

Abstract
In vivo and in vitro studies revealed that nitroalkenes serve as protective mediators in the lung by inducing the cytoprotective enzyme heme oxygenase-1 (HO-1). LNO2 increased HO-1 mRNA, protein and activity in cultured pulmonary epithelial cells treated with 5 to 50 μM LNO2 and in lungs of rats injected intraperitoneally with 2.6 mg/kg LNO2 twice daily for 20 days. Western blotting revealed that HO-1 protein increased significantly within 4 hours of in vitro LNO2 addition and was preceded by an increase in HO-1 mRNA, consistent with transcriptional regulation of HO-1 expression by LNO2. LNO2 also de-phosphorylated and activated eukaryotic initiation factor 2α (eIF 2α) a key translational regulatory protein, indicating that increased translation may also contribute to LNO2-induced increases in HO-1. Exposure of cells to LNO2 activated ERK and JNK, as evidenced by increased phosphorylation. Downstream targets of ERK and JNK, Elk-1 and c-Jun respectively, were also phosphorylated in response to LNO2 exposure. However, inhibitor studies revealed that only the ERK pathway is necessary for the LNO2-mediated increase in HO-1 mRNA and protein. These data reveal that LNO2 induces pulmonary epithelial HO-1 expression and downstream adaptive responses to inflammation via both transcriptional and translational regulatory mechanisms.
Introduction
Pulmonary inflammation is a life-threatening complication of diverse pathogenic processes that can result in a number of complications and in pathologies such as acute respiratory distress syndrome (ARDS) [1,2]. Enhanced production of reactive oxygen species (ROS) and secondary oxides of nitrogen derived from nitric oxide (NO) contribute significantly to the extensive oxidation and nitration reactions that occur during inflammatory lung injury [3]. Inflammation is also associated with the release of pro-inflammatory cytokines including tumor necrosis-factor (TNF)-α, interleukin (IL)-1, -4, -6, -8, -10, and -13 [2] and the infiltration and activation of inflammatory cells. These host defense responses can be both consequences and causes of the accelerated formation of oxidizing and nitrating species during acute lung injury (ALI) [4,5].
The signaling reactions leading to the resolution of inflammation are in part mediated by the redox reactions that represent one element of tissue inflammatory responses to injury and invading pathogens. For example, increased gene expression of antioxidant enzymes such as Mn superoxide dismutase (MnSOD) occur in rat lungs in response to an acute exposure to LPS [6]. Other redox defenses, such as peroxiredoxins I and II, are induced in response to oxidative inflammatory mediators [7].
Recently the NO and nitrite (NO2-)-dependent nitration of unsaturated fatty acids has been observed and fatty acid nitroalkene derivatives have been detected in the red cells, plasma and urine of healthy humans [8-10]. Current data supports that nitrated fatty acids function as pluripotent endogenous signaling molecules [11,12]. For example, platelet and neutrophil function is inhibited by nitrolinoleic acid (LNO2) via non-cGMP-dependent mechanisms [12,13]. Thrombin-induced platelet aggregation is inhibited by LNO2-mediated attenuation of cAMP-dependent Ca+2 mobilization and activation of vasodilator-stimulated phospho-protein (VASP) phosphorylation at serine 157 [12]. Current evidence supports that there is a dual regulation of platelet adenylyl cyclase and phosphodiesterase 3A activities by LNO2 [12]. More recently, it has been observed that nitrated fatty acids undergo Nef-like decay reactions in aqueous milieu, yielding NO, explaining the ability of LNO2 to mediate cGMP-dependent vessel relaxation [14,15]. Further, both LNO2 and OA-NO2 serve as potent endogenous PPAR-ligands [10,16]. Finally, high levels of LNO2 has been found in the pulmonary edema fluid of patients with ARDS [17] suggesting that it may be both a marker of pathology and a player in the signaling for the resolution of pulmonary inflammation. In aggregate, there are multiple reactivities whereby nitrated fatty acids may serve to modulate pulmonary inflammatory responses.
Preliminary gene array analysis with HUVEC cells revealed that low concentrations of LNO2 inhibit the expression of multiple inflammatory-related genes encoding cell adhesion molecules, cytokines and cytokine receptors. In contrast, gene expression of HO-1, PGHS-2, VEGF, the 10, 40, 60, 70 and 90 kD heat shock proteins were up-regulated, with HO-1 the most extensively-induced gene (B. Freeman, unpublished data). This finding was replicated in aortic endothelial cells [18]. Because of the profound impact that HO-1 plays in the response of tissues to pathogenic and inflammatory stimuli [19], we examined cellular HO-1 responses to LNO2 in more detail, specifically, the signaling pathways involved. Herein, we report that LNO2 exerts MAPK-dependent induction of HO-1 and propose that HO-1 induction represents a key mechanism whereby nitrated fatty acids serve as protective mediators in the lung.
Methods
Materials
Nitrated oleic acid and linoleic acid were synthesized as previously described [8,10]. SB202190 (p38 inhibitor), PD98059 (MEK inhibitor) and SP600125 (JNK inhibitor) were purchased from Calbiochem (San Diego, CA). All chemicals were at least of analytical grade and from Sigma-Aldrich unless otherwise noted. Formaldehyde (20% TEM grade ampules) was from Tousimis Corporation (Rockville, MD). All antibodies were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA) unless otherwise noted. Alexa-Fluor 594-conjugated goat anti-rabbit IgG and Alexa-Fluor 488-conjugated goat anti-rabbit IgG were purchased from Molecular Probes. Rabbit polyclonal anti-HO-1 was from Stressgen (Victoria, BC, Canada), and anti-phospho-eIF 2α, -eEF 2α and -4E were from Cell Signaling (Danver, MA).
Animals
All experimental work on animals was approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham and University of Pittsburgh (IACUC Protocol #050307444, 06XX07679, and 0605735). Adult male Sprague Dawley rats and C57Bl/6 mice (25-30 g) were housed under temperature and humidity-controlled conditions. Rats were injected intraperitioneally (i.p) twice daily with 2.6 mg/kg i.p. of LNO2 dissolved in PBS in 0.5% ethanol, or vehicle alone. After 20 days, rats were killed with a lethal injection of sodium pentobarbital and the lungs were harvested. LNO2 bioavailability and biodistribution was studied using mice that received a single injection of 160 mg/kg i.p. and were killed one hour later.
Mass Spectrometric Analysis
Lipids were extracted and LNO2 levels were measured in serum and harvested lung tissue as published previously [8,10,20]. Samples were analyzed by electrospray ionization triple quadrupole mass spectrometry (HPLC ESI MS/MS) in the multiple reaction monitoring mode to detect nitrated linoleic acid as previously [8], with some modifications. Lipids were resolved on a 20 × 2 mm C18 cartridge column (Phenomenex) using an HPLC gradient consisting of (A) H2O containing 0.1% acetic acid and (B) acetonitrile containing 0.1% acid. Gradient conditions were as follows: 0-1.5 min, 35% B; 1.5-10 min, 100% B; 10-12 min, 100% B; 12-12.1 min, 35% B, followed by three min equilibration. During extraction [13C18] LNO2 was added as an internal standard to quantitate free LNO2 in treated and control samples.
Cell Culture and treatments
A549 cells are a human pulmonary epithelial cell line that was purchased from the ATCC (Manassas, VA). Cultures were maintained in DMEM supplemented with 1% penicillin/streptomycin and 10% FCS in a humidified incubator at 37°C with 5% CO2. L2 cells are a rat-derived pulmonary epithelial cell line that was obtained from the ATCC. Cultures were maintained in F12K nutrient mixture supplemented with 1% penicillin /streptomycin and 10% FBS in a humidified incubator 37°C with 5% CO2. All cells were grown to 95% confluence and then treated with various agents. LNO2 and OA-NO2 were dissolved in methanol for addition to cell cultures at a final concentration of up to 50 μM LNO2 in 10% serum/F12K (L2 cells) and 5 μM LNO2 when in 1% serum/DMEM (A549 cells). The final concentration of methanol did not exceed 0.025% (vol/vol).
MAPK inhibitors SB202190 (p38) and PD98059 (MEK) were dissolved in DMSO. The final concentration of DMSO did not exceed 0.05%. The JNK inhibitor SP600125 was dissolved in water. The final concentrations of methanol and DMSO were 0.025 and 0.05%, respectively and vehicle controls were included in all experiments. Cells were pre-treated with 25 μM SB202190, 50 μM PD98059, 20 μM SP600125, or with the dilution buffer (containing DMSO or water) for 2 h, and then incubated with 5-50 μM LNO2 for various lengths of time as outlined in the results. Following treatments, cells were washed once with phosphate buffered saline (PBS) and harvested with a cell scraper in PBS. Cells were centrifuged at 500 × g for 5 min, trace PBS removed and cell pellets either processed immediately or stored at -80°C.
Immunohistochemistry
Cells were washed with PBS for 5 min and fixed in 3% formaldehyde in PBS at room temperature (RT) for 20 min. After 4 × 5 min washes with PBS, cells were permeabilized using 0.1% Triton X-100 in PBS for 20 min, and then blocked with 10% goat serum in 1% bovine serum/PBS for 1-2 hr at RT. Cells were incubated overnight at 4°C with rabbit anti-HO-1diluted 1:1000 in 10% goat serum, and washed 4 times for 5 min with PBS. Cells were incubated with Alexa Fluor 594-conjugated goat anti-rabbit IgG, diluted 1:100 in 10% goat serum, for 1 h at RT in the dark and washed 4 times for 5 min in PBS. Chambers were removed and cover slips mounted using Vectashield Hard Set Mounting Medium with 1.5 μg/mL DAPI. Rat lung tissues were formalin fixed, paraffin-embedded and processed as described previously [18]. A 1:200 dilution of Alexa Fluor 488-conjugated goat anti-rabbit IgG was used as the secondary antibody. Images were acquired using a Leitz DMRXA2 fluorescence microscope, using a 40X objective and Simple PCI imaging software (Compix, Inc, Irvine, CA).
Western blotting analysis
Western blotting was done as previously described [21,22]. Briefly, whole cell lysates were prepared using the M-PER reagent and nuclear fractions were prepared using the Pierce M-PER and NE-PER reagents respectively (Pierce, Rockford IL) and proteins measured using an adaptation of the Bradford method. Between 20-50 μg of protein was resolved by electrophoresis under denaturing conditions on a 10% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA), transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, San Francisco, CA) and then blocked in 5% non-fat dry milk (NFDM) at room temperature for 1 h. Membranes were then incubated overnight at 4°C with the appropriate primary antibody in 5% NFDM in Tris-buffered saline, with 0.05% Tween-20 (T-TBS). After washing with T-TBS, the membranes were incubated with the appropriate HRP-coupled secondary antibody at room temperature for 2 h. After repeated washes with T-TBS, membranes were incubated with ECL Plus reagent (Amersham Biosciences, Piscataway, NJ) for 5 min. Blots were exposed to ECL Hyperfilm (Amersham), and quantitation of chemiluminescence was done using an Alpha Inotech quantification software (Alpha Inotech Corp, San Leandro, CA).
RNA Purification and Northern Hybridization
RNA purification and Northern hybridization was done as previously with the following modifications [23]. Briefly, after treatments, cells were removed from culture dishes with a rubber policeman, and scraped into 1.5 ml microfuge tubes. RNA was isolated in a guanidine thiocyanate solution (4 M guanidine, 0.5% sarcosyl, 25 mM sodium citrate, pH 7.0, 100 μM β-mercaptoethanol). A 10% volume of NaOAc, pH 4.0 was added to the lysate, and the RNA was purified via acid phenol/chloroform extraction, precipitated in isopropanol overnight, washed in 70% EtOH, and resuspended in water. RNA was precipitated a second time with 1 M NH4OAc and absolute EtOH at -80C for 1 h, washed with 70% EtOH and resuspended in water.
3 μg of total RNA was denatured in 50% formamide, 1% formaldehyde, MOPS buffer (20 mM MOPS pH 7.0, 8 mM NaOAc, 1 mM EDTA). Loading dyes and ethidium bromide were added and samples were loaded onto a 1% agarose, 5% formaldehyde gel containing MOPS buffer. After electrophoresis, the RNA was transferred via capillary blot onto a Hybond XL membrane. The membrane was UV crosslinked (50 mJ/cm2) and hybridized in 260 mM Na2HPO4 pH 7.2, 7% SDS, 1 mM EDTA, 1% BSA at 65°C overnight with 50 ng of [α-32P] dATP labeled HO-1 cDNA probe. The membrane was washed in 3 changes of 20 mM Na2HPO4, pH 7.2, 1% SDS, 1 mM EDTA at 70°C and exposed to film. GAPDH was used as a loading control. Each mRNA species was measured in quadruplicate and each experiment was repeated at least 3 times.
HO activity analysis
HO activity was measured as previously [24-26]. A549 and L2 cells were grown to confluence, rinsed twice with PBS, scraped with a rubber policeman, and centrifuged at 2000 × g for 10 min. The pellet was resuspended in 0.1 M KPO4 and 2 mM MgCl2, frozen (80°C) and thawed 3 times to break the cell membrane. The cells were then sonicated at 4° C and an aliquot reserved for protein determination using a modification of the Bradford Assay. The remaining sample was centrifuged at 12 000 × g at 4° C for 20 min, and the supernatant added to the reaction mixture containing 3 mg rat liver cytosol, 20 μM hemin, 2 mM glucose 6-phosphate, 0.2 U glucose 6-phosphate dehydrogenase, and 0.8 mM β-NADPH and incubated at 37°C for 60 min in the dark. One mL of chloroform was then added to the extract, and the change in optical density of the aqueous phase was measured at 464-530 nm. The concentration of bilirubin produced in 60 min was calculated using an extinction coefficient of 40 mM-1cm-1 for bilirubin.
Statistical Analysis
Statistical analyses, where appropriate, were done either using a student’s t-test or a one-way ANOVA with a Tukey’s test post hoc. Differences were considered significant at p<0.05. Results are representative of at least 2-4 independent experiments.
Results
The potential HO-1-mediated signaling actions of nitrated fatty acids in lung tissue were evaluated using human (A549) and rat (L2) pulmonary epithelial cells. The relative abilities of LNO2 and OA-NO2 to induce HO-1 in pulmonary cells in vitro were determined by western blotting (Fig. 1). LNO2 induced a 3-fold increase in HO-1 protein expression whereas OA-NO2 had no effect (Fig 1). Based on these findings, LNO2 was the focus of the remainder of these studies.
Figure 1. LNO2 but not OA-NO2 induces HO-1 expression in pulmonary epithelial cells.
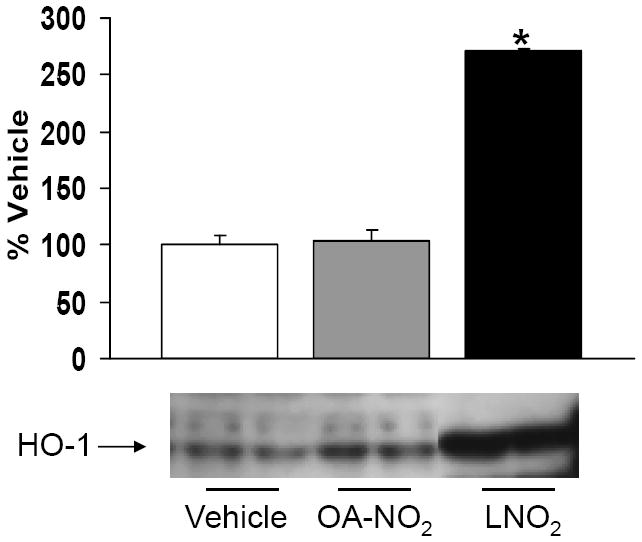
A549 cells were exposed to 5 μM OA-NO2, LNO2 or the vehicle control for 6 h and then HO-1 protein was visualized via Western blotting. Semi-quantitative analysis was done using densitometry and changes in HO-1 with each nitrated fatty acid were expressed relative to the vehicle control, which was arbitrarily set to 100%. Rapid, significant (*, p<0.05) induction of HO-1 was observed in response to LNO2; OA-NO2 did not increase HO-1 protein (representative results are shown as a composite figure derived from the same Western blot).
LNO2 (5 and 50 μM) induced a profound increase in HO-1 protein expression, as evidenced by both immunohistochemical analysis (Fig. 2) and Western blotting in A549 cells and L2 cells (Fig. 4 and 5). In contrast, up to 50 μM linoleic acid did not induce HO-1 protein in rat pulmonary L2 cells and human A549 cells (Fig. 2).
Figure 2. LNO2 induces HO-1 in cultured cells and in rat lungs in vivo.
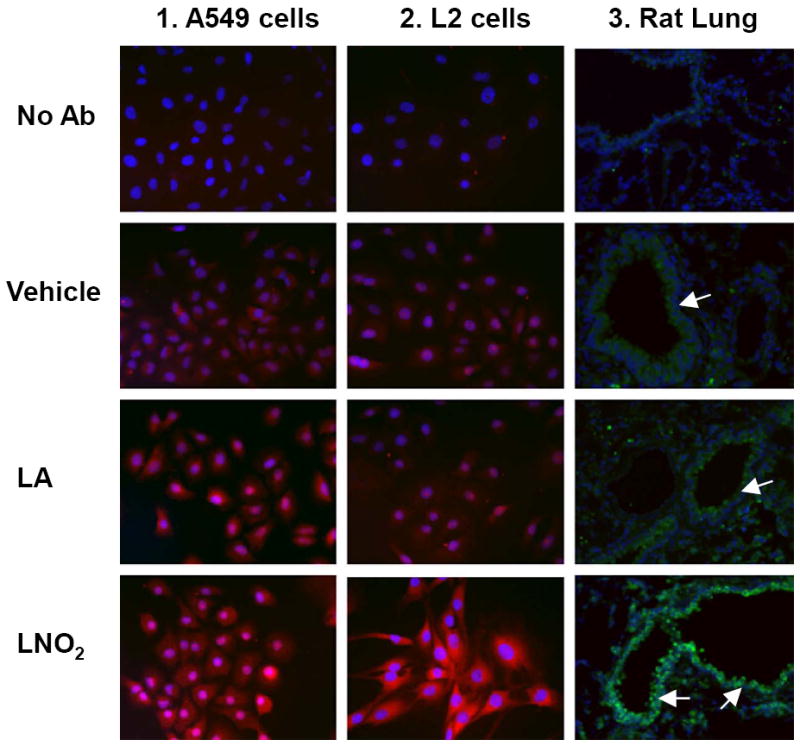
Human pulmonary epithelial cells (A549 cells) and rat pulmonary epithelial cells (L2 cells) were treated with LNO2 (5 and 50 μM). HO-1 protein was visualized by Alexa Fluor 594 (red) or Alexa Fluor 488 (green) with a fluorescence microscope, 40X objective. Nuclei were counterstained blue with DAPI (blue). Treatment groups for Panels 1 and 2 correspond to no primary antibody, vehicle control (MeOH), linoleic acid (LA), and LNO2. In Panel C, rats were injected with 2.6 mg/kg i.p. of LNO2 twice daily for 20 days. Treatment groups correspond to no primary antibody, vehicle, LA and LNO2. Airways are marked with arrows.
Figure 4. LNO2 induces HO-1 in pulmonary cells in a time-dependent manner.
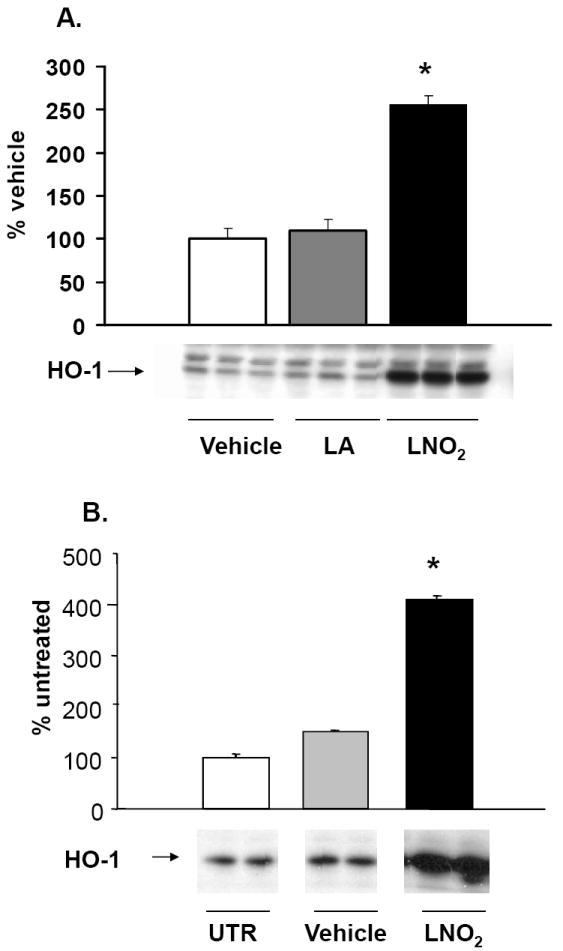
Human pulmonary cells (A549) and L2 cells were treated with 5-50 μM LNO2 for 4 h. Changes in HO-1 protein were visualized using Western blotting with anti-HO-1 and semi-quantitative analysis performed using densitometry. There was a statistically significant increase (*, p<0.05) in HO-1 protein following LNO2 treatment in A549 cells (A) relative to vehicle and to native fatty acid control. A similar increase in HO-1 was observed in L2 cells (B; representative results are shown as a composite figure derived from the same Western blot).
Figure 5. Native LNO2 is required for HO-1 induction.
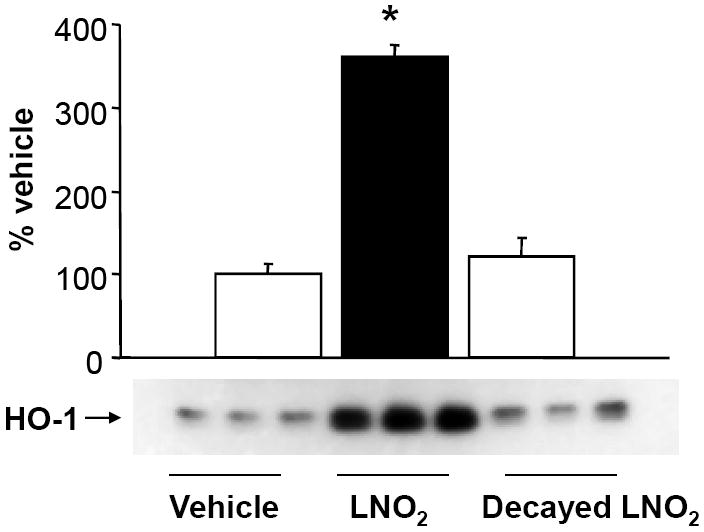
A549 cells were incubated for 4 h with either 5 μM LNO2 in 1% serum, or LNO2 that had been allowed to decay in media for 2 h. Changes in HO-1 protein was visualized and measured as described above in the Methods. Decayed LNO2 caused no induction of HO-1 protein relative to the vehicle control.
To determine if the HO-1-inducing actions of LNO2 observed in vitro translated to an in vivo response, rats were injected with 2.6 mg/kg twice daily for 20 days and lung HO-1 protein expression was evaluated. LNO2 treatment induced extensive increases in HO-1 protein in airway epithelial cells whereas control animals injected with linoleic acid showed no increases in HO-1 expression (Fig. 2). This LNO2–dependent induction of lung HO-1 expression in vivo also indicates that systemic increases in LNO2 concentration can induce cell signaling responses in tissues remote from the site of LNO2 administration. To confirm that exogenous administration of LNO2 can cause systemic increases in LNO2 levels, LNO2 levels were measured in serum and lung tissue following i.p. injection of mice (Fig. 3A, 3B), and following chronic administration in rat lungs (Fig. 3C). LNO2 was below the level of detection basally in mouse serum and lung tissues but following i.p. injection of LNO2 380 ± 11 nM and 369 ± 67 pmol/g tissue were measured in serum and lung tissue respectively (n=3). The lack of quantifiable basal levels of LNO2 in mouse serum differs from previous findings in human samples [8]. Following chronic administration of LNO2, treated rats had significantly elevated levels of LNO2 in their lung tissue compared to control animals (treated, 8.77±2.44 pmol/g tissue; control, 0.13±0.02 pmol/g tissue).
Figure 3. Exogenous LNO2 increases serum and tissue levels of LNO2.
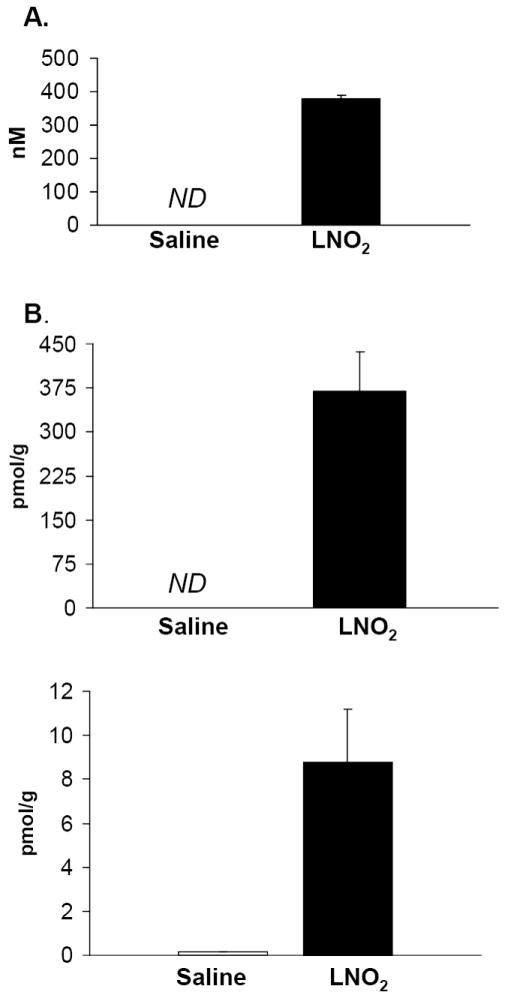
Mice were given a single i.p injection of LNO2 (160 mg/kg) and LNO2 was measured in serum and lung tissue one hour later (3A & 3B). LNO2 was below the level of quantification (ND, on graph) in saline-injected mice, but significant amounts (380 nM and 369 pmol/g tissue) were measured following injection in serum and lung tissue respectively (n=3). Lungs from rats chronically administered LNO2 (2.6 mg/kg twice daily for 20 days; 3C) also demonstrated significant increases of LNO2 (n=3).
LNO2 decays in aqueous milieu, with close to 90% of detectable LNO2 lost in the first hour [14]. This raises the possibility that LNO2 decay products might serve as the proximal signaling mediator responsible for increased HO-1 expression. To address this possibility, LNO2 was allowed to decay in complete media for 2 h at 25 °C. Decayed LNO2 did not induce HO-1 protein expression in vitro (Fig. 5), indicating that native LNO2 is required to initiate reactions responsible for increased HO-1 expression by pulmonary epithelial cells. In order to determine the mechanism of increased HO-1 protein, Northern blots were done to assay for changes in mRNA. Northern analysis showed consistently significant increases in HO-1 mRNA expression following exposure to LNO2 (Fig. 6). As increased HO-1 protein expression may not necessarily translate into increased HO catalytic activity, changes in HO activity were measured (Fig. 7). LNO2 (5-50 μM) induced a 2-8 fold increase in HO activity in both human A549 and rat L2 cells.
Figure 6. LNO2 increases HO-1 mRNA expression in pulmonary epithelial cells.
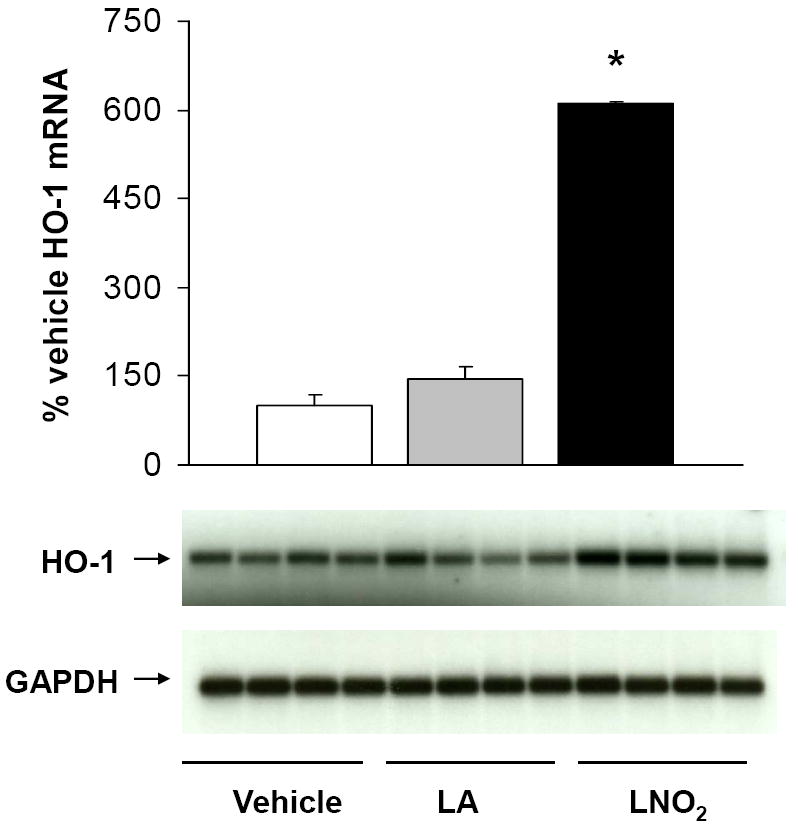
Human A549 cells were incubated with 5 μM LNO2 in 1% serum-containing media for 2 h and mRNA measured via Northern blotting. HO-1 mRNA was normalized to GAPDH and expressed as an increase over basal HO-1 message. There was a statistically significant increase (*, p<0.05) in HO-1 mRNA in following LNO2 treatment relative to both vehicle and native fatty acid controls.
Figure 7. LNO2 increases HO-1 activity in pulmonary epithelial cells.
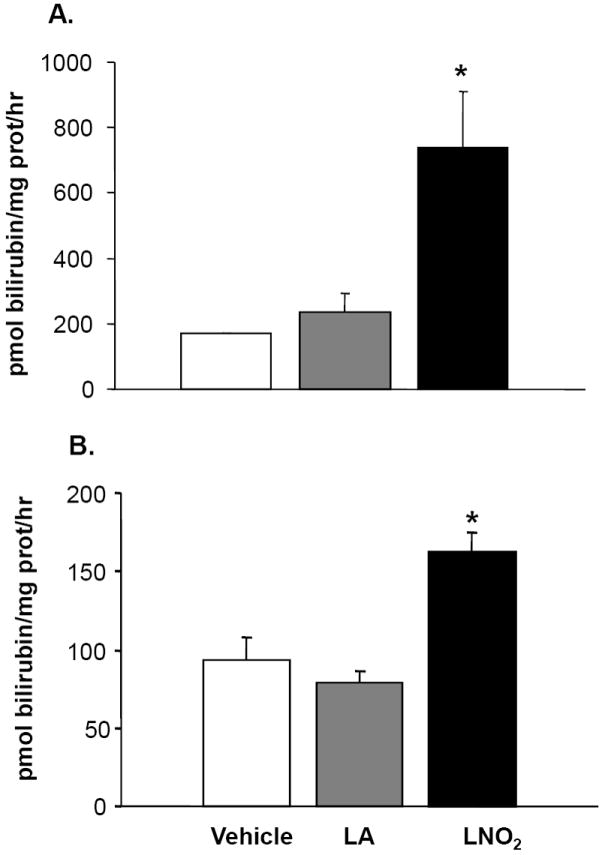
A. L2 cells were incubated with 50 μM LNO2 in 10% serum for 8 h and B. A549 cells were incubated with 5 μM LNO2 in 1% serum for 8 h and HO-1 activity determined. There was a statistically significant increase (*, p<0.05) in HO-1 activity following LNO2 treatment in both L2 and A549 cells relative to both vehicle and native fatty acid control.
As previous studies indicate that HO-1 mRNA and protein induction is MAPK regulated, the link between LNO2-induced HO-1 expression and activation of MAPK signaling cascades in pulmonary cells was explored. LNO2 treatment increased phosphorylation of the JNK, ERK and p38 in L2 cells (Fig. 8A and 8B, and data not shown), and ERK and JNK phosphorylation in A549 cells (Fig. 8C and data not shown) but had no effect on p38 MAPK phosphorylation (data not shown). In L2 cells, JNK and ERK phosphorylation peaked at 2 h, but was still significantly elevated at 4 h post-LNO2 treatment. c-Jun phosphorylation, one of the downstream targets of JNK phosphorylation, was also significantly increased at 2 and 4 h post-LNO2 addition (Fig. 9, A and B). In human A549 cells, a similar pattern of ERK activation was observed, with significant increases at 2 and 4 h post-treatment (Fig. 8C), with one of its downstream targets Elk-1, also being phosphorylated (Fig. 9C). However, relative to the rat L2 cells, there was a rapid LNO2-dependent turn-on, turn-off of the JNK pathway, with c-Jun phosphorylation being increased 2-fold at 2 h post-incubation, but returning to basal levels by 4 h post-incubation (Fig. 9B). In all cases, control treatment of cells with similar concentrations of linoleic acid showed no effect on ERK, JNK and p38 MAPK.
Figure 8. LNO2 activates the JNK and ERK MAPK pathways in pulmonary epithelial cells.
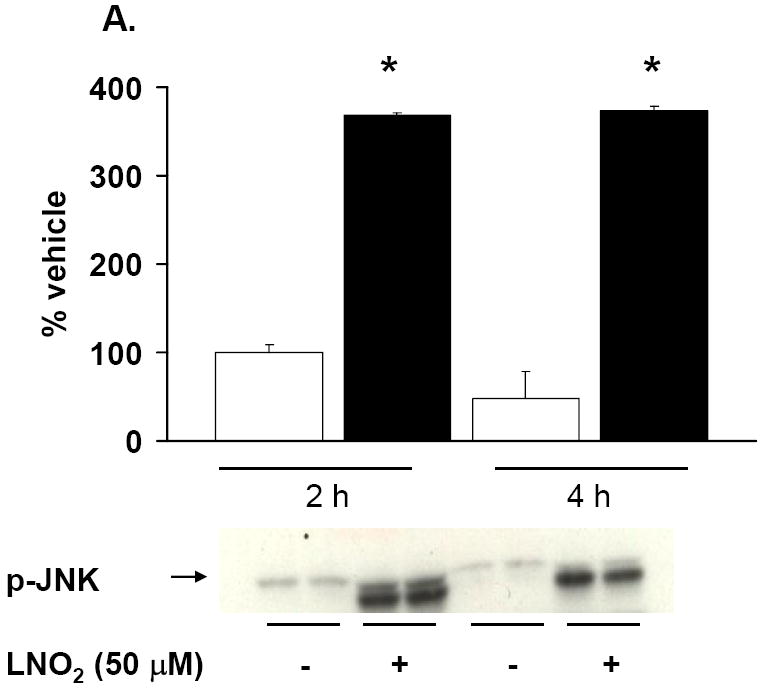
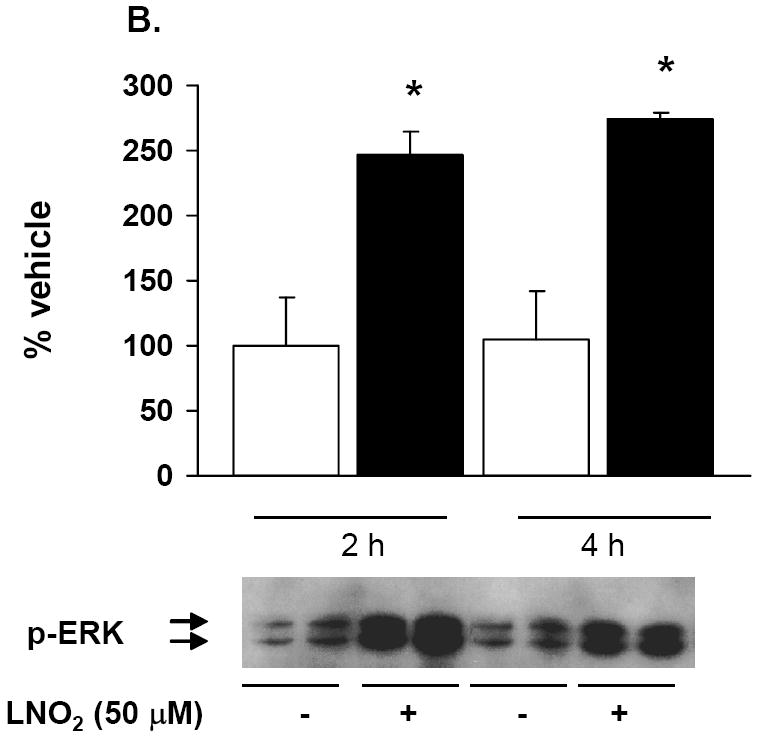
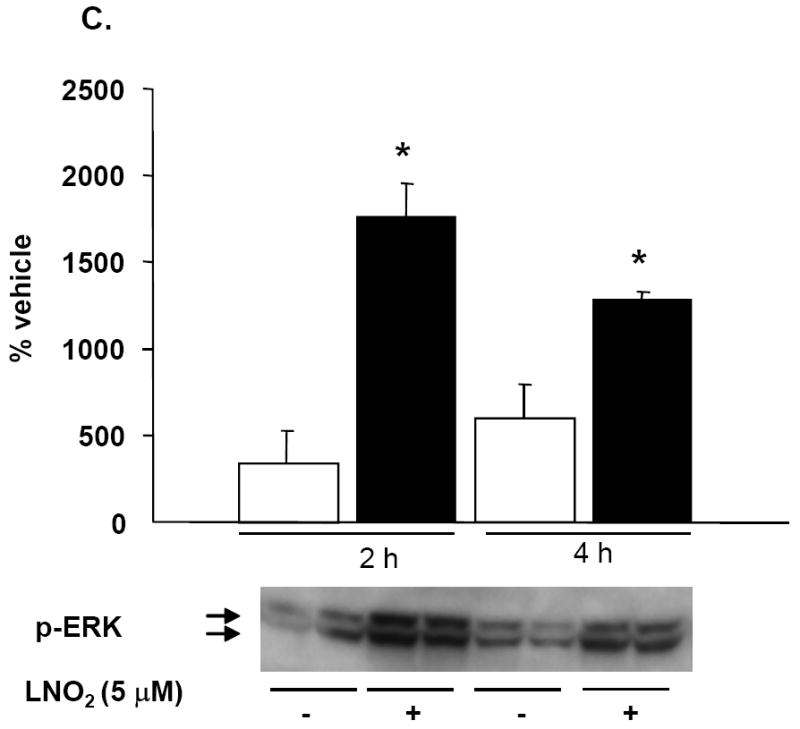
L2 (A, B) and A549 (C) cells were incubated with 50 and 5 μM LNO2, respectively, for up to 4 h and JNK (A) and ERK (B, C) phosphorylation were visualized via Western blotting with phospho-specific antibodies and semi-quantitative measurements of increased phosphorylation was determined via densitometry. There was a statistically significant increase in JNK (L2) and ERK (L2 and A549) phosphorylation following LNO2 treatment relative to the vehicle control.
Figure 9. LNO2 increases transcription factor activation.
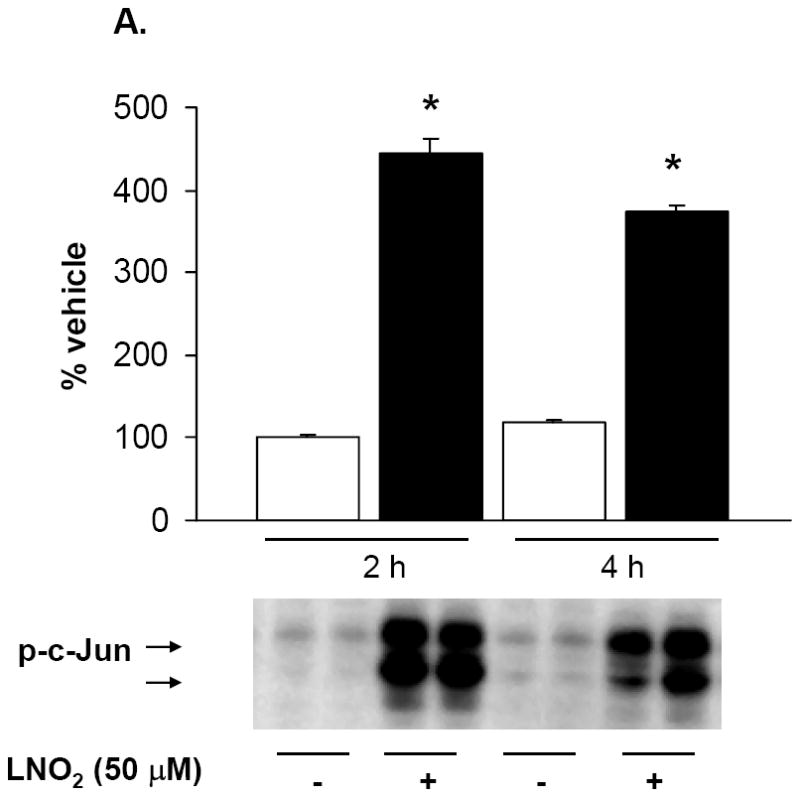
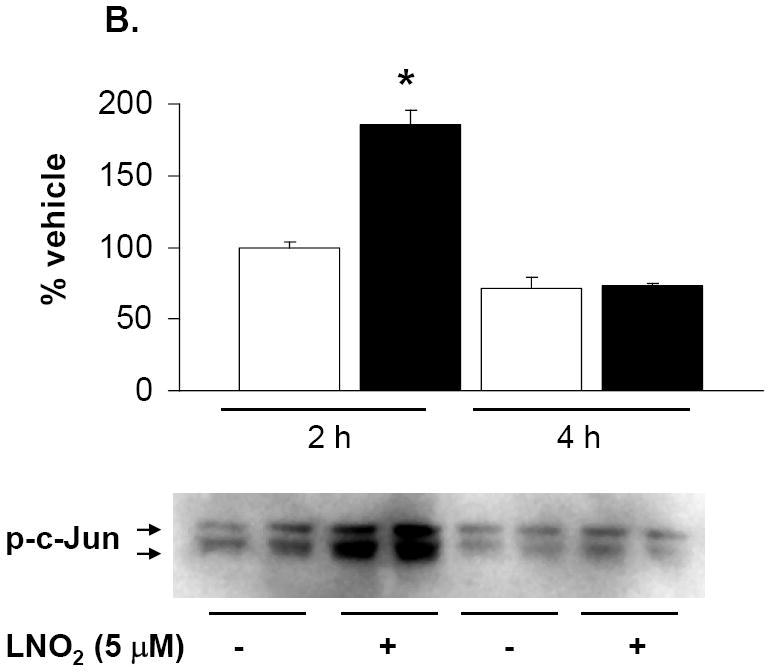
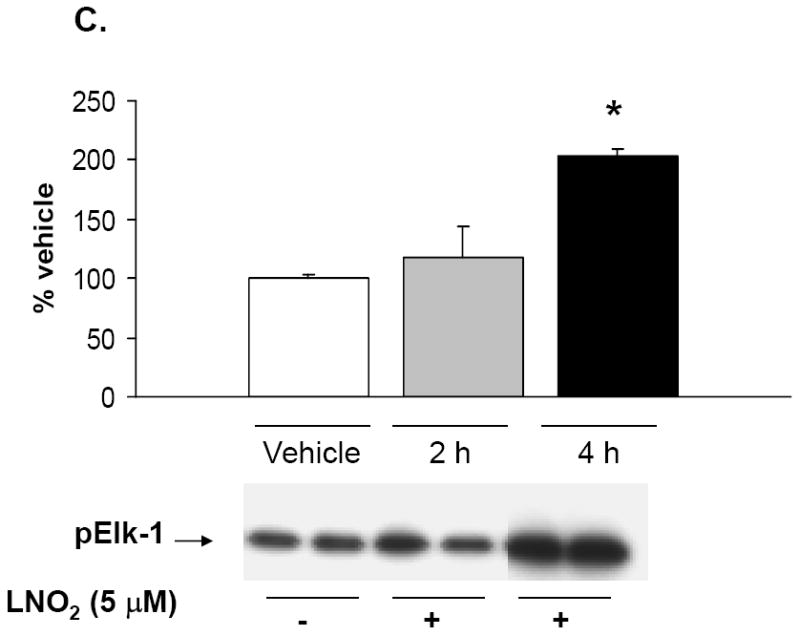
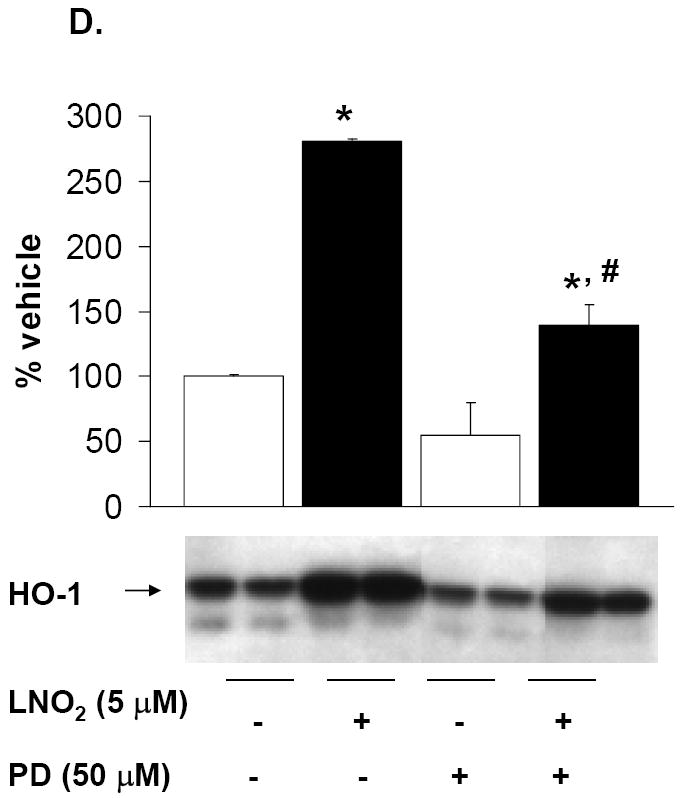
Rat L2 and human A549 cells were incubated with 5-50 μM LNO2 in complete media for up to 4 h. Cells were fractionated into cytoplasmic and nuclear fractions, and Western blotting with phospho-specific antibodies was done on the appropriate cellular fraction. Increases in c-Jun phosphorylation were measured in L2 (A) and A549 (B) cells. There was a statistically significant increase in c-Jun (A) and (B) phosphorylation following LNO2 treatment relative to the vehicle control (*, p<0.05). Changes in phosphorylation of Elk-1 (C, composite image), a downstream target of ERK was also visualized and quantitated in A549 cells, and was found to increase significantly 4h post-incubation (*, p<0.05). The MEK inhibitor PD98059 partially inhibited the LNO2-mediated increase in HO-1 protein in A549 cells (D). Representative results are shown in (D) as a composite figure derived from the same Western blot.
Cells treated with the MAPK inhibitors SB202190 (p38 pathway inhibitor) and JNKi (JNK pathway inhibitor) did not inhibit LNO2-mediated increases in HO-1 protein expression (data not shown). Addition of the MEK and ERK pathway inhibitor PD98059 to cells prior to LNO2 addition partially inhibited LNO2 induction of HO-1 protein expression (Fig. 9D).
Northern analysis revealed that a transcriptional component contributed to LNO2 induction of HO-1 protein and activity. Since inhibition of MAPK signaling, already recognized as a mechanism contributing to increased HO-1 mRNA expression, only partially reversed LNO2-induced HO-1 expression, additional mechanisms were investigated that might account for the impact of LNO2 on HO-1 expression. Focus was directed towards translational proteins regulated via phosphorylation switches and/or MAPK/redox regulation. Changes in eukaryotic elongation factor 2 (eEF 2), eukaryotic initiation factor 2 alpha (eIF 2α) and eukaryotic initiation factor 4E (eIF 4E) were measured by western blotting using phospho-specific antibodies. A significant decrease in the phosphorylation (hence activation) of eIF 2α (Fig. 10) was observed 2 h following cell exposure to LNO2. No changes in eEF 2 or eIF 4E were observed (data not shown).
Figure 10. LNO2 decreases the phosphorylation of eIF 2α in A549 cells.
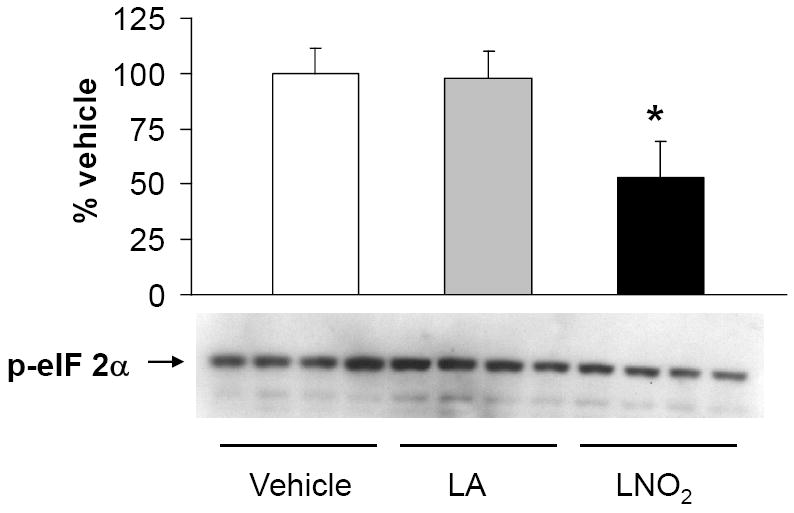
A549 cells were treated with 5 μM linoleic acid (LA), LNO2 or an equal volume of vehicle (MeOH) in 1% serum for 2 h. Western blotting with phospho-specific eIF 2α Semi-quantitative of the changes in eIF 2α phosphorylation was done by densitometry. There was a statistically significant decrease in phosphorylation (de-activation) of eIF 2α following LNO2 treatment relative to the vehicle control (*, p<0.05).
Discussion
Nitrated unsaturated fatty acids, including LNO2, have been identified as pluripotent cell signaling molecules [8-10,16] These species are capable of undergoing a Nef-like reaction in aqueous milieu to release NO and induce cGMP-dependent cell signaling responses [14]. An important component of nitrated fatty acid signaling, however, is receptor-dependent and non-cGMP-dependent [8,12,16].
Herein we report that LNO2 induces the cytoprotective gene product HO-1 in cultured pulmonary epithelial cells and in rat lungs in vivo. Moreover, we show that in cultured pulmonary epithelial cells this response is regulated via MAPK-dependent mechanisms. This supports the hypothesis that increased LNO2 levels generated as a byproduct of pulmonary inflammatory nitration reactions elicit an adaptive cytoprotective response by signaling for the up-regulation of the cytoprotective gene product HO-1.
HO-1 is a key oxidative and inflammatory defense enzyme that lends tissue resistance to a diversity of inflammatory and metabolic stresses, ranging from hypothermia and arthritis to LPS-induced acute lung injury [27-31]. The cytoprotective actions of HO-1 derive from the catabolism of pro-oxidative heme and/or the oxidant-protective properties of the products of heme catabolism, biliverdin and carbon monoxide [32-34]. Indeed, the salutary effects of HO-1 in the lung are well established. Polymorphisms in the 5’ flanking region of the HO-1 gene have been linked to decreased HO-1 inducibility and increased susceptibility to the development of emphysema and lung adenocarcinoma in smokers [35]. HO-1 induction has also been shown to restore alveolar fluid clearance following hemorrhagic shock [36]. In cystic fibrosis, in airway epithelial cells, overexpression of HO-1 is protective against Pseudomonas aeruginosa infection [37]. An exception to the generally-protective role of HO-1 is found under acute hyperoxic conditions. HO-1 knockout mice show significantly decreased levels of markers of oxidative lung injury relative to wild-type controls [38]. The basis of damage under acute hyperoxic conditions appears to be generation of toxic iron intermediates that, under the unique conditions of hyperoxia, contribute to lung damage. The cellular consequences of HO-1 induction are not necessarily the same within a hyperoxic environment as in an inflammatory disease state such as ARDS.
Herein, in vivo and in vitro model systems were employed to define the mechanisms underlying LNO2-mediated induction of HO-1. LNO2 induces HO-1 protein both in vitro and in vivo. In these studies, LNO2 was injected i.p. and distal effects on HO-1 activation were observed in the lung. A single i.p. injection of LNO2 produced detectable levels of LNO2 in both mouse serum and in lung tissue and in rat lung tissue following chronic administration. This demonstrates that exogenous administration of LNO2 can increase LNO2 systemically and further that an i.p. injection can increase LNO2 in remote organs. LNO2 has also been found adducted to GSH in vivo and to protein thiols ex vivo, with both reactions being reversible in the presence of alternative thiol acceptor species [39,40]. It is thus postulated that similar adducts occurred herein to form a NO2-FA “reserve” that can impact HO-1 synthesis in the lung for extended periods of time. Herein, linoleic acid alone did not increase HO-1 expression, supporting that LNO2 or an active metabolite is responsible for the increase in pulmonary HO-1 expression.
Nitrated oleic acid (OA-NO2) did not increase HO-1 in human pulmonary epithelial cells. The basis for this difference between LNO2 and OA-NO2 is not currently known. However, it is hypothesized that small, structural differences render this selectivity. Consistent with this, PPARγ exhibits selective molecular recognition for specific LNO2 positional isomers in the PPARγ ligand binding domain [41]. Specific differences (and similarities) in signaling actions and regulation of gene regulation of these and other NO2-FA derivatives are an active area of examination by our own and other groups.
In vitro, the analysis of A549 responses to LNO2 have the advantage of being made in a human cell line, but have the disadvantage of examining cell responses in an adenocarcinoma–derived cell line. Thus, in some respects, L2 cell responses to LNO2 may reflect a more “normal” response. However, differences in the regulation of HO-1 in human and rodent cells at both the MAPK and the promoter level exist [42-44]. In aggregate, examination of the differentiated responses of both A549 and L2 lung cell lines to LNO2-dependent HO-1 expression provides a comprehensive picture of the inflammatory regulation of HO-1 signaling in the pulmonary epithelium.
Previous studies of the regulation of HO-1 expression in lung showed increased HO-1 transcription as the predominant mechanism underlying increases in HO activity. Indeed, transcriptional regulation of HO-1 protein in L2 cells occurs in response to the lipid peroxidation by-product 4-hydroxy-2,3, nonenal (HNE) and in A549 cells in response to TGF-β1 [25,45]. Herein, a significant increase (approx. 2-fold) in HO-1 mRNA was also observed in response to LNO2 addition. However, the more robust LNO2–induced increases in HO-1 protein and activity that are observed suggested additional regulatory mechanisms operating in concert with increased mRNA synthesis. Thus, the impact of LNO2 on some of the key proteins in the translational machinery (eEF 2, eIF 4E and eIF 2α) was considered. eEF 2 and eIF 2α are both activated by de-phosphorylation [46,47], whereas eIF 4E is activated via phosphorylation of serine 209 [48]. Of these proteins, only a change in the dephosphorylation and activation of eIF 2α was observed in response to LNO2 (Fig. 9). De-phosphorylation of eIF 2α is required for the initiation of translation and in many systems as phosphorylation of eIF 2α serves as a brake that prevents new protein synthesis [49]. Stress conditions such as hypoxia and ischemia-reperfusion have been shown to increase phosphorylation of eIF 2α as a means of shutting down protein synthesis as a response to stress [50,51]. Mechanistically, the kinases responsible for phosphorylating eIF 2α are coordinately regulated by specific cellular stresses [50-52]. In contrast, following exposure to LNO2, eIF 2α is de-phosphorylated and this precedes maximal induction of the HO-1 protein. This finding argues against LNO2 representing a specific cellular “stress”, as stress invariably blocks protein synthesis via the phosphorylation of eIF 2α. In aggregate, these data are consistent with the hypothesis that LNO2 contributes to the resolution of pulmonary inflammation.
HO-1 has previously been shown to be regulated by activation of the MAPK signaling cascades. The MAPK are activated by dual phosphorylation on specific tyrosine and threonine residues [53] and are activated by inflammatory and oxidative mediators [54-56]. MAPK phosphorylate and activate specific transcription factors, thereby regulating the transcription of many genes. Exposure to LNO2 induced profound activation of the ERK and JNK MAPK in both rat and human lung cells, and temporally preceded increases in cellular HO-1 mRNA and protein synthesis. As significant differences in the rodent and human HO-1 promoter exist [44] the relevance of specific MAPK pathways with respect to downstream activation of gene expression must be considered separately. The human HO-1 gene has been shown to be regulated by all three MAPK in a variety of cell types, but little is known about the MAPK-mediated regulation of HO-1 in pulmonary epithelial cells. In human aortic smooth muscle cells, HO-1 induction by oxidized LDL is regulated by ERK, JNK and p38 [52]. In human endothelial cells, both inhibitor-based reporter gene studies revealed that activation of the ERK and JNK pathways is required for HO-1 mRNA induction [57]. In contrast, in human hepatic myofibroblasts, transient GSH depletion causes increased HO-1 mRNA stability that is p38-dependent [58] and in human papillary thyroid carcinoma cells (KAT5), MAPK inhibitor studies revealed that activation of both the ERK and p38 MAPK pathways are required for increased HO-1 protein expression [59]. There are conflicting reports about the role of p38 MAPK in HO-1 regulation in pulmonary epithelial cells. In human A549 cells, TGF-β1 has previously been reported to activate HO-1 transcription via the p38 pathway [45]. However, even within this cell type, the mechanism of HO-1 induction appears to be agonist specific. In another report using A549 cells, blocking the p38 pathway with the inhibitor SB203580 had no effect on hemin-induced HO-1 activity [60]. This suggests both p38 dependent and independent activation of HO-1 message and protein in human pulmonary epithelial cells. Herein, it was determined that LNO2 activates the ERK and JNK MAPK in human A549 cells, but that only ERK activation is required for increased HO-1 synthesis. It is postulated that the electrophilic reactivity’s of LNO2 mediate these actions of LNO2 by reacting with nucleophilic amino acids of protein kinases and phosphatases, thereby regulating multiple points in the MAPK signaling cascade.
The rat HO-1 gene has also been shown to be regulated by all three MAPK. In cultured rat hepatocytes, transcriptional upregulation of HO-1 occurs via both the JNK and p38 pathways [61]. In rat pulmonary aortic endothelial cells, hypoxia increased HO-1 message, and increased the phosphorylation of both ERK and p38 MAPK. However, when these pathways were blocked with specific inhibitors, hypoxia-induced HO-1 mRNA expression was further enhanced [62] shedding doubt on whether there is a causal connection between MAPK activation and HO-1 induction in this cell type. In a previous report of HO-1 induction in rat L2 cells in response to the lipid peroxidation by-product HNE, JNK, ERK and p38 were all activated by the agonist, but only ERK activation was causally linked with increased HO-1 expression [25]. Herein, we observed that LNO2 activates JNK, ERK and p38 MAPK in rat L2 cells and that inhibition of the ERK pathway only partially limits HO-1 expression. This finding draws attention once again to the fact that MAPK activation may be species and cell-type specific, and even within a given cell type varies depending on the agonist.
Previous work has established that LNO2 increases Nrf-2 nuclear translocation and identified this as a seminal signaling event in inhibiting VSMC proliferation [63]. This present study has additionally identified the ERK pathway as an important signaling event in LNO2-induced HO-1 upregulation in pulmonary epithelial cells. These two signaling mechanisms are not mutually exclusive and are not unexpected. It has previously been shown that Nrf-2 nuclear translocation and subsequent Nrf-2-driven upregulation of Phase II gene expression can be blocked by specific ERK pathway (MEK) inhibitors [64]. Thus, the current findings, and those reported previously point to an integrated mechanism of LNO2-mediated, ERK-dependent Nrf-2 nuclear translocation. Detailed analysis of this interaction is beyond the scope of this current work and in a focus of future studies.
In summary, we reveal via in vitro and in vivo studies that LNO2 can induce adaptive, pulmonary-protective responses via the induction of HO-1. These findings are consistent with the concept that LNO2, produced as a by-product of NO-dependent oxidative inflammatory reactions, mediates the resolution of inflammation via the induction of the cytoprotective gene products such as HO-1.
Acknowledgments
This work was supported by National Institutes of Health Grants HL58115, HL64937 and HL077100 (B.A.F), HL68157 (A.A), HL081332 (L.B.W.), AHA 0665418U (F.J.S.), ADA 7-06-JF-06 (P.R.S.B.), an AHA South-East Affiliate, Beginning Grant-in-Aid and a Parker B. Francis Fellowship in Pulmonary Research (KEI).
Abbreviations
- eIF 2α
eukaryotic initiation factor 2 alpha
- ERK
Extracellular signal-regulated kinase
- HO-1
Heme oxygenase-1
- JNK
c-Jun N-terminal kinase
- LNO2
Nitro-linoleic acid
- MAPK
Mitogen activated protein kinases
- OA-NO2
Nitro-oleic acid
- ROS
Reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Wong HR. ARDS. The future. Critical Care Clinics. 2002;18:177–196. doi: 10.1016/s0749-0704(03)00072-1. [DOI] [PubMed] [Google Scholar]
- 2.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. American Journal of Physiology Lung, Cellular and Molecular Physiology. 2005;288:L3–L15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34:1507–1516. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 4.Lamb N, Gutteridge J, Baker C, Evans T, Quinlan G. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Critical Care Medicine. 2005;27:1738–1744. doi: 10.1097/00003246-199909000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Lamb N, Quinlan G, Westerman S, Gutteridge J, Evans T. Nitration of proteins in bronchoalveolar lavage fluid from patients with acute respiratory distress syndrome receiving inhaled nitric oxide. Am J Respir Crit Care Med. 1999;160:1031–1034. doi: 10.1164/ajrccm.160.3.9810048. [DOI] [PubMed] [Google Scholar]
- 6.Feng N, Chu S, Wang D, Hsu K, Lin C, Lin H. Effects of various antioxidants on endotoxin-induced lung injury and gene expression: mRNA expressions of MnSOD, interleukin-1beta and iNOS. The Chinese Journal of Physiology. 2004;47:111–120. [PubMed] [Google Scholar]
- 7.Bast A, Wolf G, Oberbaumer I, Walther R. Oxidative and nitrosative stress induces peroxiredoxins in pancreatic beta cells. Diabetologia. 2002;45:867–876. doi: 10.1007/s00125-002-0846-1. [DOI] [PubMed] [Google Scholar]
- 8.Baker P, Schopfer F, Sweeney S, Freeman B. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proceedings of the National Academy of Sciences. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chemical Research in Toxicology. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 10.Baker P, Lin Y, Schopfer F, Woodcock S, Groeger A, Batthyany C, Sweeney S, Long M, Iles K, Baker L, Branchaud B, Chen Y, Freeman B. Fatty acid transduction of nitric oxide signaling: Identification of unsaturated fatty acid nitro derivatives and PPAR receptor-dependent signaling activity. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coles B, Bloodsworth A, Clark S, Lewis M, Cross A, Freeman B, O’Donnell V. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circulation Research. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 12.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O’Donnell VB. Nitrolinoleate inhibits platelet activation by attenuating calcium mobilization and inducing phosphorylation of vasodilator-stimulated phosphoprotein through elevation of cAMP. J Biol Chem. 2002;277:5832–5840. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 13.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O’Donnell VB. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circulation Research. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 14.Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O’Donnell VB, Eiserich JP, Freeman BA. Nitrolinoleate, a nitric oxide-derived mediator of cell function: synthesis, characterization, and vasomotor activity. Proceedings of the National Academy of Sciences. 2002;99:15941–15946. doi: 10.1073/pnas.232409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schopfer F, Baker P, Giles G, Chumley P, Batthyany C, Crawford J, Patel R, Hogg N, Branchaud B, Lancaster J, Freeman B. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 16.Schopfer F, Lin Y, Baker P, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen Y, Freeman B. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proceedings of the National Academy of Sciences. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iles KE, Cole MP, Saini D, Wright M, Powell P, Baker P, Matthay M, Agarwal A, Freeman BA. Fatty acid transduction of nitric oxide signaling: Inflammation induces the generation and adaptive mediator actions of LNO2 in the lung. Free Radical Biology and Medicine. 2006;41(S1):S34. [Google Scholar]
- 18.Wright M, Schopfer F, Baker P, Vidyasagar V, Powell P, Chumley P, Iles K, Freeman B, Agarwal A. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proceedings of the National Academy of Sciences. 2006;103:4299–4304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jozkowicz A, Dulak J. Effects of protoporphyrins on production of nitric oxide and expression of vascular endothelial growth factor in vascular smooth muscle cells and macrophages. Acta Biochim Pol. 2003;50:69–79. [PubMed] [Google Scholar]
- 20.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian Journal of Medical Science. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 21.Iles K, Dickinson D, Watanabe N, Iwamoto T, Forman H. AP-1 activation through endogenous H(2)O(2) generation by alveolar macrophages. Free Radic Biol Med. 2002;32:1304–1313. doi: 10.1016/s0891-5849(02)00840-7. [DOI] [PubMed] [Google Scholar]
- 22.Iles KE, Nagy LE. Chronic ethanol feeding increases the quantity of G alpha s-protein in rat liver plasma membranes. Hepatology. 1995;21:1154–1160. [PubMed] [Google Scholar]
- 23.Dickinson D, Iles K, Watanabe N, Iwamoto T, Zhang H, Krzywanski D, Forman H. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 24.Visner GA, Lu F, Zhou H, Liu J, Kazemfar K, Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation. 2003;107:911–916. doi: 10.1161/01.cir.0000048191.75585.60. [DOI] [PubMed] [Google Scholar]
- 25.Iles K, Dickinson D, Wigley A, Welty N, Blank V, Forman H. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic Biol Med. 2005;39:355–364. doi: 10.1016/j.freeradbiomed.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balla G, Jacob H, Balla J, Rosenberg M, Nath K, Apple F, Eaton J, Vercellotti G. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 27.Fernandez P, Guillen MI, Gomar F, Alcaraz MJ. Expression of heme oxygenase-1 and regulation by cytokines in human osteoarthritic chondrocytes. Biochem Pharmacol. 2003;66:2049–2052. doi: 10.1016/s0006-2952(03)00543-4. [DOI] [PubMed] [Google Scholar]
- 28.Grasso S, Scifo C, Cardile V, Gulino R, Renis M. Adaptive responses to the stress induced by hyperthermia or hydrogen peroxide in human fibroblasts. Experimental Biology and Medicine (Maywood) 2003;228:491–498. doi: 10.1177/15353702-0322805-12. [DOI] [PubMed] [Google Scholar]
- 29.Hartsfield CL, Alam J, Choi AM. Transcriptional regulation of the heme oxygenase 1 gene by pyrrolidine dithiocarbamate. FASEB J. 1998;12:1675–1682. doi: 10.1096/fasebj.12.15.1675. [DOI] [PubMed] [Google Scholar]
- 30.Inoue S, Suzuki M, Nagashima Y, Suzuki S, Hashiba T, Tsuburai T, Ikehara K, Matsuse T, Ishigatsubo Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Human Gene Therapy. 2001;12:967–979. doi: 10.1089/104303401750195926. [DOI] [PubMed] [Google Scholar]
- 31.Oguro T, Hayashi M, Numazawa S, Asakawa K, Yoshida T. Heme oxygenase-1 gene expression by a glutathione depletor, phorone, mediated through AP-1 activation in rats. Biochem Biophys Res Commun. 2003;221:259–265. doi: 10.1006/bbrc.1996.0583. [DOI] [PubMed] [Google Scholar]
- 32.Herget J, Palecek F, Preclik P, Cermakova M, Vizek M, Petrovicka M. Pulmonary hypertension induced by repeated pulmonary inflammation in the rat. Journal of Applied Physiology. 1981;51:755–761. doi: 10.1152/jappl.1981.51.3.755. [DOI] [PubMed] [Google Scholar]
- 33.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annual Reviews of Pharmacology and Toxicology. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 34.Tenhunen R, Marver H, Schmid R. The enzymatic catabolism of hemoglobin: stimulation of microsomal heme oxygenase by hemin. The Journal of Laboratory and Clinical Medicine. 1970;75:410–421. [PubMed] [Google Scholar]
- 35.Kikuchi A, Yamaya M, Suzuki S, Yasuda H, Kubo H, Nakayama K, Handa M, Sasaki T, Shibahara S, Sekizawa K, Sasaki H. Association of susceptibility to the development of lung adenocarcinoma with the heme oxygenase-1 gene promoter polymorphism. Human Genetics. 2005;116:354–360. doi: 10.1007/s00439-004-1162-2. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Pespeni M, Roux J, Dennery P, Matthay M, Pittet J. HO-1 induction restores c-AMP-dependent lung epithelial fluid transport following severe hemorrhage in rats. FASEB J. 2005;19:287–289. doi: 10.1096/fj.04-2254fje. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, Lu F, Latham C, Zander D, Visner G. Heme oxygenase-1 expression in human lungs with cystic fibrosis and cytoprotective effects against Pseudomonas aeruginosa in vitro. Am J Respir Crit Care Med. 2005;170:633–640. doi: 10.1164/rccm.200311-1607OC. [DOI] [PubMed] [Google Scholar]
- 38.Dennery P, Visner G, Weng Y, Nguyen X, Lu F, Zander D, Yang G. Resistance to hyperoxia with heme oxygenase-1 disruption: role of iron. Free Radicals in Biology and Medicine. 2003;34:124–133. doi: 10.1016/s0891-5849(02)01295-9. [DOI] [PubMed] [Google Scholar]
- 39.Baker L, Baker P, Golin-Bisello F, Schopfer F, Fink M, Woodcock S, Branchaud B, Radi R, Freeman B. Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J Biol Chem. 2008;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batthyany C, Schopfer F, Baker P, Durán R, Baker L, Huang Y, Cerveñansky C, Branchaud B, Freeman B. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Zhang J, Schopfer F, Martynowski D, Garcia-Barrio M, Kovach A, Suino-Powell K, Baker P, Freeman B, Chen Y, Xu H. Molecular recognition of nitrated fatty acids by PPAR gamma. Nature Structural and Molecular Biology. 2008;15:865–867. doi: 10.1038/nsmb.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill-Kapturczak N, Thamilselvan V, Liu F, Nick HS, Agarwal A. Mechanism of heme oxygenase-1 gene induction by curcumin in human renal proximal tubule cells. American Journal of Physiology: Renal Physiology. 2001;281:F851–F859. doi: 10.1152/ajprenal.2001.281.5.F851. [DOI] [PubMed] [Google Scholar]
- 43.Hill-Kapturczak N, Sikorski E, Voakes C, Garcia J, Nick HS, Agarwal A. An internal enhancer regulates heme- and cadmium-mediated induction of human heme oxygenase-1. American Journal of Physiology: Renal Physiology. 2003;285:F515–F523. doi: 10.1152/ajprenal.00137.2003. [DOI] [PubMed] [Google Scholar]
- 44.Sikorski E, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. American Journal of Physiology Renal Physiology. 2004;286:F425–F441. doi: 10.1152/ajprenal.00297.2003. [DOI] [PubMed] [Google Scholar]
- 45.Ning W, Song R, Li C, Park E, Mohsenin A, Choi A, Choi M. TGF-beta1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. American Journal of Physiology Lung, Cellular and Molecular Physiology. 2002;283:L1094–L1102. doi: 10.1152/ajplung.00151.2002. [DOI] [PubMed] [Google Scholar]
- 46.Jørgensen R, Merrill A, Andersen G. The life and death of translation elongation factor 2. Biochemical Society Transactions. 2006;34:1–6. doi: 10.1042/BST20060001. abstract. [DOI] [PubMed] [Google Scholar]
- 47.Rose A, Broholm C, Kiillerich K, Finn S, Proud C, Rider M, Richter E, Kiens B. Exercise rapidly increases eukaryotic elongation factor 2 phosphorylation in skeletal muscle of men. Journal of Physiology. 2005;569:223–228. doi: 10.1113/jphysiol.2005.097154. abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joshi B, Cai A, Keiper B, Minich W, Mendez R, Beach C, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads R. Phosphorylation of eukaryotic protein synthesis initiation factor 4E at Ser-209. J Biol Chem. 1995;270:14597–14603. doi: 10.1074/jbc.270.24.14597. abstract. [DOI] [PubMed] [Google Scholar]
- 49.Shao J, Grammatikakis N, Scroggins B, Uma S, Huang W, Chen J, Hartson S, Matts R. Hsp90 regulates p50(cdc37) function during the biogenesis of the activeconformation of the heme-regulated eIF2 alpha kinase. J Biol Chem. 2001;276:206–214. doi: 10.1074/jbc.M007583200. [DOI] [PubMed] [Google Scholar]
- 50.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters B. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Molecular and Cellular Biology. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montie H, Kayali F, Haezebrouck A, Rossi N, Degracia D. Renal ischemia and reperfusion activates the eIF2 alpha kinase PERK. Biochemica Biophysica Acta. 2005;1741:314–324. doi: 10.1016/j.bbadis.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Anwar A, Li F, Leake D, Ishii T, Mann G, Siow R. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: Role of mitogen-activated protein kinases and Nrf2. Free Radicals in Biology and Medicine. 2005;39:227–236. doi: 10.1016/j.freeradbiomed.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 53.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 54.Lu F, Zander DS, Visner GA. Increased expression of heme oxygenase-1 in human lung transplantation. Journal of Heart and Lung Transplantation. 2002;21:1120–1126. doi: 10.1016/s1053-2498(02)00423-0. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki H, Tashiro S, Sun J, Doi H, Satomi S, Igarashi K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J Biol Chem. 2003;278:49246–49253. doi: 10.1074/jbc.M306764200. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Zhu X, Zhang G, Ling T. Protective effect of hemoglobin-induced heme oxygenase-1 on injured lungs caused by limb ischemia-reperfusion in rats. Chinese Journal of Traumatology. 2002;5:86–91. [PubMed] [Google Scholar]
- 57.Kiemer A, Bildner N, Weber N, Vollmar A. Characterization of heme oxygenase 1 (heat shock protein 32) induction by atrial natriuretic peptide in human endothelial cells. Endocrinology. 2003;144:802–812. doi: 10.1210/en.2002-220610. [DOI] [PubMed] [Google Scholar]
- 58.Li L, Julien B, Grenard P, Teixeira-Clerc F, Mallat A, Lotersztajn S. Molecular mechanisms regulating the antifibrogenic protein heme-oxygenase-1 in human hepatic myofibroblasts. Journal of Hepatology. 2004;41:407–413. doi: 10.1016/j.jhep.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 59.Chen G, Liu Z, Vlantis A, Tse G, Leung B, van Hasselt C. Heme oxygenase-1 protects against apoptosis induced by tumor necrosis factor-alpha and cycloheximide in papillary thyroid carcinoma cells. Journal of Cellular Biochemistry. 2004;92:1246–1256. doi: 10.1002/jcb.20157. [DOI] [PubMed] [Google Scholar]
- 60.Bundy R, Hoare G, Kite A, Beach J, Yacoub M, Marczin N. Redox regulation of p38 MAPK activation and expression of ICAM-1 and heme oxygenase-1 in human alveolar epithelial (A549) cells. Antioxidants and Redox Signaling. 2005;7:14–24. doi: 10.1089/ars.2005.7.14. [DOI] [PubMed] [Google Scholar]
- 61.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003;278:17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 62.Ryter S, Xi S, Hartsfield C, Choi A. Mitogen activated protein kinase (MAPK) pathway regulates heme oxygenase-1 gene expression by hypoxia in vascular cells. Antioxidants and Redox Signaling. 2002;4:587–592. doi: 10.1089/15230860260220085. [DOI] [PubMed] [Google Scholar]
- 63.Ichikawa T, Zhang J, Chen K, Liu Y, Schopfer F, Baker P, Freeman B, Chen Y, Cui T. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endrocrinology. 2008;149:4086–4094. doi: 10.1210/en.2007-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicological Sciences. 2003;73:124–134. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]