

Abstract
Centromere protein B (CENP-B) binds constitutively to mammalian centromere repeat DNA and is highly conserved between humans and mouse. Cenpb null mice appear normal but have lower body and testis weights. We demonstrate here that testis-weight reduction is seen in male null mice generated on three different genetic backgrounds (denoted R1, W9.5, and C57), whereas body-weight reduction is dependent on the genetic background as well as the gender of the animals. In addition, Cenpb null females show 31%, 33%, and 44% reduced uterine weights on the R1, W9.5, and C57 backgrounds, respectively. Production of “revertant” mice lacking the targeted frameshift mutation but not the other components of the targeting construct corrected these differences, indicating that the observed phenotype is attributable to Cenpb gene disruption rather than a neighbouring gene effect induced by the targeting construct. The R1 and W9.5 Cenpb null females are reproductively competent but show age-dependent reproductive deterioration leading to a complete breakdown at or before 9 months of age. Reproductive dysfunction is much more severe in the C57 background as Cenpb null females are totally incompetent or are capable of producing no more than one litter. These results implicate a further genetic modifier effect on female reproductive performance. Histology of the uterus reveals normal myometrium and endometrium but grossly disrupted luminal and glandular epithelium. Tissue in situ hybridization demonstrates high Cenpb expression in the uterine epithelium of wild-type animals. This study details the first significant phenotype of Cenpb gene disruption and suggests an important role of Cenpb in uterine morphogenesis and function that may have direct implications for human reproductive pathology.
The centromere is essential for proper chromosome movements during mitosis and meiosis. An increasing number of centromere proteins have now been identified but little is known about the precise roles of these proteins, especially in whole animals (Choo 1997a; Craig et al. 1998; Dobie et al. 1999). Recent gene disruption studies in mice have produced null mutations in three different centromere proteins. Mutations in two of these proteins, Cenpc and Incenp, caused early embryonic lethality (Kalitsis et al. 1998; Cutts et al. 1999). The third protein, Cenpb, was nonessential as null mice appeared healthy (Hudson et al. 1998; Kapoor et al. 1998; Perez-Castro et al. 1998) except for lower body and testis weights (Hudson et al. 1998).
The biological role of CENP-B has intrigued researchers for many years. CENP-B is a constitutive and abundant centromere-specific protein, and is highly conserved in mammals (Earnshaw et al. 1987a; Sullivan and Glass 1991; Haaf and Ward 1995; Bejarano and Valdivia 1996; Yoda et al. 1996). The protein shows an overall 96% nucleotide sequence similarity between humans and mouse, with a surprisingly high level (95% and 83%, respectively) of homology even in the 5′ and 3′ untranslated mRNA sequences (Earnshaw et al. 1987b; Sullivan and Glass 1991). The protein binds centromeric human α-satellite and mouse minor satellite DNA via a 17-bp consensus CENP-B box motif (Pietras et al. 1983; Rattner 1991). Through its dimerization properties, the protein is thought to be involved in the assembly of the large arrays of centromeric repeats (Muro et al. 1992; Yoda et al. 1992). The presence of this protein on both the active and inactive centromeres of mitotically stable pseudodicentric human chromosomes (Earnshaw et al. 1989; Page et al. 1995; Sullivan and Schwartz 1995) indicates that CENP-B binding is not immediately associated with centromere activity. The absence of this protein on the Y chromosome in humans and mouse (Earnshaw et al. 1987a), on the centromeres of African green monkey (known to be composed largely of α-satellite DNA containing little or no binding sites for CENP-B) (Goldberg et al. 1996), as well as on an increasing number of human marker chromosomes containing analphoid neocentromeres (Voullaire et al. 1993; Choo 1997b; Depinet et al. 1997; du Sart et al. 1997), suggests that the role of this protein is dispensable.
In this study, we have further investigated the Cenpb null mice and present evidence that the phenotype of these mice was specifically related to Cenpb gene disruption, excluding the possibility that the phenotype might have been due to a neighboring gene effect. We describe the influence of the null mutation on body and testis weights in different genetic backgrounds and report a previously unrecognized link between Cenpb deficiency and severe female reproductive dysfunction resulting from abnormality of the uterine epithelium. Our data further indicate a role of genetic modifiers in this reproductive dysfunctional phenotype.
RESULTS
Generation of Control Mice Carrying the Targeted Selectable Marker Cassette but not the Frameshift Mutation
To confirm that the phenotype in Cenpb null mice was caused by Cenpb gene disruption, as distinct from a consequence of the gene targeting event exerting an effect on neighboring genes, we generated mice (designated “targeted control” or o/o mice) carrying the internal ribosome entry site (IRES)–neomycin selectable marker element in the 3′ noncoding region of the Cenpb gene but lacking the translational frameshift mutation caused by the 26-mer oligonucleotide designated D/TAA (5′-GTACCTAGGTATACTTTTAAACTGAC-3′) introduced into the 5′ coding region of the Cenpb gene in the null mice (Fig. 1A). This linker introduced a DraI site, a frameshift mutation, and three stop codons in all three reading frames, of which TAA was in-frame with Cenpb translation, disrupting not only the critical amino-terminal 125-amino acid centromere DNA-binding domain, but also removing all remaining carboxy-terminal regions including the dimerization domain (Hudson et al. 1998). Heterozygous (+/o) embrionic stem (ES) cell lines carrying the targeted control allele have been produced previously (Hudson et al. 1998). In this study, these cell lines were microinjected into C57BL/6 blastocysts to produce germline chimeras. Through selective breeding, wildtype (+/+), heterozygous (+/o), and homozygous targeted control (o/o) mice were generated and identified by PCR screening (Fig. 1B). The analysis of 128 offspring (generation 2) from 11 heterozygous breeding pairs gave the expected Mendelian ratio of 26 +/+, 73 +/o, and 29 o/o, suggesting no obvious viability bias among the different genotypes.
Figure 1.

Strategies for production and screening of Cenpb disrupted and targeted control mice. (A) Intronless wild-type Cenpb gene showing the 1.8-kb coding region (solid box) and polyadenylation p(A) site. Recombination at the sites shown by the dotted crosses results in the incorporation of the targeting construct containing a translational frameshift oligonucleotide (D/TAA) at the 5′ coding region and an IRES-selectable marker cassette (shaded box) within the 3′-untranslated region (i). This recombination event results in the disruption of the Cenpb gene. An alternative recombination event at sites indicated by the solid crosses, one of which is 3′ of the D/TAA frameshift mutation, results in the introduction of the IRES-selectable marker cassette but not the frameshift mutation (ii). This targeted allele served as a control for any positional effect the inserted IRES-selectable marker cassette may have on the phenotype of the mice. The Cenpb wild-type gene (+), null allele (−), and targeted control allele (o) allele were detected as described previously (Hudson et al. 1998) as 5.3-, 0.7-, and 7-kb bands (broken lines), respectively, by DraI (D) digestion and Southern blot hybridization using a 5′-probe (open box) (see Hudson et al. 1998). Once the heterozygous ES cell lines carrying the + and o alleles were identified, they were used for the production of the targeted control (o/o) mice. Subsequent genotype screening for these mice was based on PCR analysis using primers B4top and B1comp, which gave a 281-bp product for the + allele, and primers Neo1 and B1comp which gave a 320-bp product for the o allele. (B) PCR analysis of mouse progeny from a +/o × +/o cross using a combination of the primers B4top, B1comp, and Neo1, showing the +/+ (lanes 5,7), +/o (lanes 2,3,4,6), and o/o (lane 1) genotypes.
Figure 2 shows immunofluorescence analysis of fibroblast cell lines derived from +/+ and o/o littermates using an anti-CENP-B monoclonal antibody (Hudson et al. 1998). The results indicated the presence of Cenpb proteins on the centromeres in both the cell lines. The highly variable signals detected on different chromosomes were quite typical for this protein (Hudson et al. 1998). These results therefore established that Cenpb gene expression in the o/o animals was normal and had not been noticeably affected by the insertion of the IRES/selectable marker cassette. These animals therefore served as “revertant” controls for the Cenpb null mutation.
Figure 2.

Immunofluorescence analysis of fibroblast cell lines derived from +/+ (A) and o/o (B) mice. Metaphase chromosomes (DAPI-stained and pseudocolored red) were prepared from fibroblasts cultures of mouse tail tissues and stained with a monoclonal anti-CENP-B antibody (green). (1) Merged immunofluorescence signals; (2) split image for the anti-CENP-B (green) signals.
Body and Testis Weights of Cenpb Null Mice on Different Genetic Backgrounds
In a previous study, we generated Cenpb null mice that were maintained on a mixed genetic background (Simpson et al. 1997; Hudson et al. 1998). These mice were designated R1−/− here to distinguish them from two new Cenpb null mouse strains, W9.5−/− and C57−/−, produced on different genetic backgrounds for this study. W9.5−/− was on a mixed but different background to that of R1−/− and represented an independent gene-targeting event to R1−/− (Hudson et al. 1998). C57−/− was a congenic strain (Markel et al. 1997; Simpson et al. 1997) generated by backcrossing the R1+/− mice to C57BL/6 animals for eight generations.
We previously described a significant reduction of between 15%–20% in the body weights of 10-week-old adult male and female R1−/− animals up to 33 weeks old (Hudson et al. 1998). We show here that this trend could be extended to much older R1−/− animals of up to 90 to 100 weeks of age (Fig. 3A,B). A similar body weight reduction was observed for the W9.5−/− females (Fig. 3D) but was absent from the W9.5−/− males (Fig. 3C) and the C57−/− animals of both sexes (Fig. 3E,F). When testis weights were determined, a statistically significant, 14%–26% reduction was seen in all three genetic backgrounds (Table 1A). This reduction in testis size did not have any measurable effect on male fertility in animals up to 2 years old. Longevity for the R1 mice [100 weeks of follow-up; hazard ratio of 1.02 (P = 1.0) for −/− versus +/+ males (n = 27 and n = 44, respectively); hazard ratio of 0.79 (P = 0.7) for −/− versus +/+ females (n = 26 and n = 28, respectively)], W9.5, and C57 mice (40 weeks of follow-up) were normal in the different backgrounds.
Figure 3.

Total body weight of male (A,C,E) and female (B,D,F) mice in different genetic backgrounds: (A,B) RI; (C,D)W9.5; and C57 (E,F) C57. At least four animals were used for each time point. Age of the animals was determined from birth. (▴) +/+ animals; (○) −/− animals.
Table 1.
Total Testis, Uterus, and Ovary Weight and Number of Embryos Flushed from the Oviducts of Mice on Different Genetic Backgrounds
| Genetic background | Av. weight ±s.d. +/+ | −/− | Percent reduction | t-test P-value |
|---|---|---|---|---|
| A. Testis | ||||
| R1 | 208.2 ± 29.4 (n = 15) | 154.0 ± 37.6 (n = 16) | 26 | <0.001 |
| W9.5 | 191.9 ± 14.3 (n = 6) | 156.1 ± 17.1 (n = 7) | 19 | 0.002 |
| C57 | 186.8 ± 15.3 (n = 13) | 159.9 ± 17.9 (n = 12) | 14 | <0.001 |
| B. Uterus | ||||
| R1 | 114.9 ± 23.0 (n = 6) | 79.7 ± 27.8 (n = 7) | 31 | 0.032 |
| W9.5 | 122.8 ± 31.7 (n = 9) | 82.5 ± 19.0 (n = 5) | 33 | 0.024 |
| C57 | 94.5 ± 14.3 (n = 10) | 52.9 ± 20.1 (n = 7) | 44 | <0.001 |
| C. Ovary | ||||
| R1 | 10.4 ± 3.8 (n = 6) | 11.2 ± 1.8 (n = 7) | N.R. | 0.635 |
| W9.5 | 8.7 ± 2.1 (n = 9) | 8.8 ± 2.4 (n = 5) | N.R. | 0.878 |
| C57 | 7.3 ± 2.2 (n = 10) | 8.0 ± 1.8 (n = 7) | N.R. | 0.513 |
| D. No. of embryos | ||||
| R1 | 7.8 ± 5.0 (n = 6) | 4.7 ± 3.3 (n = 7) | N.R. | 0.198 |
| W9.5 | 5.9 ± 3.5 (n = 9) | 3.4 ± 3.1 (n = 5) | N.R. | 0.210 |
| C57 | 4.9 ± 2.9 (n = 10) | 4.6 ± 3.6 (n = 7) | N.R. | 0.838 |
Ten-week-old +/+ and −/− mice were used. Uteri, ovaries, and embryos were collected at day 0.5 of vaginal plug using previously unmated females. Weight is in mg. (N.R.) Not relevant, as P-value is insignificant.
To further investigate the reasons for the observed body weight reduction, various organs from 4-, 6-, 8-, 10- and 24-week-old, age-matched R1−/− and R1+/+ males and females (n = 4 for each category) were weighed. The organs included stomach, small and large intestines, liver, salivary gland, spleen, pancreas, kidney, thymus, brain and olfactory bulb, lung, adrenal, heart, testes, epididymis, bulbourethral gland, ovary, uterus, ovarian fat pads, subcutaneous fat pad, and subrenal fat pad. In addition, whole-body compositions were analyzed in 20-week-old R1−/− and R1+/+ animals of both sexes in terms of their dry weight, ash weight, moisture, protein, and fat. Nose–rump length was also measured. When the results were expressed as a percentage of fresh body wet weight, no significant difference was seen between the test and control groups for all the measurements, except for the testes (Table 1A) and the uterus (see below). These results suggested that the organs and body composition of the R1−/− animals were overall proportionally smaller than those of the R1+/+ animals. Plasma leptin levels [3.7 ± 2.4 ng/ml for the −/− mice (n = 17) and 4.6 ± 3.5 ng/ml for the +/+ mice (n = 21); P = 0.358] were not significantly different between 6-month-old female R1 and W9.5 Cenpb null and wild-type animals, suggesting that hypophagia (suppressed food intake) was unlikely to be responsible for reduced body weight in the −/− animals.
To determine whether cells deficient in Cenpb have an altered growth rate, we compared the population doubling times of three independently derived −/− ES cell lines (one in R1 and two in W9.5 backgrounds) (Hudson et al. 1998) with those of a +/+ and +/− cell line from each background. No significant difference was observed between the various cell lines over 400 cell divisions (data not shown). Karyotyping of the −/− cell lines at the late doubling passages also revealed no abnormality when compared with +/+ and +/− cells. This suggested that the Cenpb null ES cell lines grew normally and that their growth rate, unlike that previously described for the telomerase-deficient ES cells (Niida et al. 1998), did not deteriorate with increasing doubling times over the period tested.
Cenpb Null Mice Showed Reduced Uterus Weights
The uteri of 10-week-old [day 0.5 vaginal plug (VP)] previously unmated −/− animals in the R1, W9.5, and C57 backgrounds were weighed and respectively found to be 31%, 33%, and 44%, smaller than those of their corresponding wild-type siblings (Table 1B and Fig. 4A). Table 2A shows the results for the total uterus weights of 8-, 9-, 10-, and 24-week-old previously unmated female R1 mice (day 0.5 VP) as well as the total uterus weights of 4-, 6-, 8-, and 10 week-old C57 mice. No statistically significant difference was observed in the 8-week-old +/+ and −/− R1 animals, with a trend toward a difference emerging at 9 weeks and a significant difference seen at 10 and 24 weeks. Similar trends were observed in the uteri of the W9.5 animals (data not shown). With the C57 null females, however, smaller uteri were observed in −/− animals compared with +/+ mice at a significantly earlier timepoint of 6 weeks (Table 2A). These results indicated a dramatic slowdown in uterine growth during the 8- to 10-week postnatal period for R1 and W9.5 and the 4- to 6-week postnatal period for C57 null mice.
Figure 4.

Uterine and pregnancy problems in Cenpb null mice. (A) Size comparison of uteri from 10-week-old R1+/+ and R1−/− mice, showing the left and right uterine horns. The sizes of the ovaries and oviducts attached to these horns were normal in both animals. (B) Nine-month-old, 1-week overdue W9.5−/− pregnant female (first pregnancy), showing a full-term dead fetus (arrowhead) attributable to placental necrosis in the right horn (rh) and fetal growth arrest/resorption (arrow) in the left horn (lh; shown in circle). (C,D) Twelve-month-old, 10-day-overdue R1−/− pregnant female (fourth pregnancy), showing external and internal views of a necrotic fetus in the right horn, and absence of fetuses in the left horn. (E,F) Nine-month-old, 4-day-overdue R1−/− pregnant female (first pregnancy), showing external and internal views of decomposed fetal content in the left horn, and absence of fetuses in the right horn.
Table 2.
Uterus Weight in Female +/+ and −/− R1 and C57 Mice at Different Ages and 17β-Estradiol and Progesterone Levels in R1 Animals
| Age (weeks) | +/+ | −/− | Percent reduction | t-test P-value |
|---|---|---|---|---|
| A. Uterus | ||||
| R1 | ||||
| 8 | 100.6 ± 29.7 (n = 8) | 83.3 ± 32.6 (n = 8) | N.R. | 0.285 |
| 9 | 113.2 ± 34.6 (n = 6) | 73.9 ± 26.4 (n = 6) | 35 | 0.051 |
| 10 | 114.9 ± 23.0 (n = 6) | 79.7 ± 27.8 (n = 7) | 31 | 0.032 |
| 24 | 162.7 ± 50.3 (n = 7) | 100.0 ± 44.8 (n = 7) | 39 | 0.030 |
| C57 | ||||
| 4 | 8.0 ± 2.9 (n = 7) | 7.1 ± 1.3 (n = 3) | N.R. | 0.599 |
| 6 | 82.5 ± 28.2 (n = 8) | 49.9 ± 5.2 (n = 7) | 40 | 0.010 |
| 8 | 98.9 ± 32.5 (n = 7) | 63.1 ± 8.9 (n = 5) | 36 | 0.040 |
| 10 | 94.5 ± 14.3 (n = 10) | 52.9 ± 20.1 (n = 7) | 44 | <0.001 |
| B. 17β-Estradiol (R1) | ||||
| 6 | 26.0 ± 15.6 (n = 7) | 19.8 ± 7.2 (n = 10) | 0.286 | |
| 8 | 27.3 ± 14.7 (n = 6) | 33.3 ± 18.8 (n = 8) | 0.536 | |
| 10 | 33.6 ± 22.1 (n = 8) | 32.9 ± 11.1 (n = 12) | 0.927 | |
| 24 | 39.8 ± 10.8 (n = 6) | 42.5 ± 18.2 (n = 4) | 0.776 | |
| C. Progesterone (R1) | ||||
| 6 | 6.2 ± 5.3 (n = 15) | 11.5 ± 12.4 (n = 13) | 0.144 | |
| 8 | 10.4 ± 12.7 (n = 5) | 4.0 ± 2.6 (n = 8) | 0.184 | |
| 10 | 4.0 ± 3.8 (n = 15) | 2.5 ± 3.3 (n = 5) | 0.533 | |
| 24 | 11.7 ± 8.6 (n = 9) | 6.7 ± 3.5 (n = 7) | 0.165 | |
Uteri were collected at day 0.5 of vaginal plug except for the prepubertal uteri from 4-week-old virgin C57 mice. Sera were collected from virgin females for measurement of 17β-estradiol (pm/liter) and progesterone (nm/liter) levels. (N.R.) Not relevant, as P-value is insignificant.
Further Evidence that the Cenpb Null Phenotype was Directly Related to Cenpb Gene Disruption
The phenotype of the targeted control (o/o) mice was ascertained. At the gross level, these animals were phenotypically indistinguishable from their +/+ wild-type littermates. Table 3 compares the body, testis, and uterus weights of (as well as the ovary weight and number of eggs produced by; see below) these animals. No statistically significant difference was observed between the two genotypes. These results provided evidence that the correction of the frameshift mutation has allowed the o/o animals to revert back to a wild-type phenotype and that the IRES/selectable marker cassette (which was retained in the o/o animals) was not responsible for the phenotype observed in the −/− mice. A corollary of this was that the phenotype seen in our Cenpb null mice was a direct consequence of the disruption of the Cenpb gene itself.
Table 3.
Total Body and Organ Weight (mg), and Number of Embryos Flushed from the Oviducts of Mice
| Av. weight ± s.d. | t-test P-value | ||
|---|---|---|---|
| +/+ | o/o | ||
| Body (grams) | |||
| male | 29.5 ± 2.1 (n = 8) | 29.1 ± 2.5 (n = 6) | 0.743 |
| female | 22.2 ± 1.3 (n = 6) | 20.9 ± 2.1 (n = 8) | 0.209 |
| Testis (mg) | 207.4 ± 26.4 (n = 8) | 196.6 ± 30.1 (n = 6) | 0.485 |
| Uterus (mg) | 111.3 ± 32.8 (n = 6) | 114.2 ± 19.5 (n = 8) | 0.843 |
| Ovary (mg) | 10.7 ± 0.6 (n = 6) | 11.0 ± 3.7 (n = 8) | 0.814 |
| No. of embryos | 6.7 ± 4.2 (n =6) | 6.3 ± 4.8 (n = 8) | 0.869 |
Ten-week old +/+ and o/o littermates on W9.5 genetic background were used. Uteri, ovaries, and embryos were collected at day 0.5 of vaginal plug from previously unmated females.
Compromised Reproduction in Cenpb Null Females
When 10-week-old virgin R1−/− females (n = 10) were crossed with stud males in an ongoing breeding program, little difference was observed in the first three to four litters compared with control R1+/+ and R1+/− females, indicating normal reproduction in young R1−/− females. A progressive deterioration (see below) in reproductive performance, however, was observed with increasing maternal age until this failed totally in all the R1−/− females by 9 months, when the R1+/− and R1+/+ females have continued to be reproductively competent at or long after this age.
Next, we crossed a cohort of R1−/− (n = 3) and R1+/+ (n = 4) 9-month-old virgin females with C57BL/6 normal stud males in a breeding program lasting for 7 months. One pregnancy occurred in each of the R1−/− females but the animals sickened because of being overdue and required autopsy (see below). In comparison, 20 normal pregnancies occurred in the control group, which resulted in 87 healthy pups over the same period. These results, together with those described above, indicated that the observed reproductive problems were age-related rather than a consequence of prior pregnancies.
A similar reproductive phenotype was seen in 8- to 9-week-old −/− females in the W9.5 background. A cohort of 9-month-old W9.5−/− (n = 8) and W9.5+/+ (n = 8) virgin females was mated with C57BL/6 or ARC SWISS stud males over a 15-week period. Two females in the −/− group achieved a pregnancy but both were distressed because of failure of spontaneous labor at expected delivery date (post maturity) and required autopsy (discussed below). The remaining six −/− animals failed to show any visible sign of pregnancy. In contrast, the +/+ group of animals produced a total of 10 pregnancies that yielded 48 healthy pups over this period.
Compared with the animals in the R1 and W9.5 backgrounds, a significantly more severe reproductive phenotype was apparent in the −/− animals on the C57 congenic background. The reproductive performance of 8- to 15-week-old C57−/− (n = 5) females was assessed over a period of 5 months. Pregnancies were observed in all five animals. Only one of these pregnancies went to normal term and birth (four healthy pups); subsequent to this healthy litter, this female failed to become visibly pregnant again. Two of the pregnant females required autopsy because of postmaturity in one case and complication during delivery (dislocated fetal torso entrapped within the birth canal) in the other case. The fourth female had a slow delivery (>24 hr) that resulted in four live and one dead pup; in her second pregnancy, this female sickened because of being overdue and was culled. The fifth animal produced two small litters of two and three well-formed but dead pups at birth; this female has since failed to become pregnant again. Therefore, the five young C57−/− females together gave rise to only eight healthy pups over a 5-month mating period. In stark contrast, 74 healthy pups resulted from a cohort of six, age-matched, control C57+/+ females over a shorter mating duration of 3 months. These results suggested that the reproductive fitness of the C57 Cenpb null female mice was severely compromised, and to a much greater extent compared with those seen in the R1 and W9.5 Cenpb null mice.
Overall, pregnancy problems in the C57−/−, R1−/−, and W9.5−/− females manifested either as a failure of the animals to become visibly pregnant despite detection of vaginal plug or, as occurred most frequently with mice that did achieve visible pregnancy, the animals sickened because of being overdue (by up to 10 days) or difficulty with delivery. Autopsy of these sickened mice revealed dead fetuses in all cases. Where discernible, fetal development appeared normal (Fig. 4B,D). Causes of fetal death in utero included placental necrosis (Fig. 4B), fetal growth arrest or resorption (Fig. 4B), fetal necrosis (Fig. 4C,D) and decomposition (Fig. 4E,F). Because the progeny of crosses between the −/− females and normal stud males must all have a +/− genotype, the observed fetal problem could not have been related to any possible complication caused by the presence of Cenpb null embryos in the litter. Microbacterial tests of necrotic or decomposed tissues revealed profuse growth of a variety of opportunistic bacteria including Flavobacterium, Meningosepticum (water bug), Haemophilus influenzae (fecal/genital bug), Gram-negative rods including Escherichia coli (gut bug), and Gram-positive cocci.
Normal Ovarian and Hormonal Functions in Cenpb Null Females
To further investigate the causes for the compromised reproductive phenotype seen in the Cenpb null mice, we compared the total ovary weight in previously unmated wild-type and Cenpb null littermates at day of vaginal plug. No significant difference was observed for the animals in the R1, W9.5 and C57 background (Table 1C). The ovaries of Cenpb null mice on all three genetic backgrounds were able to produce a normal number of fertilized eggs (Table 1D). Direct measurement of the serum 17β-estradiol and progesterone levels in nonmated animals in all three genetic backgrounds also indicated no significant difference between the Cenpb null and wild-type animals (Table 2B,C; W9.5 and C57 data not shown). These results suggested that defective egg production or female reproductive hormones were unlikely causes for the observed reproductive problems in the Cenpb null female mice.
Defective Uterine Epithelium
We next explored the possibility that a primary defect could have occurred in the uterus, affecting its ability to support implantation and/or fetal growth. This was investigated by direct histological examination of the uterine tissues.
Histology of uterine sections indicated no major abnormality in 10-week-old R1−/− mice. In 6- to 9-month-old R1−/− and 10 week-old C57−/− animals, the myometrium and endometrium were relatively normal, but gross abnormality of the epithelium was detected. In the wildtype animals, the tall columnar cells of the uterine luminal epithelium consisted of elongated nuclei that were primarily basally situated (Fig. 5A), whereas the cells of the endometrial glandular epithelium consisted of nuclei that were more ovoid and centrally located (Fig. 5C). In the Cenpb null mice, the columnar cell morphology and basal nuclear appearance of the luminal epithelium was severely disrupted and replaced by highly disorganised and apoptotic cells (Fig. 5B). A similar phenotype was also apparent in the epithelium of the endometrial glands (Fig. 5D). Other abnormalities (not shown) included fewer endometrial glands, significantly increased leukocyte infiltration, hemorrhage, ulceration, and infection. It was also evident that the severity of these phenotypes was significantly greater in the C57−/− animals compared with those of the Cenpb null animals in the other genetic backgrounds.
Figure 5.
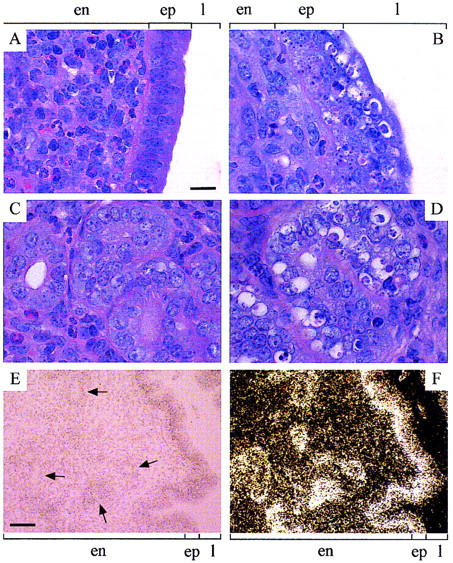
Histology (A–D) and in situ hybridization (E,F) of uterine sections. (A,C) Ten-week-old, hematoxylin-and eosin-stained C57+/+ uterus (day 0.5 VP), showing normal morphology of the endometrial epithelium lining the uterine lumen (A) and endometrial glands (C). (B,D) ten-week-old, hematoxylin- and eosin-stained C57−/− uterus (day 0.5 VP), showing highly disorganized and apoptotic (clear) epithelial cells lining the uterine lumen (B) and endometrial glands (D). Apoptosis of the clear cells (containing condensing chromatin) was confirmed by TUNEL assay (Gavrieli et al. 1992) (data not shown). Scale bar for A–D, 20 μm. (E,F) Bright-field (hematoxylin-stained) and dark-field views, respectively, of 10-week-old R1+/+ uterus hybridized with a 35S-labeled mouse Cenpb antisense riboprobe, showing mRNA signals (dark brown grains in E and white grains in F) throughout the endometrium (and the myometrium; not included in picture) with maximal mRNA expression in the epithelial lining of the uterine lumen and endometrial glands (selected examples of which are indicated by arrows). Scale bar for E and F, 50 μm. (en) Endometrium; (ep) epithelium, (l) uterine lumen.
High Cenpb Expression in Uterine Epithelium
In situ hybridization was used to determine the Cenpb mRNA expression pattern of the normal uterine tissues. Using a Cenpb-specific antisense riboprobe, expression was observed throughout the uterine section. A disproportionately higher level of expression was seen in the epithelial lining of the uterine lumen and endometrial glands compared with the endometrial and myometrial layers (Fig. 5E,F). No significant hybridization was obtained with the Cenpb sense control probe in any of these tissues (not shown).
DISCUSSION
Centromere proteins are important components for the proper execution of mitosis and meiosis but relatively little is known about their roles, or the consequences of their defects, in whole animals. The role of Cenpb is particularly intriguing since despite its conservation, cellular abundance, and specificity to the centromere, a number of lines of evidence point to this protein being nonessential for cell division and growth both in tissue culture and in the animal (referenced in Introduction). In a previous study, we demonstrated that Cenpb null mice in a mixed (R1) genetic background have a lower body and testis weight but otherwise appear normal (Hudson et al. 1998). Here, we have extended the analysis to mice on two new genetic backgrounds (mixed W9.5 and congenic C57). The results indicate that a reduction in testis weight is also seen in these backgrounds. On the other hand, body-weight reduction is apparent in the W9.5−/− females but not in the W9.5−/− males or the C57−/− males and females. This observation suggests the presence of genetic modifiers (Banbury 1997; Threadgill et al. 1997) that may influence body-weight development in a Cenpb null milieu. Furthermore, in the W9.5 genetic background, the modifier effect appears to be gender-dependent. The recognition of these genetic modifier effects offers a possible explanation for the lack of body-weight phenotype in Cenpb null mice produced by two other groups on different genetic backgrounds (Kapoor et al. 1998; Perez-Castro et al. 1998). Further studies setting up a backcross or F1 intercross (e.g., MacPhee et al. 1995; Rozmahel et al. 1996) with C57 Cenpb null mice followed by phenotypic analysis of offspring should enable the identification of the number and chromosomal locations of any likely genetic modifiers that affect Cenpb expression.
Our data further indicate that the uteri of 10-week-old −/− animals in all the three genetic backgrounds of R1, W9.5, and C57 were significantly smaller (by 31%, 33%, and 44%, respectively) than their wild-type littermates. It could be argued that our gene targeting strategy, by introducing the IRES/selectable marker cassette into the 3′ noncoding region of the Cenpb gene, might have resulted in the observed phenotype in these animals attributable to an inadvertent effect of this cassette on neighboring genes. This possibility, however, now appears unlikely as the newly generated targeted control (o/o) animals, in which the IRES/selectable marker cassette is present but not the frameshift mutation, do not show such a phenotype. It can be inferred from these control studies that the observed phenotype of our Cenpb null mice is a direct consequence of a disruption of the Cenpb gene.
Evidence is presented that Cenpb null females are compromised reproductively. The severity of this abnormality is subjected to the influence of genetic modifiers. This modifier effect is particularly stark when the reproductive performance of the R1−/− females is directly compared with their congenic C57−/− derivatives. In this comparison, although the R1−/− females are reproductively competent (but showing progressive age-dependent deterioration) up to 6–9 months of age, reproduction in the C57−/− females fails totally or is severely affected at an early postpubertal age between 8–10 weeks. At present, it is unclear whether the putative genetic modifiers underlying female reproductive competence and those controlling the body weight are related.
Although our data have indicated a failure of the uteri of the Cenpb null mice to reach a normal size, this is unlikely, on its own, to be the major cause of the observed severe reproductive dysfunction. This is evident from the relatively normal reproductive performance of young R1−/− females despite their smaller uteri. Our data point to a disruption in the normal morphogenesis of the uterine epithelial tissue as the likely primary cause. The epithelium is a vital component of the uterus. During pregnancy, this tissue remodels itself to prepare the uterus to become receptive to the developing blastocyst. This remodelling, which is critically dependent on the integrity of the polarised epithelial cell phenotype (Denker 1990; Glasser and Mulholland 1993), provides the embryo with a secure place for nutrition, growth, and differentiation (including the development of a functional placenta) (Denker 1990; Glasser et al. 1991; Giudice 1997). In the reproductively dysfunctional Cenpb null mice, the epithelial cells of the uterine lumen and endometrial glands have become grossly disorganized and apoptotic. In particular, the columnar morphology and the basal nuclear positioning of the luminal epithelium have been seriously disrupted. These disruptions are expected to acutely compromise the proper functioning of the uterine epithelium and offer an explanation for the range of pregnancy problems seen in the affected animals.
The mechanism whereby Cenpb deficiency leads to the degeneration of the uterine epithelial cells remains to be determined. It is known that during pregnancy or the periodic oestrus cycle-induced remodelling of the uterus, active mitoses occur especially in the endometrial epithelial layer (Bronson et al. 1996; Kimura et al. 1978). The observed high-expression level of Cenpb in the normal uterine epithelial cells is consistent with an important role of this protein in the modulation of the mitotic activities of these cells. We have previously proposed a model whereby the function of Cenpb may be replaced by a functionally redundant protein in the Cenpb null mice (Hudson et al. 1998). The present investigation indicates that such a redundant protein if it exists is incapable of fully substituting for the role of Cenpb. Further studies should elucidate what this role may be.
In humans, one-third of normal pregnancies ends in spontaneous abortion, with two-thirds of these occurring before clinical detection of pregnancy (Wilcox et al. 1988). Abnormality in uterine remodelling to make it receptive as well as supportive of the developing blastocyst has been cited as a principal cause of pregnancy wastage (Glasser 1998). Because CENP-B is highly conserved between mouse and humans, it would be of clinical importance to determine whether CENP-B expression is altered in pathological conditions associated with female infertility or aberrant female reproductive performance. In addition, these studies may shed light on conditions like metritis (inflammation of the uterus) and pyometra (uterine infection), which are considerable problems in veterinary medicine without known causal links (Santschi et al. 1995; Dhaliwal et al. 1998; Lawler 1998; Rajala and Grohn 1998; Smith et al. 1999). The observations described in this study have broad implications for understanding uterine morphogenesis, centromere function, as well as human and animal reproductive pathology, and warrant further detailed study.
METHODS
Generation and PCR Screening of Cenpb Targeted Control Mice
For chimeric mouse production, W9.5 +/o ES cells (Hudson et al. 1998) were microinjected into C57BL/6 blastocysts, followed by breeding of the resulting chimeras to C57BL/6 mice to generate heterozygous progeny (generation 1). Heterozygous (+/o) offspring were bred to obtain wild-type (+/+), +/o, and homozygous (o/o) progeny (generation 2) that were used for subsequent analysis. The W9.5o/o mice have incorporated the IRES-selectable marker cassette but not the 26-mer (D/TAA) translational frameshift oligonucleotide at the 5′ region of the Cenpb-coding sequence (Hudson et al. 1998). Targeted W9.5+/o ES cell lines were originally identified by Southern analysis (Hudson et al. 1998) (see Fig. 1). For PCR genotyping of cell line and mouse tail DNA, the following primers were used: B4top (5′-CTTTCCTCCCCATTAGTCCC-3′) and B1comp (5′-ACGCTGTCTTCTTTTAGCC-3′), which gave a 281-bp product for the wild-type (+) allele; or Neo1 (5′-CCTCGTGCTTTACGGTATCG-3′) and B1comp, which gave a 320-bp product for the targeted control (o) allele. PCR conditions were 95°C for 2 min, followed by 35 cycles at 95°C for 30 sec, 60°C for 1 min, and 72°C for 1 min, in a 20-μl volume containing 50–200 ng genomic DNA, 0.66 units of Taq polymerase, 200 μm dNTPs, and 200 ng of each primer in dH2O.
Generation of Cenpb Null Mice on Different Genetic Backgrounds
Cenpb null mice were produced previously by microinjecting gene-targeted ES cells derived from the R1 cell line into C57BL/6 blastocysts to obtain chimeras that were subsequently mated to C57BL/6 mice (Hudson et al. 1998). Heterozygous (+/−) offspring (generation 1) were bred to obtain homozygous Cenpb null (−/−) mice, +/−, and wild-type (+/+) littermates (generation 2). These mice were maintained on a mixed genetic background of R1 (129/SvJ × 129/Sv-+p+Tyr−c MgfSl-J/+) (Simpson et al. 1997) and C57BL/6, in which 129/SvJ has been shown previously to be an impure inbred 129 strain (Threadgill et al. 1997). These animals were designated R1−/− here to distinguish them from Cenpb null mice created on two other genetic backgrounds in this study. R1 progeny (generation 2 or 3) from heterozygous brother/sister or cousin matings were used for subsequent analysis.
One of the new mouse strains, denoted W9.5−/−, was produced by microinjecting a previously Cenpb gene-targeted +/− W9.5 (originally derived from a 129/Sv blastocyst; Buzin et al.1994) ES cell-derived line (Hudson et al. 1998) into C57BL/6 blastocysts to obtain chimeras from which were mated to C57BL/6 mice. Heterozygous progeny (generation 1) were bred to generate homozygous Cenpb −/−, +/−, and +/+ littermates (generation 2). The W9.5 mouse strain was maintained on a mixed background of 129/Sv and C57BL/6 by intercrossing W9.5+/− progeny from generation 1 or 2 via brother/sister or cousin matings. The resulting W9.5−/− and W9.5+/+ progeny (generation 2 and 3) were used for analysis. The third mouse strain, designated C57−/−, was congenic on a C57BL/6 genetic background. This was produced by mating first generation +/− progeny derived from the R1/C57BL/6 chimeras described above to C57BL/6 mice. Heterozygous progeny were backcrossed to C57BL/6 for a further seven generations. At generations 8 and 9, +/− progeny were intercrossed to generate the C57−/− congenic and C57 +/+ control mice that were used for analysis in this study.
Immunocytochemistry
Immunofluorescence staining using anti-CENP-B monoclonal antibody was performed as described previously (Hudson et al. 1998) on colcemid-arrested mouse fibroblastic cell lines grown from mouse tail biopsies using the scratch technique (Fowler 1984).
Organ Weighing, Body Composition, Longevity, Reproductive Function, and Hormonal Tests
For organ wet-weight and body-composition determination (Clark and Tarttelin 1976), an average of five age-matched R1−/− and R1+/+ animals of each sex were used. Survival/longevity analysis was performed by comparing R1−/− males (n = 27) with R1+/+ males (n = 44), and R1−/− females (n = 26) with R1+/+ females (n = 28), using Kaplan Meier plot; hazard ratios and significance values were calculated by the Cox proportional hazard regression method (Stata Corp 1997). Reproductive performance of mice was examined by setting up appropriate breeding pairs for mating and observing for vaginal plug (day 0.5 VP). Plugged mice were closely monitored during pregnancy and parturition. Plasma leptin, serum progesterone, and 17β-estradiol evels were measured using Linco Mouse Leptin RIA Kit, Bayer Diagnostics Progesterone Kit, and Sorin 17β-estradiol RIA Kit, respectively.
Histology and Tissue In Situ Hybridization
Tissue preparation, histology, and in situ hybridization were as described previously (Edmondson et al. 1995; Hudson et al. 1998). For in situ hybridization, a 700-bp PstI fragment located at nucleotide positions 1391–2095 of the Cenpb sequence (EMBL accession no. X55038) was cloned into Bluescript (Stratagene) in both orientations. The resulting sense and antisense clones were linerized with ClaI and riboprobes were labeled with 35S-CTP using T7 polymerase.
Acknowledgments
We thank S. Gazeas, A. Sylvain and J. Ladhams for care of mice; R. O'Dowd and P. Farmer for technical assistance; R. Wolfe for statistical analysis; A. Thorburn for leptin measurement; B. Leury for body composition determination; C. Print for helpful discussions; C.W. Chow for histological tissue preparation and advice; and the Department of Biochemistry of the Royal Children's Hospital for 17β-estradiol and progesterone assays. This work was approved by the Royal Children's Hospital Animal Ethics Committee and was supported by the National Health and Medical Research Council of Australia. K.H.A.C. is a Principal Research Fellow of the Council.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL CHOO@CRYPTIC.RCH.UNIMELB.EDU.AU; FAX 61-3-9348 1391.
REFERENCES
- Banbury Conference Consortium. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Bejarano LA, Valdivia MM. Molecular cloning of an intronless gene for the hamster centromere antigen CENP-B. Biochim Biophys Acta. 1996;1307:21–25. doi: 10.1016/0167-4781(96)00039-5. [DOI] [PubMed] [Google Scholar]
- Bronson FH, Dagg CP, Snell GD. Reproduction. In: Green EL, editor. Biology of the laboratory mouse. New York, NY: McGraw-Hill; 1966. pp. 187–204. [Google Scholar]
- Buzin CH, Mann JR, Singer-Sam J. Quantitative RT-PCR assays show Xist RNA levels are low in female adult mouse tissue, embryos and embryoid bodies. Development. 1994;120:3529–3536. doi: 10.1242/dev.120.12.3529. [DOI] [PubMed] [Google Scholar]
- Choo KHA. The centromere. Oxford, UK: Oxford University Press; 1997a. [Google Scholar]
- ————— Centromere DNA dynamics: Latent centromeres and neocentromere formation. Am J Hum Genet. 1997b;61:1225–1233. doi: 10.1086/301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RG, Tarttelin MF. An accurate method for the preparation and analysis of the composition of animal tissue. Physiol Behav. 1976;17:351–352. doi: 10.1016/0031-9384(76)90086-x. [DOI] [PubMed] [Google Scholar]
- Craig JM, Earnshaw WC, Vagnarelli P. Mammalian centromere: DNA sequence, protein composition, and role in cell cycle progression. Exp Cell Res. 1998;246:249–262. doi: 10.1006/excr.1998.4278. [DOI] [PubMed] [Google Scholar]
- Cutts SM, Fowler KJ, Kile BT, Hii L, O'Dowd RA, Hudson DF, Saffery R, Kalitsis P, Earle E, Choo KHA. Defective chromosome segregation, microtubule bundling, and nuclear bridging in Inner centromere protein (Incenp) gene-disrupted mice. Hum Mol Genet. 1999;8:1145–1155. doi: 10.1093/hmg/8.7.1145. [DOI] [PubMed] [Google Scholar]
- Denker HW. Trophoblast-endometrial interactions at embryo implantation: A cell biological paradox. Troph Res. 1990;4:3–29. [Google Scholar]
- Depinet TW, Zackowski JL, Earnshaw WC, Kaffe S, Sekhon GS, Stallard R, Sullivan BA, Vance GH, VanDyke DL, Willard HF, et al. Characterization of neo-centromeres in marker chromosomes lacking detectable alpha-satellite DNA. Hum Mol Genet. 1997;6:1195–1204. doi: 10.1093/hmg/6.8.1195. [DOI] [PubMed] [Google Scholar]
- Dhaliwal GK, Wray C, Noakes DE. Uterine bacterial flora and uterine lesions in bitches with cystic endometrial hyperplasia. Vet Rec. 1998;143:659–661. [PubMed] [Google Scholar]
- Dobie KW, Hari KL, Maggert KA, Karpen GH. Centromere proteins and chromosome inheritance: a complex affair. Curr Opin Genet Dev. 1999;9:206–217. doi: 10.1016/S0959-437X(99)80031-8. [DOI] [PubMed] [Google Scholar]
- du Sart D, Cancilla MR, Earle E, Mao J, Saffery R, Tainton KM, Kalitsis P, Martyn J, Barry AE, Choo KHA. A functional neo-centromere formed through activation of a latent human centromere and consisting of non-alpha-satellite DNA. Nat Genet. 1997;16:144–153. doi: 10.1038/ng0697-144. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Machlin PS, Bordwell BJ, Rothfield NF, Cleveland DW. Analysis of anticentromere autoantibodies using cloned autoantigen CENP-B. Proc Natl Acad Sci. 1987a;84:4979–4983. doi: 10.1073/pnas.84.14.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Sullivan KF, Machlin PS, Cooke CA, Kaiser DA, Pollard TD, Rothfield NF, Cleveland DW. Molecular cloning of cDNA for CENP-B, the major human centromere autoantigen. J Cell Biol. 1987b;104:817–829. doi: 10.1083/jcb.104.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma. 1989;98:1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- Edmondson SR, Werther GA, Russell A, LeRoith D, Roberts CT, Beck F. Localization of growth hormone receptor/binding protein messenger ribonucleic acid (mRNA) during rat fetal development: Relationship to insulin-like growth factor-I mRNA. Endocrinology. 1995;136:4602–4609. doi: 10.1210/endo.136.10.7664680. [DOI] [PubMed] [Google Scholar]
- Fowler KJ. Storage of skin biopsies at −70 degrees C for future fibroblast culture. 1984. J Clin Pathol. 1984;37:1191–1193. doi: 10.1136/jcp.37.10.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice LC. Intimate conversations: The implanting conceptus and its maternal host. Organ. 1997;VIII:48–51. [Google Scholar]
- Glasser SR. The uterus: Idiot savant or Rosetta Stone? J Assist Reprod Genet. 1998;15:168–173. doi: 10.1023/A:1023035915993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser SR, Mulholland J. Receptivity is a polarity dependent special function of hormonally regulated uterine epithelial cell. Microscopy Res Tech. 1993;25:106–120. doi: 10.1002/jemt.1070250204. [DOI] [PubMed] [Google Scholar]
- Glasser SR, Mulholland J, Julian J, Mani S. Blastocyst-endometrial relationships: Reciprocal interactions between uterine epithelial and stromal cells and blastocysts. Troph Res. 1991;5:229–280. [Google Scholar]
- Goldberg IG, Sawhney AF, Pluta AF, Warburton PE, Earnshaw WC. Surprising deficiency of CENP-B binding sites in African green monkey alpha satellite DNA: Implications for CENP-B function at centromeres. Mol Cell Biol. 1996;16:5156–5168. doi: 10.1128/mcb.16.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf T, Ward DC. Rabl orientation of CENP-B box sequences in Tupaia belangeri fibroblasts. Cytogenet Cell Genet. 1995;70:258–262. doi: 10.1159/000134047. [DOI] [PubMed] [Google Scholar]
- Hudson DF, Fowler KJ, Earle E, Saffery R, Kalitsis P, Trowell H, Hill J, Wreford NG, deKretser DM, Cancilla MR, Howman E, Hii L, Cutts SM, Irvine DV, Choo KHA. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J Cell Biol. 1998;141:309–319. doi: 10.1083/jcb.141.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalitsis P, Fowler KJ, Earle E, Hill J, Choo KHA. Targeted disruption of mouse centromere protein C gene leads to mitotic disarray and early embryo death. Proc Natl Acad Sci. 1998;95:1136–1141. doi: 10.1073/pnas.95.3.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Montes de Oca Luna R, Liu G, Lozano G, Cummings C, Mancini M, Ouspenski I, Brinkley BR, May GS. The cenpB gene is not essential in mice. Chromosoma. 1998;107:570–576. doi: 10.1007/s004120050343. [DOI] [PubMed] [Google Scholar]
- Kimura J, Obata T, Okada H. Steroidal control mechanism of cell proliferation in mouse uterine epithelium. Endocrinol Jpn. 1978;25:7–12. doi: 10.1507/endocrj1954.25.7. [DOI] [PubMed] [Google Scholar]
- Lawler DF. Diagnosis and treatment of inflammatory uterine disease in cats. Vet Med. 1998;93:750–753. [Google Scholar]
- Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS, Moore KJ. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat Genet. 1997;17:280–284. doi: 10.1038/ng1197-280. [DOI] [PubMed] [Google Scholar]
- MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell. 1995;81:957–966. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Muro Y, Masumoto H, Yoda K, Nozaki N, Ohashi M, Okazaki T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J Cell Biol. 1992;116:585–596. doi: 10.1083/jcb.116.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Matsumoto T, Satoh H, Shiwa M, Tokutake Y, Furuichi Y, Shinkai Y. Severe growth defect in mouse cells lacking the telomerase RNA component. Nat Genet. 1998;19:203–206. doi: 10.1038/580. [DOI] [PubMed] [Google Scholar]
- Page SL, Earnshaw WC, Choo KHA, Shaffer LG. Further evidence that CENP-C is a necessary component of active centromeres: studies of a dic(X;15) with simultaneous immunofluorescence and FISH. Hum Mol Genet. 1995;4:289–294. doi: 10.1093/hmg/4.2.289. [DOI] [PubMed] [Google Scholar]
- Perez-Castro AV, Shamanski FL, Meneses JJ, Lovato TL, Vogel KG, Moyzis RK, Pedersen R. Centromeric protein b null mice are viable with no apparent abnormalities. Dev Biol. 1998;201:135–143. doi: 10.1006/dbio.1998.9005. [DOI] [PubMed] [Google Scholar]
- Pietras D, Bennett K, Siracusa L, Woodworth-Gutai M, Chapman V, Gross K, Kane-Haas C, Hastie N. Construction of a small Mus musculus repetitive DNA library: Identification of a new satellite sequence in Mus musculus. Nucleic Acids Res. 1983;11:6965–6983. doi: 10.1093/nar/11.20.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala PJ, Grohn YT. Effects of dystocia, retained placenta, and metritis on milk yield in dairy cows. J Dairy Sci. 1998;81:3172–3181. doi: 10.3168/jds.s0022-0302(98)75883-7. [DOI] [PubMed] [Google Scholar]
- Rattner JB. The Structure of the mammalian centromere. BioEssays. 1991;13:51–56. doi: 10.1002/bies.950130202. [DOI] [PubMed] [Google Scholar]
- Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, et al. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- Santschi EM, Adams SB, Robertson JT, Bowes RM, Mitten LA, Sojka JE. Ovariohysterectomy in six mares. Vet Surg. 1995;24:165–171. doi: 10.1111/j.1532-950x.1995.tb01311.x. [DOI] [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nature Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Smith KC, Parkinson TJ, Long SE. Abbattoir survey of acquired reproductive abnormalities in ewes. Vet Rec. 1999;144:491–496. doi: 10.1136/vr.144.18.491. [DOI] [PubMed] [Google Scholar]
- Stata Corp. Stata statistical software: Release 5.0. College Station, TX: Stata Corp.; 1997. [Google Scholar]
- Sullivan BA, Schwartz S. Identification of centromeric antigens in dicentric Robertsonian translocations: CENP-C and CENP-E are necessary components of functional centromeres. Hum Mol Genet. 1995;4:2189–2197. doi: 10.1093/hmg/4.12.2189. [DOI] [PubMed] [Google Scholar]
- Sullivan KF, Glass CA. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma. 1991;100:360–370. doi: 10.1007/BF00337514. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Yee D, Matin A, Nadeau JH, Magnuson T. Genealogy of the 129 inbred strains: 129/SvJ is a contaminated inbred strain. Mam Genome. 1997;8:390–393. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
- Voullaire LE, Slater HR, Petrovic V, Choo KHA. A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: Activation of a latent centromere? Am J Hum Genet. 1993;52:1153–1163. [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, O'Connor JF, Baird DD, Schlatterer JP, Canfield RE, Armstrong EG, Nisula BC. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189–194. doi: 10.1056/NEJM198807283190401. [DOI] [PubMed] [Google Scholar]
- Yoda K, Kitagawa K, Masumoto H, Muro Y, Okazaki T. A human centromere protein, CENP-B, has a DNA binding domain containing four potential α-helices at the NH2 terminus, which is separable from dimerizing activity. J Cell Biol. 1992;119:1413–1427. doi: 10.1083/jcb.119.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda K, Nakamura T, Masumoto H, Suzuki N, Nitagawa K, Nakano M, Shinjo A, Okazaki T. Centromere protein B of African Green monkey cells: Gene structure, cellular expression, and centromeric localization. Mol Cell Biol. 1996;16:5169–5177. doi: 10.1128/mcb.16.9.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]