

Abstract
Genetic modifiers make an important contribution to neurological disease phenotypes. Significant progress has been made by studying genetic modifiers in model organisms. The ability to study complex genetic interactions in model systems contributes to our understanding of the genetic factors that influence neurological disease. This will lead to the development of novel therapeutic strategies and personalized treatment based on genetic risk.
Introduction
Genetic modifiers play a significant role in influencing the clinical severity of neurological disease. Modifier loci that segregate independently from the primary mutation can influence penetrance, age of onset, progression or severity of disease. Identification of genetic modifiers is important for understanding the pathophysiology of inherited disease and may suggest genes and pathways that contribute to complex disease. Modifier loci have been mapped for several neurological diseases in human and mouse, including tremor, dystonia, epilepsy, motoneuron degeneration, and Huntington’s disease [1–14]. Isolating modifier genes in humans is very challenging; however, genetic screens in model organisms can facilitate modifier identification and suggest candidate genes for follow-up study in human patients.
Mouse Models
Due to the availability of rich genomic resources and numerous inbred strains, mouse models are a tractable mammalian system for isolating genetic modifiers. Further, genetic engineering of mouse mutants enables the creation of sophisticated models that can recapitulate the human disease. A forward genetics approach can be used to identify genetic modifiers by systematically crossing a mutant onto different inbred strain backgrounds and comparing the phenotype. Modifiers can act to improve or exacerbate the mutant phenotype (Figure 1). The responsible genes can then be identified by conventional positional cloning or quantitative trait loci (QTL) mapping.
Figure 1.
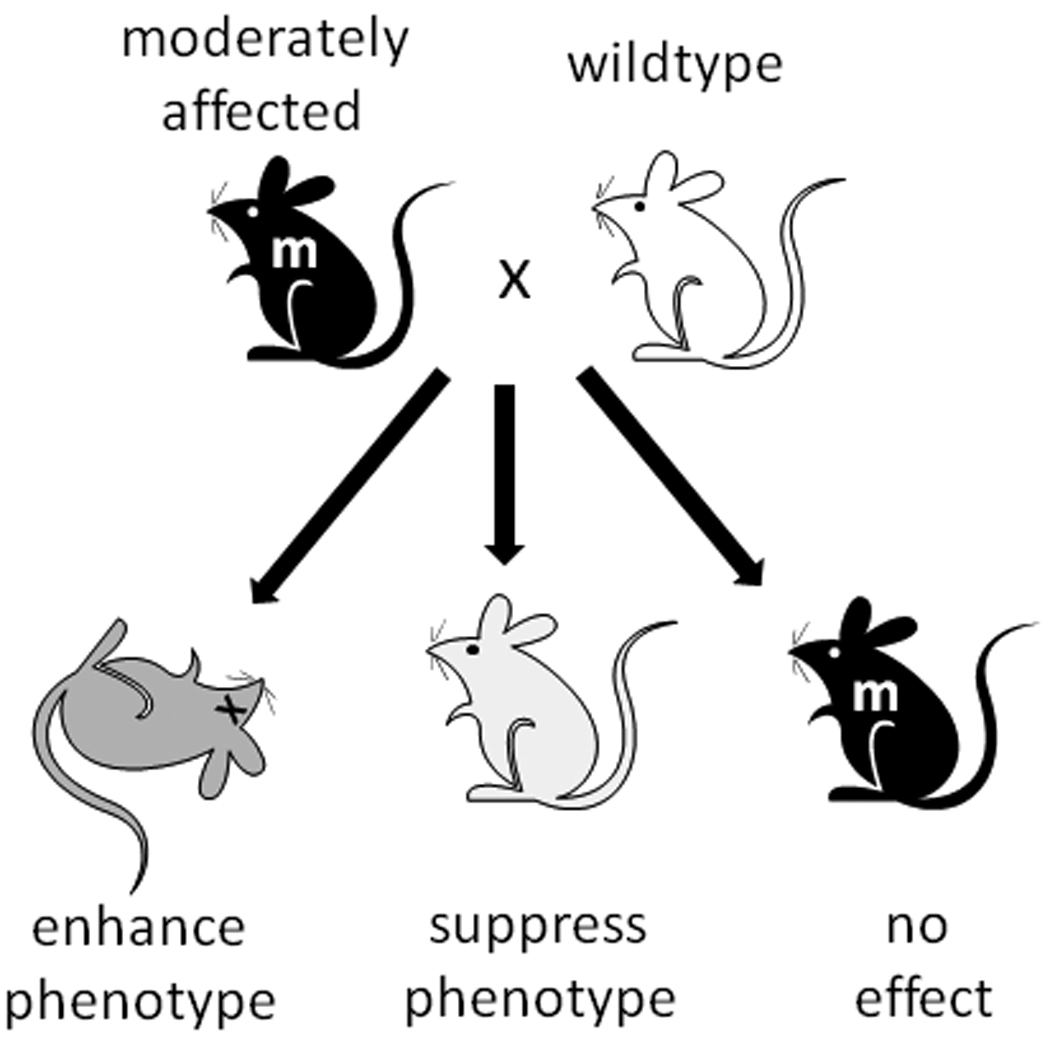
Strain-dependent variation of phenotype in mouse models due to genetic modifiers. To search for modifier effects, a mouse mutant (m; black) can be crossed to a wildtype mouse from a different inbred strain background (white). Modifiers can act as enhancers that exacerbate the phenotype or suppressors that abrogate the phenotype. Alternatively, an inbred strain that does not carry modifier variants will have no effect on the phenotype.
Scnm1
One of the first neurological modifier genes identified was Scnm1 (sodium channel modifier 1), a single gene modifier of the mouse neurological mutant, medJ. The Scn8amedJ mutation is a deletion in a splice donor site that results in partial exon skipping [15]. On the C3H inbred strain background, medJ/medJ mice exhibit a progressive movement disorder with dystonia and ataxia, and live a normal lifespan. On the susceptible C57BL/6 strain background, medJ/medJ mice exhibit a severe phenotype, with progressive paralysis and lethality by one month of age [13;16]. The difference in phenotype severity correlates with the amount of correctly spliced transcript, with 10% correctly spliced in C3H and only 5% in B6. The dramatic effect on the Scn8amedJ phenotype was mapped to a single gene, Scnm1, by positional cloning in an F2 intercross [3;13]. Sequencing of Scnm1 in the 2 strains revealed that the severely affected C57BL/6 strain has a mutation that results in a premature stop codon. Scnm1 functions as an auxiliary spliceosomal protein and facilitates splicing of non-consensus splice sites [14;17;18]. The identification of Scnm1 as a disease modifier highlighted splicing proteins as an important class of potential modifiers. Approximately 10% of human disease-causing mutations are predicted to affect pre-mRNA splicing; thus, variation in genes involved in splicing regulation may play a significant role in modifying disease [19;20].
Epilepsy
A similar approach has also been applied to study epilepsy. Although most epilepsy exhibits complex inheritance, modifiers of rare monogenic epilepsy are likely to contribute to susceptibility in disease with more complex inheritance. Mutations in the voltage-gated sodium channels SCN1A and SCN2A are the most frequent cause of monogenic epilepsy. Family members carrying the same mutation exhibit a high degree of variable expressivity and penetrance, suggesting a role for genetic modifiers. Similarly, in mouse epilepsy models with sodium channel mutations, phenotype severity varies depending on the inbred strain background. Scn1a+/− knock-out mice are a model for Dravet syndrome, a severe infant-onset epileptic encephalopathy. Heterozygous Scn1a+/− mice exhibit a dramatic difference in phenotype severity on different inbred strain backgrounds. On the 129 inbred strain background over 90% of mice survive to 3 months of age, while on the C57BL/6 strain background only 20% survive to 3 months [21]. The Scn2aQ54 transgenic epilepsy model also shows strain-dependent differences in phenotype onset and severity. On the resistant C57BL/6 background fewer than 15% of mice exhibit spontaneous seizures during the first month of life, while on a (C57BL/6 × SJL)F1 background over 80% have seizure onset by one month of age. This dramatic difference in the age of onset is reflected in survival, with fewer than 25% of Scn2aQ54 on the (C57BL/6 × SJL)F1 background surviving to 6 months of age, compared to over 75% on the C57BL/6 background [1].
We exploited the strain background difference to isolate genetic modifiers of epilepsy in the Scn2aQ54 transgenic mouse model. QTL mapping identified two modifier loci that are responsible for the strain difference in Scn2aQ54 mice: Moe1 (modifier of epilepsy 1) on chromosome 11 and Moe2 on chromosome 19. Standard QTL mapping does not have sufficient resolution for gene identification. Several different strategies can be used for fine mapping to higher resolution, including selective phenotyping, recombinant progeny testing, recombinant inbred segregation testing, haplotype analysis with in silico mapping, advance intercross lines, genetically heterogenous stocks and interval-specific congenic strains (reviewed in [22–25]). Generation of interval-specific congenic strains produces genetically identical individuals carrying a small chromosome interval from one strain (donor) at the modifier locus on the other strain background (recipient). The QTL interval can be dissected by generating several lines with varying donor-derived segments that are then used to determine which segment confers the modified phenotype (Figure 2). We used an interval-specific congenic approach to fine map the Moe2 locus to a 5 Mb region on chromosome 19 and identified the voltage-gated potassium channel Kcnv2 as a strong candidate modifier [2].
Figure 2.

Fine mapping of modifier QTL using interval-specific congenic strains. The QTL interval from the severely affected strain (donor; grey) is introgressed on the resistant strain background (recipient; black) by selective genotyping and breeding for multiple generations (≥5). Several lines are generated that carry varying donor-derived segments in the QTL interval. Each interval-specific congenic line is tested to determine the effect on the mutant phenotype. In this example, the modifier is localized to the region of overlap between the two lines that confer the modified phenotype.
Huntington’s Disease
Even in the highly penetrant, monogenic Huntington’s disease (HD), modifier genes are believed to influence disease expression [26;27]. Although length of the CAG repeat accounts for approximately 70% of the variance in the age of onset, the remaining variance exhibits a high degree of heritability, suggesting a role for genetic modifiers [26;28;29]. Several modifier loci influencing age of onset have been mapped in human patients [5;8;26]. Further evidence for genetic modifiers influencing HD come from mouse models which exhibit phenotype variation depending on the inbred strain background [9;30]. The HdhQ111 knock-in model shows variable phenotype severity on the C57BL/6, FVB and 129 inbred strain backgrounds by several measures, including intergenerational repeat instability, somatic repeat instability, nuclear accumulation of full-length mutant huntingtin and intranuclear N-terminal huntingtin inclusions [9]. Similarly, using the YAC128 transgenic model, variable phenotype severity was demonstrated on the C57BL/6, 129 and FVB strain backgrounds [30].
Strain background effects have also been reported in other mouse models of neurological disorders, including Parkinson’s, Alzheimer’s, amyotrophic lateral sclerosis (ALS), Charcot-Marie-Tooth, Rett Syndrome and spinocerebellar ataxia [31–37].
Lower-Model Organisms
Due to the late onset of age-related neurodegeneration, most studies have used lower model organisms to screen for potential genetic modifiers. Models with shorter generation times like yeast and C.elegans expedite screening of late-onset phenotypes like HD, Parkinson’s, ALS and Alzheimer’s. Transgenic models recapitulating a feature of the human disease process have been used for modifier screens. Protein misfolding models in C.elegans have been used for genome-wide screens to identify modifiers of α-synuclein inclusion formation, tau-induced pathology, polyglutamine aggregation, and mutant SOD1 aggregation [38–41]. Yeast models have been used to screen for modifiers of toxicity due to α-synuclein and huntingtin misfolding [42–45]. These studies have been reviewed in detail elsewhere [27;46]. Drosophila models have been used to screen for modifiers of presenilin function, polyglutamine disease and frontotemporal dementia (FTD) [47–49]. A recent screen for modifiers of non-coding polyglutamine repeat instability in Drosophila found novel modifiers that independently influence different aspects of repeat instability and identified a protein with homology to CNOT2 as a modifier of germ-line repeat expansion [49]. Ahmad and colleagues developed a Drosophila model of FTD3 and identified serpin5 as a modifier in a genome-wide screen [47]. Genes identified in these lower-model organism screens suggest proteins and pathways that may influence the disease process, which can be subsequently tested in mammalian systems.
Candidate Gene Approach
Reverse genetic, hypothesis-driven approaches are another way to identify potential genetic modifiers. Hypotheses can be generated by screens in lower model organisms as discussed above or can be based on our current knowledge of disease mechanisms and pathways. The discovery of VEGF as a modifier of ALS came from such a reverse-genetics approach [50]. Hypotheses can be tested by crossing mouse mutants together. Between the wide selection of spontaneous and engineered mouse mutants already available (http://www.findmice.org) and the International Knock-out Mouse Consortium, which is working to generate null alleles of all protein coding genes in the mouse (http://www.knockoutmouse.org), this is possible in most cases. For example, based on the hypothesis that the potassium M-current may influence epilepsy susceptibility, we crossed our Scn2aQ54 epilepsy model with a subclinical Kcnq2 mutant allele. The Scn2aQ54;Kcnq2V182M/+ double mutants exhibited a dramatic exacerbation, changing the phenotype from a mild, late-onset syndrome to severe, juvenile-onset epilepsy with premature lethality [51]. Conversely, a genetic interaction may suppress the primary phenotype. Glasscock et al [52] hypothesized that absence epilepsy due to P/Q-type calcium-channel dysfunction in tottering mice could be improved by increasing excitability at axon terminals. To test this hypothesis, they crossed the tottering mutant with mice deficient in the voltage-gated potassium channel Kcna1 (Kv1.1). The absence epilepsy phenotype was masked in the double mutant mice, supporting their hypothesis that some ion channel variants can interact to suppress hyperexcitability [52].
Sensitized Screens for Modifiers
Sensitized N-ethyl-N-nitrosourea (ENU) mutagenesis screens offer another unbiased, forward-genetic approach for identifying potential modifier genes. Sensitized screens have been widely applied in Drosophila and C. elegans to identify functional interaction between genes and elucidate physiologic pathways. This can also be applied in mice and provides a complementary approach to QTL analysis, which is based on existing inbred strain differences. Mutagenesis increases the pool of potential modifier genes by saturating the genome with ENU-induced point mutations. Sensitized ENU mutagenesis screens in mice have been used successfully to isolate modifiers of Waardenburg syndrome and thrombocytopenia [53;54]. We are currently using a sensitized ENU mutagenesis screen to identify suppressors of the severely affected Scn2aQ54;Kcnq2V182M/+ double mutants described above [51].
FUTURE DIRECTIONS AND CONCLUSIONS
Studies of neurological disease in model organisms have demonstrated the significant influence of genetic modifiers on neurological phenotype expression. The ability to study more complex multigenetic interactions in model systems may increase our understanding of the balance of genetic interactions that contribute to complex neurological disease in humans. Identification of modifier genes and analysis of the underlying pathophysiology will help to elucidate the molecular pathways involved in neurological diseases and suggest novel targets for therapeutic intervention. This knowledge may also improve the utility of molecular diagnosis and enable personalized treatment based on genetic risk factors.
Acknowledgements
Thank you to Nicole Hawkins and Benjamin Jorge for critical reading of the manuscript. Related work in my lab is supported by NIH grant NS-053792.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Bergren SK, Chen S, Galecki A, Kearney JA. Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm.Genome. 2005;16:683–690. doi: 10.1007/s00335-005-0049-4. [DOI] [PubMed] [Google Scholar]
- 2. Bergren SK, Rutter ED, Kearney JA. Fine mapping of an epilepsy modifier gene on mouse Chromosome 19. Mamm.Genome. 2009;20:359–366. doi: 10.1007/s00335-009-9193-6.. This study reports the fine mapping and candidate gene analysis of a modifier locus in a mouse model of spontaneous epilepsy. A likely modifier candidate was nominated based on sequence analysis and gene function.
- 3.Buchner DA, Trudeau M, George AL, Jr, Sprunger LK, Meisler MH. High-resolution mapping of the sodium channel modifier Scnm1 on mouse chromosome 3 and identification of a 1.3-kb recombination hot spot. Genomics. 2003;82:452–459. doi: 10.1016/s0888-7543(03)00152-6. [DOI] [PubMed] [Google Scholar]
- 4.Buchner DA, Trudeau M, Meisler MH. SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science. 2003;301:967–969. doi: 10.1126/science.1086187. [DOI] [PubMed] [Google Scholar]
- 5.Djousse L, Knowlton B, Hayden MR, Almqvist EW, Brinkman RR, Ross CA, Margolis RL, Rosenblatt A, Durr A, Dode C, et al. Evidence for a modifier of onset age in Huntington disease linked to the HD gene in 4p16. Neurogenetics. 2004;5:109–114. doi: 10.1007/s10048-004-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum.Mol.Genet. 2006;15:1187–1194. doi: 10.1093/hmg/ddl034. [DOI] [PubMed] [Google Scholar]
- 7.Kunst CB, Messer L, Gordon J, Haines J, Patterson D. Genetic mapping of a mouse modifier gene that can prevent ALS onset. Genomics. 2000;70:181–189. doi: 10.1006/geno.2000.6379. [DOI] [PubMed] [Google Scholar]
- 8.Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dode C, Morrison PJ, Suchowersky O, Ross CA, Margolis RL, et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am.J.Hum.Genet. 2003;73:682–687. doi: 10.1086/378133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloret A, Dragileva E, Teed A, Espinola J, Fossale E, Gillis T, Lopez E, Myers RH, MacDonald ME, Wheeler VC. Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington's disease knock-in mice. Hum.Mol.Genet. 2006;15:2015–2024. doi: 10.1093/hmg/ddl125. [DOI] [PubMed] [Google Scholar]
- 10.Metzger S, Bauer P, Tomiuk J, Laccone F, Didonato S, Gellera C, Mariotti C, Lange HW, Weirich-Schwaiger H, Wenning GK, et al. Genetic analysis of candidate genes modifying the age-at-onset in Huntington's disease. Hum.Genet. 2006;120:285–292. doi: 10.1007/s00439-006-0221-2. [DOI] [PubMed] [Google Scholar]
- 11.Metzger S, Bauer P, Tomiuk J, Laccone F, Didonato S, Gellera C, Soliveri P, Lange HW, Weirich-Schwaiger H, Wenning GK, et al. The S18Y polymorphism in the UCHL1 gene is a genetic modifier in Huntington's disease. Neurogenetics. 2006;7:27–30. doi: 10.1007/s10048-005-0023-z. [DOI] [PubMed] [Google Scholar]
- 12.Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, Soehn AS, Propping P, Freudenberg-Hua Y, Freudenberg J, Tong L, et al. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum.Mol.Genet. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- 13.Sprunger LK, Escayg A, Tallaksen-Greene S, Albin RL, Meisler MH. Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Hum.Mol.Genet. 1999;8:471–479. doi: 10.1093/hmg/8.3.471. [DOI] [PubMed] [Google Scholar]
- 14.Ulbrich M, Schmidt VC, Ronsiek M, Mussmann A, Bartsch JW, Augustin M, Jockusch H, Schmitt-John T. Genetic modifiers that aggravate the neurological phenotype of the wobbler mouse. Neuroreport. 2002;13:535–539. doi: 10.1097/00001756-200203250-00035. [DOI] [PubMed] [Google Scholar]
- 15.Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J.Biol.Chem. 1996;271:17576–17581. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- 16.Kearney JA, Buchner DA, De HG, Adamska M, Levin SI, Furay AR, Albin RL, Jones JM, Montal M, Stevens MJ, et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6) Hum.Mol.Genet. 2002;11:2765–2775. doi: 10.1093/hmg/11.22.2765. [DOI] [PubMed] [Google Scholar]
- 17. Howell VM, Jones JM, Bergren SK, Li L, Billi AC, Avenarius MR, Meisler MH. Evidence for a direct role of the disease modifier SCNM1 in splicing. Hum.Mol.Genet. 2007;16:2506–2516. doi: 10.1093/hmg/ddm206.. In this follow-up study, the authors confirm that Scnm1 is a component of the spliceosome. Scnm1 was identified as a modifier of the mouse neurological mutant Scn8amedJ and was shown to be required for efficient splicing of a mutated splice site.
- 18.Howell VM, De HG, Bergren S, Jones JM, Culiat CT, Michaud EJ, Frankel WN, Meisler MH. A targeted deleterious allele of the splicing factor SCNM1 in the mouse. Genetics. 2008;180:1419–1427. doi: 10.1534/genetics.108.094227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nissim-Rafinia M, Kerem B. The splicing machinery is a genetic modifier of disease severity. Trends Genet. 2005;21:480–483. doi: 10.1016/j.tig.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754.. This paper reports a new mouse model of Dravet syndrome, a severe epileptic encephalopathy. The authors describe a striking variation in phenotype depending on the genetic strain background. This is reminiscent of the variable expressivity seen in patients.
- 22.Darvasi A. Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet. 1998;18:19–24. doi: 10.1038/ng0198-19. [DOI] [PubMed] [Google Scholar]
- 23.Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev.Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- 24.McPeek MS. From mouse to human: fine mapping of quantitative trait loci in a model organism. Proc Natl Acad Sci U S A. 2000;97:12389–12390. doi: 10.1073/pnas.240463597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shalom A, Darvasi A. Experimental designs for QTL fine mapping in rodents. Methods Mol.Biol. 2002;195:199–223. doi: 10.1385/1-59259-176-0:199. [DOI] [PubMed] [Google Scholar]
- 26. Gusella JF, MacDonald ME. Huntington's disease: the case for genetic modifiers. Genome Med. 2009;1:80. doi: 10.1186/gm80.. This review details the evidence from humans and model systems that supports a role for genetic modifiers in Huntington's Disease.
- 27.Rubinsztein DC. Functional genomics approaches to neurodegenerative diseases. Mamm.Genome. 2008;19:587–590. doi: 10.1007/s00335-008-9130-0. [DOI] [PubMed] [Google Scholar]
- 28.Djousse L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, Margolis R, Rosenblatt A, Durr A, Dode C, et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am.J.Med Genet A. 2003;119A:279–282. doi: 10.1002/ajmg.a.20190. [DOI] [PubMed] [Google Scholar]
- 29.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, Graham RK, Leavitt BR, Hayden MR. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol.Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 32. Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis.Model.Mech. 2009;2:359–373. doi: 10.1242/dmm.002527.. This paper reports a new mouse model of a Charcot-Marie-Tooth type 2D. The authors describe phenotype variation depending on the genetic strain background.
- 33.Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, et al. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum.Mol.Genet. 1997;6:1951–1959. doi: 10.1093/hmg/6.11.1951. [DOI] [PubMed] [Google Scholar]
- 34.Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J.Neurol.Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Krezowski J, Knudson D, Ebeling C, Pitstick R, Giri RK, Schenk D, Westaway D, Younkin L, Younkin SG, Ashe KH, et al. Identification of loci determining susceptibility to the lethal effects of amyloid precursor protein transgene overexpression. Hum.Mol.Genet. 2004;13:1989–1997. doi: 10.1093/hmg/ddh210. [DOI] [PubMed] [Google Scholar]
- 36.Lorenzetti D, Watase K, Xu B, Matzuk MM, Orr HT, Zoghbi HY. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus. Hum.Mol.Genet. 2000;9:779–785. doi: 10.1093/hmg/9.5.779. [DOI] [PubMed] [Google Scholar]
- 37.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum.Mol.Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- 39.Nollen EA, Garcia SM, van HG, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS.Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, III, et al. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009;106:1392–1397. doi: 10.1073/pnas.0813045106.. This study describes strain-dependent phenotype variation in the SOD1-YFP transgenic model of ALS.
- 42.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 45. Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet. 2009;41:316–323. doi: 10.1038/ng.337.. This study uses an integrative approach, combining data from genetic screens and mRNA expression profiling in yeast to define “major response pathways” involved in α-synuclein toxicity.
- 46. van Ham TJ, Breitling R, Swertz MA, Nollen EA. Neurodegenerative diseases: Lessons from genome-wide screens in small model organisms. EMBO Mol.Med. 2009;1:360–370. doi: 10.1002/emmm.200900051.. This review details the results of genome-wide screens for modifiers of neurodegeneration in lower-model organisms including yeast, C.elegans and Drosophila.
- 47. Ahmad ST, Sweeney ST, Lee JA, Sweeney NT, Gao FB. Genetic screen identifies serpin5 as a regulator of the toll pathway and CHMP2B toxicity associated with frontotemporal dementia. Proc Natl Acad Sci U S A. 2009;106:12168–12173. doi: 10.1073/pnas.0903134106.. This study describes a modifier screen in a Drosophila model of FTP3. The authors identify serpin5 as a potential modifier.
- 48.Branco J, Al-Ramahi I, Ukani L, Perez AM, Fernandez-Funez P, Rincon-Limas D, Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum.Mol.Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- 49. Jung J, van Jaarsweld MT, Shieh SY, Xu K, Bonini NM. Defining Genetic Factors that Modulate Intergenerational CAG Repeat Instability in Drosophila melanogaster. Genetics. 2010 doi: 10.1534/genetics.110.121418.. The authors used a transgenic Drosophila model expressing a long, non-coding CAG repeat to screen for modifiers of repeat length in the germ-line.
- 50.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van MI, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 51.Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, Dejonghe P, Frankel WN. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum.Mol.Genet. 2006;15:1043–1048. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- 52.Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 53.Carpinelli MR, Hilton DJ, Metcalf D, Antonchuk JL, Hyland CD, Mifsud SL, Di RL, Hilton AA, Willson TA, Roberts AW, et al. Suppressor screen in Mpl−/− mice: c-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc Natl Acad Sci U S A. 2004;101:6553–6558. doi: 10.1073/pnas.0401496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matera I, Watkins-Chow DE, Loftus SK, Hou L, Incao A, Silver DL, Rivas C, Elliott EC, Baxter LL, Pavan WJ. A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy. Hum.Mol.Genet. 2008;17:2118–2131. doi: 10.1093/hmg/ddn110.. This study describes the results of a sensitized ENU mutagenesis screen in mice to identify genetic modifiers of Waardenberg syndrome. This powerful approach has been widely used in lower model organisms, but has been used infrequently in mice.