle fiber is not feasible at the present time. It is agreed that very little Ca2+ will exchange with Mg2+ at the C-terminus during the time course of a single twitch [7]. However, in a beating heart the interval between contractions is relatively short and it becomes shorter still as the heart rate increases. Along with the shorter interval between beats and the increased action potential frequency there is an increased Ca2+ influx into the myocyte [24]. Thus as the heart rate increases the Ca2+/Mg2+ ratio at the C-terminus should increase. An important property of cardiac muscle is that as the heart rate increases the rate of relaxation increases as well [24]. This rate-dependent acceleration of relaxation plays an important role in optimizing ventricular filling in the presence of a reduced diastolic interval.
During activation at higher heart rates the formation of the cTnC·3Ca2+ complex will play the dominant role, leading to a rapid thin filament activation and force development. Relaxation begins with the dissociation of Ca2+ from site II, thus increasing the population of cTnC·2Ca2+. However, there is now agreement that the rate of relaxation is determined by processes intrinsic to the cardiac sarcomere rather than by the rate of Ca2+ removal from site II [24; 25; 26]. The mechanisms responsible for the fast relaxation kinetics are still being investigated and probably involve several different steps in the crossbridge cycle. Among the factors implicated in rapid relaxation are lengthening-induced strain on cross-bridges, leading to their more rapid detachment from actin, and phosphate-induced reversal of the crossbridge power stroke [26]. Also, evidence has been presented indicating that increase in heart rate is associated with augmented levels of myofilament protein phosphorylation and reduced myofilament Ca2+ sensitivity [27]. We suggest that the C-domain of cTnC may influence relaxation rate as a consequence of the formation of cTnC·2Ca2+ early in the relaxation phase. By reducing thin filament activation, the Ca2+ bound to sites III and IV, acting in conjunction with factors listed above, might promote a faster rate of relaxation. As suggested previously [2], variation in the Ca2+/Mg2+ binding ratio at sites III and IV has the potential to provide an on-going measure of the frequency and intensity of Ca2+ activation in cardiac muscle. To determine whether the Ca2+/Mg2+ sites do indeed serve such a function will require a more extensive investigation of the kinetic and structural correlates of C-terminal cation binding.
Acknowledgments
We would like to thank Dr. Sam Lehrer for critical reading of the manuscript.
Footnotes
This work was supported by the National Institutes of Health (grant HL-91162)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. Journal of Biological Chemistry. 1980;255:11688–93. [PubMed] [Google Scholar]
- 2.Calvert MJ, Ward DG, Trayer HR, Trayer IP. The importance of the carboxyl-terminal domain of cardiac troponin C in Ca2+-sensitive muscle regulation. J Biol Chem. 2000;275:32508–15. doi: 10.1074/jbc.M005764200. [DOI] [PubMed] [Google Scholar]
- 3.Finley NL, Howarth JW, Rosevear PR. Structure of the Mg2+-loaded C-lobe of cardiac troponin C bound to the N-domain of cardiac troponin I: comparison with the Ca2+-loaded structure. Biochemistry. 2004;43:11371–9. doi: 10.1021/bi049672i. [DOI] [PubMed] [Google Scholar]
- 4.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–92. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Swindle N, Tikunova SB. Hypertrophic cardiomyopathy-linked mutation D145E drastically alters calcium binding by the C-domain of cardiac troponin. C Biochemistry. 2010;49:4813–20. doi: 10.1021/bi100400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy E, Freudenrich CC, Lieberman M. Cellular magnesium and Na/Mg exchange in heart cells. Annu Rev Physiol. 1991;53:273–87. doi: 10.1146/annurev.ph.53.030191.001421. [DOI] [PubMed] [Google Scholar]
- 7.Robertson SP, Johnson JD, Potter JD. The time-course of Ca2+ exchange with calmodulin, troponin, parvalbumin, and myosin in response to transient increases in Ca2+ Biophysical Journal. 1981;34:559–569. doi: 10.1016/S0006-3495(81)84868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–62. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 9.Weeds AG, Taylor RS. Separation of subfragment-1 isoenzymes from rabbit skeletal muscle myosin. Nature. 1975;257:54–56. doi: 10.1038/257054a0. [DOI] [PubMed] [Google Scholar]
- 10.Swartz DR, Moss RL. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J Biol Chem. 1992;267:20497–506. [PubMed] [Google Scholar]
- 11.Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–31. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–7. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 13.Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N, Lakatta EG. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993;73:981–90. doi: 10.1161/01.res.73.6.981. [DOI] [PubMed] [Google Scholar]
- 14.Liou YM, Kuo SC, Hsieh SR. Differential effects of a green tea-derived polyphenol (-)-epigallocatechin-3-gallate on the acidosis-induced decrease in the Ca(2+) sensitivity of cardiac and skeletal muscle. Pflugers Arch. 2008;456:787–800. doi: 10.1007/s00424-008-0456-y. [DOI] [PubMed] [Google Scholar]
- 15.Li MX, Spyracopoulos L, Beier N, Putkey JA, Sykes BD. Interaction of cardiac troponin C with Ca(2+) sensitizer EMD 57033 and cardiac troponin I inhibitory peptide. Biochemistry. 2000;39:8782–90. doi: 10.1021/bi000473i. [DOI] [PubMed] [Google Scholar]
- 16.Robertson IM, Li MX, Sykes BD. Solution structure of human cardiac troponin C in complex with the green tea polyphenol, (-)-epigallocatechin 3-gallate. J Biol Chem. 2009;284:23012–23. doi: 10.1074/jbc.M109.021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKillop DFA, Geeves MA. Regulation of the interaction between actin and myosin subfragment-1 - evidence for 3 states of the thin filament. Biophysical Journal. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehrer SS, Geeves MA. The muscle thin filament as a classical cooperative/allosteric regulatory system [ published erratum appears in J Mol Biol 1998 Jun 19;279(4): 1024] J Mol Biol. 1998;277:1081–9. doi: 10.1006/jmbi.1998.1654. [DOI] [PubMed] [Google Scholar]
- 19.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci U S A. 2005;102:5038–43. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabarek Z. Insights into Modulation of Calcium Signaling by Magnesium in Calmodulin, Troponin C and Related EF-hand Proteins. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813:913–921. doi: 10.1016/j.bbamcr.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moss RL, Fitzsimons DP, Razumova MV. Regulation of the rate of force development in heart and skeletal muscles. In: Solaro RJ, Moss RL, editors. Molecular control mechanisms in striated muscle contraction. Kluwer Academic Publishers; Dordrecht: 2002. pp. 271–290. [Google Scholar]
- 22.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 23.Regnier M, Martin H, Barsotti RJ, Rivera AJ, Martyn DA, Clemmens E. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J. 2004;87:1815–24. doi: 10.1529/biophysj.103.039123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen PM. Myocardial contraction-relaxation coupling. Am J Physiol Heart Circ Physiol. 2010;299:H1741–9. doi: 10.1152/ajpheart.00759.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 2007;22:73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 26.Stehle R, Iorga B. Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. J Mol Cell Cardiol. 2010;48:843–50. doi: 10.1016/j.yjmcc.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 27.Varian KD, Janssen PM. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–9. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]