

Abstract
We studied the extent to which genotyping of simple sequence repeat polymorphisms (SSRs) in pooled DNA samples can be used to predict differences in allele frequencies between parents and their affected offspring. We also developed a simple method of correction for the effects of stutter and differential amplification on the analysis of SSRs in pooled DNA samples based on widely available software. We genotyped individually eight polymorphic microsatellite markers in 110 parent–offspring trios affected with bipolar affective disorder (BP). Analysis of pooled DNA samples predicted very accurately the differences in individual allele frequency distributions between children and their parents. The mean error was <1% (range 0%–3.2%) when marker-specific corrections for stutter and differential amplification were performed. We show that if an individual allele is significantly preferentially transmitted from parents to affected offspring, the difference in the frequency of that allele would be sufficiently large to be detected with pooling in most situations. We propose recommendations for disequilibrium mapping with pooling in which both case-control samples and trios are used in an initial screen and markers are genotyped individually only if they satisfy very relaxed criteria for statistical significance. The use of case-control samples should reduce the false-negative rate as the differences in allele frequencies between cases and controls are twice as high in the presence of the same genetic effect. The use of trios will confirm or reject any suggested differences, thus reducing the false-positive rate that can be created by hidden population stratification.
Genetic linkage analysis with realistic sized samples of <500 affected sibling pairs is likely to detect loci with genotype risk ratios (γ) of 4 or larger but not those with γ of 2 or less (Risch and Merikangas 1996; Camp 1997). Allelic association studies are a powerful alternative for locating genes of small effect in complex traits (Owen and McGuffin 1993; Risch and Merikangas 1996). Genome-wide association studies using either case-control or family-based methods are much more sensitive than standard genome-wide affected-sib-pair linkage analyses (Risch and Merikangas 1996; Camp 1997). They can detect a γ ranging from 2 to 4 with realistic sample sizes of 300–500 unrelated affected individuals under different assumptions of the mode of inheritance and different allele frequencies (Risch and Merikangas 1996; Camp 1997; Risch and Teng 1998). Such studies are likely to require extremely dense marker maps (Risch and Merikangas 1996; Kruglyak 1999) and the development of methods for high-throughput genotyping.
Much hope has been invested in the potential of single nucleotide polymorphisms (SNPs) for genome-wide association studies, because of their relative abundance as compared with simple sequence repeat markers (SSRs) as well as the fact that most are potentially amenable to digital typing methods using chip technology (Risch and Merikangas 1996; Kruglyak 1999; Lipshutz et al. 1999). Currently, a number of large-scale efforts are underway to develop high density SNP maps (Wang et al. 1998; Cargill et al. 1999), though these currently fall well short of the density required for genome-wide studies. In contrast, at the present time SSR maps are more comprehensive, and there is evidence that SSRs may detect linkage disequilibrium (LD) over larger distances than SNPs (Kendler et al. 1999) and thus be better suited than SNPs for indirect association studies.
Although SSRs do not readily lend themselves to genotyping by microarray, high-throughput genotyping is potentially possible using DNA pooling. Here, equal amounts of DNA from a number of patients are added to one pool, and similarly, equal amounts of DNA from controls are added to a second. These pools can then be genotyped, and differences between the groups detected (Pacek et al. 1993; Barcellos et al. 1997; Sheffield et al. 1997; Daniels et al. 1998; Shaw et al. 1998).
Unfortunately, PCR amplification of SSRs produces a stutter artefact, which generates additional (usually shorter) DNA fragments which correspond to 1, 2, 3, etc. shorter units. When DNA pools are genotyped, these stutter bands overlap and increase the height of shorter allele peaks. This can lead to a distortion in the estimated differences in allele frequencies between two groups analyzed with pooling. Another artifact of PCR to which SSRs are prone is differential amplification, which refers to the less efficient amplification of longer alleles. This can also distort the estimated differences.
LeDuc et al. (1995), Perlin et al. (1995), and Barcellos et al. (1997) suggested that more accurate estimation of allele frequencies can be obtained following arithmetical corrections for stutter and differential amplification. This requires the individual genotyping of a number of individuals, so that a matrix based on these observations can be prepared to perform the correction. Different markers have different stutter patterns; so a matrix needs to be constructed for each marker tested (Perlin et al. 1995; Barcellos et al. 1997). Previous work in our department has, however, shown that this time-consuming process can be avoided by simply examining the difference in the areas of two allele image patterns (AIP) produced when microsatellites are amplified in DNA pools using an automated sequencer and expressing this as a fraction of the total area of the two AIPs (the ΔAIP method; Daniels et al. 1998). This is because in many instances the purpose of our research is not to measure absolute allele frequencies but to detect differences between two groups. PCR amplification of two pools under the same conditions will result in similar stutter and differential amplification; so any differences in the image patterns are likely to be the result of different allele frequency distributions. With the use of an appropriately lax statistical criterion, this method can be used to screen for markers that warrant individual genotyping (Daniels et al. 1998; Fisher et al. 1999).
However, the ΔAIP approach has several disadvantages. First, it does not allow the results from several PCR reactions to be averaged, thus reducing error due to variation between replicate pools or PCRs. Second, it does not allow the frequency of the nontransmitted parental alleles to be estimated when trios are used. Finally, it does not allow specific differences in individual alleles to be measured that would be useful when pooled genotyping is used to attempt replication of allelic associations in multistage studies (Fisher et al. 1999). Therefore, in the present study we sought to develop a fast and simple method of correction for the effects of stutter and differential amplification based on widely available software and to determine its usefulness in detecting differences between SSR allele frequencies in DNA pools.
DNA pooling is readily applicable to case-control studies. However, such studies are susceptible to hidden population stratification even if careful matching has been performed and are likely to produce a number of false-positive results. In contrast, the use of parents or other family members as controls is robust to population stratification. Therefore, the second aim of this study was to determine the extent to which genotyping of SSRs in pooled DNA samples can be used to predict differences in allele frequencies between parents and their affected offspring.
We performed our analysis on markers in one of the candidate regions for bipolar affective disorder (BP). BP, also known as manic depression, is a common psychiatric disorder affecting ∼0.8% of the general population. It presents with episodes of mania and depression, although an episode of mania alone is sufficient for the diagnosis. A major genetic contribution to BP has been shown by family, twin, and adoption studies (Tsuang and Faraone 1990; McGuffin et al. 1994). However, the mode of inheritance, like that of other common disorders, is complex and non-Mendelian. The transmission in the majority of cases probably involves the interaction of several (perhaps many) genes together with environmental factors (Craddock et al. 1995). Linkage studies using the lod-score method have so far failed to provide definitive evidence for location of a gene for BP. We chose chromosome 4p16 because it is implicated in one of the strongest linkage results in BP: A lod score of 3.8 was reported by Blackwood et al. (1996) in a large multiply affected pedigree. Linkage to the same area was reported in a family affected with schizoaffective disorder studied in our department (Asherson et al. 1998). In addition, a genome-wide search in multiply affected pedigrees from the Old Order Amish revealed strong evidence that the same area harbors a gene that protects against the development of BP (Ginns et al. 1998). This is consistent with a hypothesis that certain alleles of the same gene can predispose to illness, whereas others can protect against it. In this study we have undertaken pooled genotyping of 53 markers on 4p and also genotyped 8 markers from the critical region individually in our sample of 110 cases of BP for which parental DNA is available. We have analyzed the results of pooled genotyping by both the ΔAIP method and by measuring peak heights with and without correction for stutter and differential amplification. We have estimated the size of the errors of the pooling approach and have proposed guidelines for the use of pooling with parental controls in large-scale LD studies.
RESULTS
We genotyped a total of 53 polymorphic SSR markers spanning the whole of the short arm of chromosome 4 in two DNA pools, one containing equal amounts of DNA from 110 individuals with BP and the other containing DNA from their 220 parents. Markers outside the implicated region were genotyped to assess the rate of positive results (presumably false positive) that our method would generate.
The use of triplicate pools provided a control for variation in pool construction, PCR, and measurement. The image patterns were highly reproducible between the three replicate pools studied in each experiment. They overlapped nearly perfectly in most experiments. When they did not, this indicated that the experiment had failed either due to PCR or to gel problems. Such experiments were repeated, and in every case we were able to produce closely overlapping images.
No markers showed differences between parents and offspring that were significant at P < 0.05 when analyzed with the ΔAIP method. To examine the accuracy of pooling, we genotyped individually the whole sample set of 110 trios for eight markers from the critical region of the chromosome: D4S2375 and D4S230 were chosen because they showed positive results in a different (case-control) study conducted at our department. DRD5 was genotyped because it is a dinucleotide repeat near a plausible candidate gene; so we wanted to have the results from individual genotyping. D4S2374 was the marker with the lowest ΔAIP P-value (0.3) among those that map within the critical region. The other four markers: MSX1, D4S431, D4S1602, and D4S3007 were chosen from the markers that map within the critical region on the basis of their size and dye type, so that all eight markers could be genotyped simultaneously on the same gel.
Figure 1 presents the allele frequencies distributions in parents and offspring as predicted from the pooled DNA samples after correction for stutter and differential amplification (left column) and from individual genotyping (right column). This demonstrates the extremely close agreement between predicted and true values for each allele and in every marker.
Figure 1.

Allele frequency distributions in parents and their children predicted from pools after correction for stutter and differential amplification (left) and actual allele frequencies produced from individual genotyping (right). (Solid bars) Affected children; (open bars) parents; (horizontal axis) allele numbers; (vertical axis) allele frequency (%). The two negative results for D4S230 are due to overcorrection for stutter of some very rare alleles (<1%). In the analysis these values are taken as zero, but we show the real results in order to highlight this potential problem.
We wanted to compare the performance of the different methods of analysis of pools with the results produced by individual genotyping. The statistical significance of the differences in allele frequencies between parents and affected offspring that were estimated from the pooled genotyping were analyzed with the ΔAIP method, as well as using CLUMP (Sham and Curtis 1995a) based on peak heights. The analysis with CLUMP was performed both with and without correction for stutter and differential amplification. The resulting P values from the three methods of analysis are presented in the second, third, and fourth columns of Table 1. The P values resulting from the analysis of the individual genotypings are presented in the last two columns of Table 1. Our preferred method for reporting the results of individual genotyping is by transmission disequilibrium test (TDT) (last column) because this test has been shown to be robust to population stratification (Spielman and Ewens 1996). For the purposes of this study we also present the P values of individual genotyping produced by comparing the allele frequencies of probands with the nontransmitted parental alleles calculated using CLUMP. This was done to present an analysis comparable with that performed on the allele frequencies estimated from pooled genotyping. None of the analyses of estimated allele frequencies or of individual genotypings approached conventional levels of statistical significance; so in this experiment the pooling approach produced neither false-positive nor false-negative predictions.
Table 1.
Results from Pools and Individual Genotyping of 110 Trios
| Marker | ΔAIPa | P values | |||
|---|---|---|---|---|---|
| Peak heightsb | Individual genotypingc | ||||
| Not corrected | Corrected | CLUMP | ETDTd | ||
| MSX1 | 1 | 0.87 | 0.45 | 0.30 | 0.34 |
| D4S2375 | 0.9 | 0.85 | — | 0.68 | 0.75 |
| D4S2374 | 0.3 | 0.34 | 0.37 | 0.78 | 0.68 |
| D4S431 | 1 | 1 | 0.75 | 0.26 | 0.15 |
| DRD5 | 1 | 0.9 | 0.76 | 0.46 | 0.30 |
| D4S1602 | 0.51 | 0.26 | 0.12 | 0.50 | 0.33 |
| D4S3007 | 0.6 | 0.9 | 0.9 | 0.4 | 0.27 |
| D4S230 | 1 | 0.68 | 0.15 | 0.85 | 0.38 |
Simulated significance levels of the ΔAIP.
Peak heights method before and after correction for stutter and differential amplification.
The allele frequencies of probands and controls (the untransmitted alleles) from the true genotyping were analyzed with CLUMP to present a statistical result that is more closely related to the one produced by the peak height method (which uses CLUMP).
Extended transmission/disequilibrium test (ETDT) from individual genotyping of the families.
To examine how well pooled genotyping will detect larger, statistically significant differences, we created artificial pools by adding DNA from one individual into a pool in amounts that would lead to an increase in the DNA concentration of that individual by 5%, 10%, and 15%. As explained in Methods, the produced changes in allele frequencies are conditional on the frequencies of the added alleles in the original pool. For this reason the results from individual genotyping do not necessarily become significant even after adding 15% DNA from one individual. The results from these “spiking” experiments are presented in Table 2. The results of pooled genotyping were analyzed with the ΔAIP method and by estimating the allele frequencies from the peak heights, both with and without correction, using CLUMP, but not with ETDT as the samples are effectively cases and controls. It can be seen that spiking produced four statistically significant differences between allele frequencies in the two groups. Two of these were detected by the ΔAIP method and when allele frequencies were estimated from peak heights but no correction was applied. However, all four were detected when simple correction for differential amplification and stutter was applied.
Table 2.
Results from the Spiking Experiment
| Marker (original pool vs. %) | P values | |||
|---|---|---|---|---|
| ΔAIP | Peak heights not corrected | Peak heights corrected | Individual genotyping | |
| D4S431 | ||||
| vs. 5% | 0.83 | 0.99 | 0.97 | 0.89 |
| vs. 10% | 0.21 | 0.31 | 0.079 | 0.22 |
| vs. 15% | <0.001 | 0.001 | <0.001 | <0.001 |
| D4S230 | ||||
| vs. 5% | 0.93 | 1 | 0.85 | 0.97 |
| vs. 10% | 0.80 | 0.97 | 0.31 | 0.39 |
| vs. 15% | 0.13 | 0.58 | 0.001 | 0.009 |
| DRD5 | ||||
| vs. 5% | 1 | 1 | 0.65 | 0.95 |
| vs. 10% | 0.98 | 0.96 | 0.15 | 0.32 |
| vs. 15% | 0.97 | 0.88 | 0.036 | 0.026 |
| D4S1602 | ||||
| vs. 5% | 0.56 | 0.57 | 0.51 | 0.91 |
| vs. 10% | 0.066 | 0.25 | 0.095 | 0.4 |
| vs. 15% | 0.01 | 0.012 | <0.001 | 0.039 |
| D4S2375 | ||||
| vs. 5% | 1 | 0.9 | — | 0.9 |
| vs. 10% | 0.92 | 0.57 | — | 0.7 |
| vs. 15% | 0.47 | 0.14 | — | 0.24 |
Presented are the values for statistical significance based on ΔAIP, from peak heights as produced by CLUMP (before and after correction), and from the true genotyping results. For each marker, three values correspond to 5%, 10%, and 15% enrichment of one individual's DNA. In bold are the significant results. D4S2375 did not require correction.
The error rates in predicting differences in individual allele frequencies (after correction for stutter and differential amplification) are presented in Figure 2a. It shows the true sizes of the differences as determined by individual genotyping (x-axis) and the predicted sizes (y-axis). The regression line shown runs remarkably close to the diagonal of the graph, indicating a nearly perfect agreement across differences of up to 12.8% (correlation coefficient 0.93, slope 0.98). The mean error rate was a surprisingly low 0.8% and was not affected by the allele frequency (data not shown). The highest error was only 3.2%, and 95% of all errors were <2%. For comparison, we present the same error rates without corrections (Fig. 2b). Although small differences were predicted with a similar accuracy, when the true differences became larger, they were consistently underestimated (correlation coefficient 0.85, slope 0.62). The mean and maximum errors were slightly higher at 1.2% and 4.3%, respectively.
Figure 2.


Comparison between true differences in allele frequencies between samples (x-axis) and predicted differences (y-axis). (○) Parent–offspring results; (♦) spiking experiments. (a) Predictions made after correction for stutter and differential amplification. (b) Predictions made with no correction.
DISCUSSION
The high reproducibility of the image patterns generated by the amplification of DNA pools has been also reported by other teams using pooled microsatellite analysis (Barcellos et al. 1997; Shaw et al. 1998) and by our previous work (Daniels et al. 1998). Very little variation between replicate PCR experiments has also been noticed previously (Daniels et al. 1998; Shaw et al. 1998). For the vast majority of markers when DNA pools from the parents and children were genotyped, the images also overlapped very closely. Two of these patterns are shown in Figure 3.
Figure 3.

Examples of overlaid image patterns from pools from parents and children.
Figure 1 demonstrates that the vast majority of differences in allele frequencies were predicted surprisingly well from the pools (left column). The mean error was only 0.8% for pools corrected for stutter and differential amplification and increased only slightly to 1.2% if correction was not performed (Fig. 2). However, there was no preferential transmission of individual alleles from parents to children for any of the markers, and consequently, the two pools looked quite similar in each experiment. We were concerned that if there were larger real differences, these might be missed by the pooling approach. Therefore, we performed our “spiking” experiments in which we constructed artificial pools from the parental pools by increasing the amount of DNA from one individual by 5%, 10%, and 15%. Table 2 shows that using peak heights with correction for stutter and differential amplification performed best in every case, predicting the point where the differences reach statistical significance. The ΔAIP method and the measurement of peak heights without correction also performed reasonably well but failed to detect significant differences for DRD5 and D4S230. Both markers had heavy stutter bands. The error rates in the “spiking” experiment are shown as diamonds in Figure 2. (The true differences ranged from ∼0% to 12.8% for reasons explained in Methods.) The differences in individual allele frequencies were predicted extremely accurately across the whole range when correction was performed (Fig. 2a). If correction was not performed (Fig. 2b), there was a clear trend for the larger differences to be underestimated. For the largest difference the error reached 4.3% and in several more instances was >3%. This applied only to the dinucleotide markers and is almost certainly attributable to stutter.
Figure 4 shows the size of the difference in allele frequencies between parents and affected offspring for an individual allele that produces statistically significant preferential transmission with α levels of 0.05 and 0.001. At least for more common alleles, these differences are likely to be large enough to be detected by the pooling method, even allowing for errors (95% of the errors were <2%). It is also clear from Figure 4 that for allele frequencies of <10%, the differences between parents and offspring that would produce a significant result become quite small and can be missed in an initial screen. This is, however, only partly a problem of low sensitivity of the pooling method because the power of the sample to detect a significant result diminishes for alleles with such a low frequency, even when using individual genotyping (see Methods). Therefore, we would miss many realistic effects (γ between 2.9 and 4) even if we perform individual genotyping when the frequency of the allele associated with disease is <10%.
Figure 4.

Allele frequency differences between parents and children for individual alleles that result in significant differences at the 0.05 and 0.001 level (bottom and top lines, respectively). The vertical axis refers to the difference in an allele frequency for a particular allele between parents and offspring, and the horizontal axis refers to the frequency of the allele in parents. These values apply for a sample of 110 trios.
In our experiments the peak heights method without correction and the ΔAIP method also performed reasonably well. However, our results suggest that they tend to underestimate some differences. This is supported by the fact that the ΔAIP method did not suggest any significant result from the genotyping of 53 markers. It is possible that none of these results would have been significant even after individual genotyping. However, we expected that approximately five markers would show differences at the P < 0.1 level just by chance, but even this level of significance was not reached by any marker. On the other hand, the tendency of these two approaches to underestimate larger differences might not be crucial as they would still detect a large difference. Therefore, the choice of any of these methods would depend on the disease under study and the size of the samples available. In the case of BP we do not expect to find genes of major effect (Berrettini 1998), so it is important to detect small differences between samples. In this situation the time spent in producing marker-specific corrections for stutter and differential amplification would be justified.
We examined >100 individuals to produce confident matrices such as the one shown in Figure 5, and even then some of the rarest alleles could not be observed. However, the size of the stutter bands for each marker appeared to have a nearly linear increase with the increasing number of repeat elements in this and the other markers analyzed (data not shown). The same observation was reported by Lipkin et al. (1998). Therefore, it should be sufficient to construct stutter bands for only a few alleles for each marker and then plot the stutter bands of unobserved alleles on the regression lines that connect them, assuming a linear increase with increasing repeat number. We have now started using only 10 samples from individuals and find this number sufficient to perform the corrections. Correction for differential amplification can also use the observations from only a few heterozygous individuals. Although this is only an approximation, small errors in producing the corrections should not bias the results significantly as even without corrections the pooling method was shown to predict a number of differences. Our method of correction (detailed in Methods) is simple and, unlike previously reported ones (Perlin et al. 1995; Barcellos et al. 1997), uses widely available software (we used Excel from the Microsoft Office package).
Figure 5.

Relationship between stutter band heights and allele sizes for marker D4S230. The sizes (vertical axis) refer to the ratio between the height of the stutter band and the height of the primary peak. The three lines correspond to stutter bands that are 1, 2, and 3 bp units shorter than the correct fragment size. Each point is estimated as the mean of 4–10 observations. Alleles 4, 5, and 7 were rare, and their stutter bands could not be plotted. The stutter bands of allele 6 appear not to fit the trend of the other alleles, but it was also rare (∼3%) and was less reliable to construct.
Based on these results, we offer the following recommendations for the use of DNA pooling with trios when used for LD mapping of common diseases. Because of the inherent small error rate in the pooling approach, more relaxed statistical criteria for following up with individual genotyping are recommended. Pooling should be used only as an initial screen to reduce the number of individual genotypings.
The image patterns can be analyzed with the ΔAIP method or, if small differences between samples are expected, with the peak heights method with correction for stutter and differential amplification as described in Methods. A very relaxed statistical criterion should be used as a cutoff point for inclusion for individual genotyping, in order not to miss positive results. This can be as high as P = 0.1, but different studies could adopt different cutoff points to suit their aims.
In addition, markers that show promising differences in a single allele should also be subjected to individual genotyping. Figure 4 can be used to define a promising difference. The biggest difference in allele frequency between parents and children for each marker (in %) can be compared with the graph, and the marker can be included for individual genotyping if it exceeds a certain cutoff point. Taking into account that 95% of the errors in our experiment are <2%, this cutoff point should probably be set at 2% below the curve regarded as significant in a particular study, for example, the P = 0.001 curve. (The graph will be different for different sample sizes.)
Tri- and tetranucleotide repeats should be preferred to dinucleotide repeats because correction for stutter will usually not be necessary. Markers with fewer alleles should be preferred, as they are less distorted by differential amplification.
The parallel use of case-control and trios samples is beneficial. When using case-control pools, the rate of false-negative results will drop because the differences in allele frequencies between patients and controls will be higher (the curves on Fig. 4 will be higher) in the presence of the same genetic effect and sample size (as shown by Risch and Teng 1998). However, more false-positive results will be generated. The use of pools of trios to replicate such results can guard against such false-positive results. One can include a marker for individual genotyping if a predicted difference is in the same direction in both pools, even if the cutoff criterion is reached only in one of the pools. Our results and those of Barcellos et al. (1997) and Shaw et al. (1998) show that analysis of pools constructed from trios will detect very small differences in individual alleles correctly. Therefore, a true result is likely to be replicated even if the effect size is smaller in the replication pool.
Individual genotyping of trios and the use of TDT should always be performed to confirm results in cases in which population stratification is likely to exist. DNA pooling with trios uses the haplotype-based haplotype relative risk (HHRR) method (Terwilliger and Ott 1992) that may not be valid in a population with substructure (Shaw et al. 1998).
Recently Risch and Teng (1998) presented theoretical evidence that DNA pooling in parent–offspring trios can be used for large-scale LD studies of complex human diseases. Our results provide practical support that the method is sufficiently accurate to be used for this purpose. This strategy has already been tested successfully by Barcellos et al. (1997) and Shaw et al. (1998). However, both teams analyzed only tetranucleotide repeat markers that, unlike dinucleotide repeats, do not show considerable PCR stutter and that are therefore easier to analyze. We have shown that the method performs well in a larger population and can use dinucleotide repeat markers that are much more abundant in the genome (Bowcock et al. 1996) and therefore likely to form the basis of genome-wide attempts to detect LD.
METHODS
Clinical Samples
The 110 probands with BP were collected through systematic screening of attenders at Lithium Clinics in Wales and the South and Midlands of England. Nine of the families responded to advertisements in the press. Only white Caucasian patients of European descent were included in the study. Ethics Committee approval for genetic linkage and association studies was obtained in all local health authorities where patients were recruited. All patients were given information and signed an informed consent for participation in these studies. Diagnosis was made on the basis of all available information that included a personal interview with the Schedule for Clinical Assessment in Neuropsychiatry (SCAN; Wing et al. 1990) and reports from the parents and hospital records. All diagnoses were reviewed by an independent clinician (N.C.), and a final consensus diagnosis according to DSM-IV (American Psychiatric Association 1994) was made. DNA was available from all 220 parents of the probands.
Preparation of Pools
Equal amounts of DNA from each patient were placed into a single tube (one pool) and from each parent into another. The DNA concentration of each sample was measured several times prior to pooling on a spectrophotometer DU640B (Beckman, USA) and gradually diluted down to 8 ng/μl. The individual samples then underwent a further round of quantitation on a fluorimeter (Fluoroscan Ascent, Life Sciences International, UK) using the 96-well-format PicoGreen method according to the manufacturer's protocol. Equal amounts of DNA were combined from the 8ng/μl dilutions. In addition, samples that did not amplify consistently in a number of previous genotypings despite adequate DNA concentration (∼5%) were excluded from the pools. When a sample was excluded, then the entire family of that individual was excluded as well and all analysis was performed only on the 110 trios included in the pools. Each pool was prepared in triplicate.
Genotyping
PCR was performed in a 12-μl volume containing 200 μm dNTPs, 0.2 μm of each primer (5′ fluorescently labeled), 1 unit of Taq DNA polymerase (Qiagen, Germany) or AmpliTaq Gold DNA polymerase (Perkin-Elmer, USA) in cases in which Qiagen Taq did not produce satisfactory amplification, and the PCR buffer supplied with the respective Taq and the appropriate Mg2+ concentrations. Genomic DNA (48 ng) was used for both pooled and individual genotyping. Cycling conditions were 4 min at 95°C, followed by 30 cycles of 30 sec at 95°C, 45 sec at the appropriate annealing temperature, and 45 sec at 72°C, followed by a final extension step of 10 min at 72°C. When AmpliTaq Gold was used, PCR consisted of 10 min at 95°C, followed by 30 cycles of 45 sec at 95°C and 45 sec at the appropriate annealing temperature. Amplifications were performed on a Perkin-Elmer 9600 thermal cycler. When pools were amplified with AmpliTaq Gold, we noticed a frequent addition of A to the PCR product that created an artifact in the image pattern. This was deleted through digestion with 0.1 μl of Klenow DNA polymerase per reaction for 2 hr at room temperature. PCR products were resolved on an ABI 373 sequencer using 6% denaturing polyacrylamide gels and analyzed by the GENESCAN and GENOTYPER software. Between 0.1 and 0.3 μl of each PCR reaction was usually needed to produce optimal image patterns for analysis of pooled genotypes (around twice the amount required for individual genotyping). We aimed to load sufficient product to place the highest allele peak in the upper half of the resolution range of the sequencer. Both over- and underloading can produce images that are unsuitable for accurate analysis.
Calculation of ΔAIP
The method was described in detail in our previous paper (Daniels et al. 1998). Briefly, the consensus images from the two pools were overlaid in GENOTYPER that scales the traces so that the heights of the largest peaks are equal (Fig. 3). Using the software program Debabelizer (Equilibrium), we measured the total area that is not shared by the two images (Dif) and the area common to both images (Com). The ΔAIP (allele image pattern difference) was estimated as ΔAIP = Dif/(Dif + Com) and provides a measure of the difference between the image patterns of the two pools. It does not directly relate to significance level. This depends on the number of alleles and size of the sample. We have used a simulation program designed by P. Holmans (Daniels et al. 1998) that takes account of these factors, as well as the approximate allele frequency distribution and provides a “simulated P value” for the presence of differences between the two pools. When performing pooling with trios, we are unable to observe the “control” population AIP. Therefore the ΔAIP method was adapted to include subtraction of the probands from parents' data.
Prediction of Allele Frequencies Using Peak Heights and Subtraction
GENOTYPER assigns values for the height of each peak of the image pattern. We used these values to estimate the approximate allele frequencies, assuming that peak height is directly proportional to the amount of DNA for that allele. Allele frequencies are thus estimated from the peak height for each allele divided by the sum of the peak heights for all alleles. As each pool was run in triplicate, we estimated the mean of the three values. Thus, we obtained the predicted allele frequencies for parents (pp) and children (pc). The allele frequency of each allele in the nontransmitted control population can be estimated as pcon = (2pp − pc). As we have no knowledge of genotypes of either population, we could not perform a TDT-based test but could perform a test based on the allele frequencies in the two populations (affected children and nontransmitted controls). This is equivalent to the HHRR method. As we were testing multiallelic markers, and some alleles were rare, we used the program CLUMP (Sham and Curtis 1995a). This assesses the significance of the result using a Monte Carlo approach, by performing repeated simulations to generate tables having the same marginal totals as the ones under consideration and counting the number of times that a χ2 value associated with the real table is achieved by the randomly simulated data. For each marker we ran 1000 simulations and estimated the nominal P-value. These estimates were performed on both uncorrected and corrected pools (see next paragraph).
Correction of Peak Heights for Stutter and Differential Amplification
The stutter patterns of a number of alleles of individually genotyped subjects were recorded by measuring the peak heights produced by GENOTYPER. The mean values of several individuals per allele were estimated (we tried to measure at least four). We measured either homozygous individuals or individuals whose alleles were so far apart that their stutter bands did not overlap. Clearly, it can be impossible to obtain values for some rare alleles, and such values are deduced as the mean of the two adjacent alleles. Different markers had different stutter patterns and produced between one and four distinct stutter bands. The heights of the bands increased progressively with the increasing number of repeat units. This increase appeared linear in the markers we tested. A typical relationship between the heights of the stutter bands of a dinucleotide repeat is shown in Figure 5.
The correction was performed progressively starting from the longest available allele whose height was left as observed. The height of the next-longest allele was reduced by the estimated size of the first stutter band of the longest allele. The height of the third-longest allele was reduced by the size of the second stutter band of the longest allele and the first stutter band of the (already corrected) second-longest allele. The process is repeated down to the shortest allele and can incorporate any number of stutter bands.
Correction for differential amplification was made by recording the relative heights of alleles of several heterozygous individuals across the whole range of alleles. This allows the calculation of the average reduction in peak height per repeat unit. The shortest allele (already corrected for stutter) is left unchanged, and larger alleles are multiplied by this increasingly higher correction factor that reached between 1.3 and 1.7 for the longest alleles of different markers. The tetranucleotide repeat D4S2375 showed no clear differential amplification, and this correction was not performed for that marker.
The corrected frequencies were normalized so that all allele frequencies summed up to 100%.
Estimation of Statistically Significant Differences
If a particular allele of a given marker is preferentially transmitted to affected offspring, the frequency of that allele will be higher in affected offspring than in parents. We estimated the size of this difference when the preferential transmission (measured by TDT) reaches different levels of statistical significance. To do this we made the simplifying assumption that the parental genotypes were in Hardy-Weinberg equilibrium (HWE). We denote the frequency of the examined parental allele as pp and the frequency of all the other alleles as qp = (1 - pp). The expected number of homozygous parents is thus Npp2, and the number of heterozygous parents is 2Npp(1 − pp), where N is the number of parents in the sample. The TDT statistic is given as
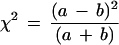 |
in which a and b are the number of transmitted and nontransmitted alleles from heterozygous parents. Clearly, b = 2Npp(1 − pp) − a. Therefore,
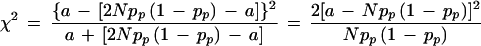 |
it follows that
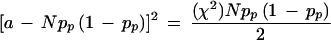 |
which simplifies to
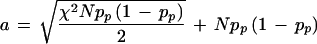 |
and to
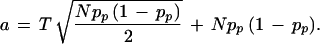 |
Thus, we can estimate the number of transmitted alleles (a) that are required to produce a given test statistic (T) on the basis of the number of parents (n) and the frequency of that allele in the parents (pp). Now, we can express the allele frequency in the affected children (pc) in that situation. It can be estimaed as the sum of the parental alleles transmitted from parents homozygous for the allele of interest (Npp2) plus the alleles transmitted from heterozygous parents (a) divided by the number of parents (N).
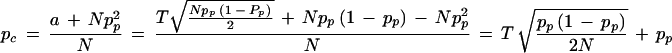 |
Hence,
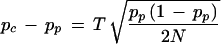 |
and
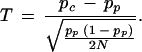 |
This formula is the same as the one proposed by Risch and Teng (1998) under HWE assumption, although it was derived via a different route of considerations:
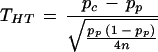 |
We should also point out that their n is the number of affected probands, whereas our N is the number of parents; so N = 2n.
Therefore, it is possible to estimate the statistical significance of the difference in any single allele by knowing only the number of parents (N), and the allele frequencies of the parents (pp) and of the affected children (pc). We have estimated the TDT statistic for T values of 1.96 and 3.29 (corresponding to χ2 values of 3.84 and 10.83 and to P-values of 0.05 and 0.001). These calculations were performed for a range of pp from 5% to 50% and a sample size of 110 families. They are presented in Figure 4 as two curves corresponding to P values of 0.05 and 0.001. The vertical axis shows the size of the difference between the frequency of that particular allele in parents and children for a sample of 110 families expressed as a percent of the total allele frequency. The horizontal axis refers to the frequency of that allele in parents.
Statistical Analysis of Individual Genotypings
For statistical analysis of transmission/disequilibrium we used the likelihood-based extended TDT (ETDT), which is designed for multiallele markers (Sham and Curtis 1995b). The program gives a multiallele P value that does not require correction. Power calculations for individual genotyping were estimated with the formulas given in the paper of Camp (1997) taking into account the corrections proposed by the same author in a later paper (Camp 1999). As an example we use the multiplicative mode of inheritance (heterozygous relative risk = γ; homozygous relative risk = γ2) in a sample of 110 families and a significance level of 0.001. For allele frequencies in the range of 0.5–0.1, our sample has 80% power to detect a γ of 2.2 to 2.9, which is a realistic value that we would like to detect. This increases, however, to 4 at an allele frequency of 0.05; that is, we require a higher genotype relative risk to have the same power when the frequency of the allele is lower.
Estimation of the Error Rate in the Pooling Approach
We have considered only alleles with >2.5% frequency in parents based on individual genotyping. The error is expressed as percent of the total for all alleles. It is estimated by comparing the size of the difference between the frequency of a single allele in parents and offspring as predicted from the pools and the difference as calculated from the true genotyping results. If the two differences are in the same direction, they are subtracted (the pool predicted the direction correctly), and if they are in the opposite direction, they are added.
Spiking Experiment
We created artificial pools by adding more DNA from one individual into a pool (the parents' pool) in amounts that would lead to an increase in the DNA concentration of that individual by 5%, 10%, and 15%. This does not automatically lead to an increase in the corresponding allele frequencies by 5%, 10%, and 15% if the individual is homozygous and by 2.5%, 5%, and 7.5% for two alleles if the individual is heterozygous. This would be the case if the added alleles were absent in the original pool. However, if they were present, a correction needs to be introduced to take account of the original allele frequency of the introduced alleles. For example, if the individual is heterozygous and one of his alleles has a frequency of 50% in the original pool, the frequency of that allele will not change, no matter how much DNA from that individual we add. The resulting allele frequency (p1) is obtained by the formula p1 = a1 + p(1 − a), where p is the original allele frequency and a is the proportion of DNA from the individual the pool was enriched with. If the individual is homozygous, a1 = a; if he is heterozygous, then a1 = a/2.
Acknowledgments
G.K. is a Wellcome Trust Advanced Fellow, N.C. is a Wellcome Trust Senior Fellow in Clinical Sciences, and M.J.O. and N.W. are supported by the Medical Research Council. This work was funded by grants 045856 and 056535 of the Wellcome Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kirov@cardiff.ac.uk; FAX 44 1222 746554.
REFERENCES
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington, DC.: American Psychiatric Association; 1994. [Google Scholar]
- Asherson P, Mant R, Williams N, Cardno A, Jones L, Murphy K, Collier DA, Nanko S, Craddock N, Morris S, et al. A study of chromosome 4p markers and dopamine D5 receptor gene in schizophrenia and bipolar disorder. Mol Psychiatry. 1998;3:310–320. doi: 10.1038/sj.mp.4000399. [DOI] [PubMed] [Google Scholar]
- Barcellos LF, Klitz W, Field LL, Tobias R, Bowcock AM, Wilson R, Nelson MP, Nagatomi J, Thomson G. Association mapping of disease loci, by use of a pooled DNA genomic screen. Am J Hum Genet. 1997;61:734–747. doi: 10.1086/515512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrettini W. Progress and pitfalls: Bipolar molecular linkage studies. J Affect Disord. 1998;50:287–297. doi: 10.1016/s0165-0327(98)00020-2. [DOI] [PubMed] [Google Scholar]
- Blackwood DHR, He L, Morris SW, McLean A, Whitton C, Thomson M, Walker MT, Woodburn K, Sharp CM, Wright AF, Shibasaki Y, St Clair DM, Porteous DJ, Muir WL. A locus for bipolar affective disorder on chromosome 4p. Nat Genet. 1996;12:427–430. doi: 10.1038/ng0496-427. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Chipperfield MA, Ceverha P, Yetman E, Phung A. Report of the DNA committee. In: Cuticchia AJ, Chipperfield MA, Foster PA, editors. Human Gene Mapping 1995: A compendium. Baltimore, MD: John Hopkins University Press; 1996. pp. 1454–1468. [Google Scholar]
- Camp NJ. Genomewide transmission/disequilibrium testing—Consideration of the genotypic relative risks at disease loci. Am J Hum Genet. 1997;61:1424–1430. doi: 10.1086/301648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Genomewide transmission/disequilibrium testing: A correction. Am J Hum Genet. 1999;64:1485–1487. doi: 10.1086/302387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalayanaraman N, Nemesh J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Craddock N, Khodel V, Van Eerdewegh P, Reich T. Mathematical limits of multilocus models: The genetic transmission of Bipolar disorder. Am J Hum Genet. 1995;57:690–702. [PMC free article] [PubMed] [Google Scholar]
- Daniels J, Holmans P, Williams N, Turic D, McGuffin P, Plomin R, Owen MJ. A simple method for analysing microsatellite allele image patterns generated from DNA pools and its application to allelic association studies. Am J Hum Genet. 1998;62:1189–1197. doi: 10.1086/301816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PJ, Turic D, Williams NM, McGuffin P, Asherson P, Ball D, Craig I, Eley T, Hill L, Chorney K, Chorney MJ, et al. DNA pooling identifies QTLs on chromosome 4 for general cognitive ability in children. Hum Mol Genet. 1999;8:915–922. doi: 10.1093/hmg/8.5.915. [DOI] [PubMed] [Google Scholar]
- Ginns EI, St Jean P, Philibert RA, Galdzicka M, Damschroder-Williams P, Thiel B, Long RT, Ingraham LJ, Dalwaldi H, Murray MA, et al. A genome-wide search for chromosomal loci linked to mental health wellness in relatives at high risk for bipolar affective disorder among the Old Order Amish. Proc Natl Acad Sci. 1998;22:15531–15536. doi: 10.1073/pnas.95.26.15531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, MacLean CJ, Ma Y, O'Neill FA, Walsh D, Straub RE. Marker-to-marker linkage disequilibrium on chromosomes 5q, 6p, and 8p in Irish high-density schizophrenia pedigrees. Am J Med Genet. 1999;88:29–33. doi: 10.1002/(sici)1096-8628(19990205)88:1<29::aid-ajmg5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- LeDuc C, Lichter MJ, Parry P. Batched analysis of genotypes. PCR Methods Applic. 1995;4:331–336. doi: 10.1101/gr.4.6.331. [DOI] [PubMed] [Google Scholar]
- Lipkin E, Mosig MO, Darvasi A, Ezra E, Shalom A, Friedmann A, Soller M. Quantitative trait locus mapping in dairy cattle by means of selective milk DNA pooling using dinucleotide microsatellite markers: Analysis of milk protein percentage. Genetics. 1988;149:1557–1567. doi: 10.1093/genetics/149.3.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz RJ, Fodor SPA, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet (Suppl.) 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- McGuffin P, Owen MJ, O'Donovan MC, Thapar A, Gottesman II. Seminars in psychiatric genetics. London, UK: Royal College of Psychaitrists; 1994. Affective disorders (chapter 6) pp. 110–127. [Google Scholar]
- Owen MJ, McGuffin P. Association and linkage: Complementary strategies for complex disorders. J Med Genet. 1993;30:638–639. doi: 10.1136/jmg.30.8.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek P, Sajantila A, Syvänen A-C. Determination of allele frequencies at loci with length polymorphism by quantitative analysis of DNA amplified from pooled samples. PCR Methods Applic. 1993;2:313–317. doi: 10.1101/gr.2.4.313. [DOI] [PubMed] [Google Scholar]
- Perlin MW, Lancia G, Ng S-K. Toward fully automated genotyping: Genotyping microsatellite markers by deconvolution. Am J Hum Genet. 1995;57:1199–1210. [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of gentic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Risch N, Teng J. The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases. I. DNA pooling. Genome Res. 1998;8:1273–1288. doi: 10.1101/gr.8.12.1273. [DOI] [PubMed] [Google Scholar]
- Sham PC, Curtis D. Monte Carlo test for association between disease and alleles at highly polymorphic loci. Ann Hum Genet. 1995a;59:97–105. doi: 10.1111/j.1469-1809.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- ————— An extended transmission disequilibrium test (TDT) for multiallele marker loci. Ann Hum Genet. 1995b;59:323–336. doi: 10.1111/j.1469-1809.1995.tb00751.x. [DOI] [PubMed] [Google Scholar]
- Shaw SH, Carrasquillo MM, Kashuk C, Puffenberger EG, Chakravarti A. Allele frequency distributions in pooled DNA samples: Applications to mapping complex disease genes. Genome Res. 1998;8:111–123. doi: 10.1101/gr.8.2.111. [DOI] [PubMed] [Google Scholar]
- Sheffield VC, Pierpont ME, Nishimura D, Beck JS, Burns TL, Berg MA, Stone EM, Patil SR, Lauer RM. Identification of a complex congenital heart defect susceptibility locus by using DNA pooling and shared segment analysis. Hum Mol Genet. 1997;6:117–121. doi: 10.1093/hmg/6.1.117. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J. A haplotype-based “haplotype relative risk” approach to detecting allelic associations. Hum Hered. 1992;42:337–346. doi: 10.1159/000154096. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Faraone SV. The genetics of mood disorders. Baltimore, MD: John Hopkins University Press; 1990. [Google Scholar]
- Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- Wing JK, Babor T, Brugha T, Burke J, Cooper JE, Giel R, Jablenski A, Regier D, Sartorius N. Schedules for clinical assessment in neuropsychiatry. Arch Gen Psychiatry. 1990;47:137–144. doi: 10.1001/archpsyc.1990.01810180089012. [DOI] [PubMed] [Google Scholar]