

Abstract
Cardiac dysfunction and mortality associated with trauma and sepsis increase with age. Mitochondria play a critical role in the energy demand of cardiac muscles, and thereby on the function of the heart. Specific molecular pathways responsible for mitochondrial functional alterations after injury in relation to aging are largely unknown. To further investigate this, 6- and 22-month-old rats were subjected to trauma-hemorrhage (T-H) or sham operation and euthanized following resuscitation. Left ventricular tissue was profiled using our custom rodent mitochondrial gene chip (RoMitochip). Our experiments demonstrated a declined left ventricular performance and decreased alteration in mitochondrial gene expression with age following T-H and we have identified c-Myc, a pleotropic transcription factor, to be the most upregulated gene in 6- and 22-month-old rats after T-H. Following T-H, while 142 probe sets were altered significantly (39 up and 103 down) in 6-month-old rats, only 66 were altered (30 up and 36 down) in 22-month-old rats; 36 probe sets (11 up and 25 down) showed the same trend in both groups. The expression of c-Myc and cardiac death promoting gene Bnip3 were increased, and Pgc1-α and Ppar-α a decreased following T-H. Eleven tRNA transcripts on mtDNA were upregulated following T-H in the aged animals, compared with the sham group. Our observations suggest a c-myc–regulated mitochondrial dysfunction following T-H injury and marked decrease in age-dependent changes in the transcriptional profile of mitochondrial genes following T-H, possibly indicating cellular senescence. To our knowledge, this is the first report on mitochondrial gene expression profile following T-H in relation to aging.
INTRODUCTION
Aging is a significant factor that contributes to mitochondrial damage and dysfunction (1–3). The progression to heart failure (independent of cause) is associated with a progressive decline in the activity of mitochondrial respiratory pathways which leads to diminished capacity for adenoside triphosphate (ATP) production (4). Mitochondrial oxidative phosphorylation is the primary energy source in the myocardial cell. The molecular mechanism of mitochondrial damage and dysfunction to cardiomyocytes and its contribution to the outcome of T-H in relation to aging are not fully known. The information concerning the molecular changes occurring in mitochondria after trauma will be valuable in both understanding the pathophysiology associated with trauma in relation to aging, and designing better intervention strategies to reduce mortality and organ dysfunction.
In a recent study using a T-H model in the rat, both the expression and the enzymatic activity of cytochrome c oxidase, a component of the mitochondrial respiratory complex, was found to be reduced significantly, along with an increase in cytosolic cytochrome c levels, decrease in mitochondrial ATP levels and increased cardiomyocyte apoptosis (5). It has been shown that cardiac output, left ventricular performance and organ blood flow in the liver, small intestine and kidney decreased significantly following T-H and resuscitation (6,7). A prolonged depression of cardiovascular function following T-H, despite fluid resuscitation was also reported by other investigators (8–10).
We recently developed a custom rodent mitochondrial gene chip with probe sets representing genes on the nuclear and mitochondrial DNA relevant to the structure and function of mitochondria. Our initial experiments demonstrated a total of 483 signature genes that were altered by hypoxia in the neonatal mouse cardiac myocytes (11). This included several transcripts on mtDNA. Pathway analysis demonstrated predominant changes in the expression of genes involved in oxidative phosphorylation, glucose and fatty acid metabolism and apoptosis. The most upregulated genes following 24-h hypoxia included HIF-1α inducible genes Bnip3, Pdk1 and Aldoc. While Bnip3 is important in the cardiomyocyte death pathway, Pdk1 enzyme is critical in conserving mitochondrial function by diverting metabolic intermediates to glycolysis.
In this manuscript, we report our findings on mitochondrial gene expression changes in rat cardiomyocytes subjected to in vitro hypoxia and age-dependent cardiac mitochondrial gene expression changes in an in vivo model of T-H. We subjected 6- and 22-month-old rats to T-H, and mitochondrial gene expression changes were studied using our custom gene chip, RoMitochip. Our findings identify possible new pathways that might regulate mitochondrial function following hemorrhage and its relationship to aging.
MATERIALS AND METHODS
Animals
Pregnant (E19) Fisher 344 rats were purchased from Charles River Laboratories (Wilmington, MA, USA) and 2-day-old pups were used for neonatal cardiomyocytes isolation (12). For T-H study, 6- and 22-month-old Fisher 344 rats were obtained through the National Institute of Aging (Bethesda, MD, USA). Several studies have characterized the cardiovascular system in the aged rat (13,14). Specifically, previous histological characterizations of aged rat heart have demonstrated fibrotic infiltration of left ventricle and decreased volume density of cardiomyocytes (15). All animal experiments were carried out in accordance with the protocol approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and were consistent with the guidelines of the National Institutes of Health.
Neonatal Cardiomyocyte Isolation and Hypoxic Exposure
Cardiomyocytes were isolated from neonatal Fisher 344 rats (Harlan Laboratories, CT, USA) as reported previously (12). After 4 d in culture, the cells were exposed to normoxia or 1% hypoxia in a CO-48 incubator (New Brunswick, NJ, USA) for 24 h. RNA was isolated and gene expression profiles were determined using the RoMitochip developed in our laboratory as described previously (11).
Trauma-Hemorrhage Procedure
This procedure was performed as described earlier (6). For surgery, the rats were fasted for 16 h prior to the procedure but allowed water ad libitum, anesthetized with isoflurane (Minrad, Bethlehem, PA, USA) and restrained in a supine position. A 5-cm midline laparotomy was performed, which was closed asceptically in two layers with sutures (Ethilon 6/0, Ethicon, Somerville, NJ, USA). Both femoral arteries and the right femoral vein were asceptically cannulated with polyethylene-10 tubing (Becton Dickinson, Sparks, MD, USA). The blood pressure was monitored constantly by attaching one of the catheters to a blood pressure analyzer (Digi-Med BPA-190, Micro-Med Inc., Louisville, KY, USA). Upon awakening, animals were bled rapidly through the other arterial catheter to a mean arterial blood pressure of 35 ±5 mmHg within 10 min. The bleeding was continued till 50% blood volume was removed in approximately 45 min and the low blood pressure was maintained for another 45 min by giving 40% of the shed blood volume as Ringer’s lactate in small volumes. At the end of the shock period, the rats were resuscitated with four times the shed blood volume in the form of lactated Ringer’s solution over 60 min. Bipuvacaine was applied to the incision sites, catheters removed and incisions were closed with sutures. The same surgical procedures were conducted on sham-operated animals, but they did not undergo hemorrhage or resuscitation. Two h following resuscitation, left ventricular performance was assessed, animals euthanized and left ventricles removed. The left ventricular performance was evaluated by anesthetizing the rats with pentobarbital sodium (~30mg/kg body wt), inserting a PE-50 catheter into the left ventricle via right carotid artery. Left ventricular contractility parameters, maximal rate of left ventricular pressure increase (+dP/dtmax) and decrease (−dP/dtmax) were determined (6). Four to five rats were used in each group.
Microarray
The RoMitochip had probe sets representing mitochondrial genes from both rat and mouse. The mouse probe sets were validated previously (11). Mitochondrial genome consists of a circular DNA (mtDNA) with about 16,000 bases and 37 transcripts (16). Of the 37 transcripts, 13 code for proteins, 2 code for ribosomal RNAs (12 and 16S), and 22 code for tRNAs. The 13 proteins on the mitochondrial DNA are: seven subunits of NADH dehydrogenase, cytochrome b, three cytochrome c oxidase subunits (1, 2 and 3), and ATP synthase 6 and 8. The gene sequences representing the rat mitochondrial genome included in the gene chip encompassed published mtDNA sequences of ten different inbred strains: WKY/NCrl (Wistar Kyoto; Accession # DQ673907); BN/NHsdMcwi (Brown Norway; Accession # AC_000022); F344/NHsd (Fisher 344; Accession # DQ673909); ACI/Eur (August × Copenhagen Irish; Accession # DQ673908); FHH/Eur (fawn hooded hypertensive; Accession # DQ673910); GK/Swe (Accession # DQ673913); GK/Far (Accession # DQ673912); T2DN/Mcwi (Accession # DQ673915); GH/OmrMcwi (Accession # DQ673911) and SS/JrHsdMcwi (Dahl Salt-sensitive; Accession # DQ673914) (17). These specific strains were chosen because complete sequences of mtDNA were available for each of them. The gene transcripts from these ten different strains were aligned and 48 different probe sets were created to represent all the 37 transcripts from these ten strains. The chip (11 micron, Affymetrix platform) contained a total of 419 probe sets representing genes from mitochondrial and nuclear DNA from the rat.
Microarray Experimentation
The microarray experiments were performed as described before (11,18). Briefly, RNA was isolated from snap frozen cardiomyocytes or snap frozen left ventricles and RNA integrity was checked by resolving on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). One hundred ng of total RNA was amplified from each RNA sample and labeled using the Affymetrix Whole-Transcript Assay (WTA) Sense Target Labeling Protocol (Affymetrix, Santa Clara, CA, USA). Eleven μg of labeled sense DNA was used for hybridization. Ribosomal RNA was not removed. The hybridized chips were washed using the Affymetrix fluidics station 450, stained, and scanned on the 3000 7G scanner as described by the manufacturer (Affymetrix). GeneChip hybridizations were carried out in the Gene Expression Shared Facility of the Comprehensive Cancer Center at the University of Alabama at Birmingham. To study the gene expression profile following in vitro hypoxia or normoxia, four chips were used per group. For in vivo experiments, one chip was used per animal and four chips were used in each group.
Data Analysis
Gene expression data was normalized using RMA (19) and quantile normalization methods. After normalization, analysis of variance (ANOVA) was applied to compare gene expression level changes. P values for pairwise comparisons were adjusted using the Tukey method in analysis of variance (ANOVA). We used statistical software R and SAS v9.13 for data analysis. For hierarchical clustering, raw data were quantile normalized, log2 transformed, and pairwise compared by t test. Additional intensity filter was applied. The selected gene lists were clustered using hierarchical clustering methods and visualized using normalized log2-transformed intensities. P value was corrected for False Discover Rate (FDR) with Benjamini-Hochberg methods using software package GeneSpring (Agilent, CA, USA).
Real-time PCR
Real-time PCR was carried out using FAM-labeled Taqmann real-time PCR primers for c-myc and β-actin (ABI, Foster City, CA, USA). The template cDNA was prepared by random priming from RNA isolated from the tissues. The results were expressed in relation to β-actin expression. The PCR reaction was carried out in an ABI 7500 thermal cycler (ABI).
Western Blot
Protein expression of c-myc, Bnip3 and Pgc1-α were analyzed by Western blot as described (20). Briefly, total proteins in tissue lysates were resolved using 4% to 12% Nupage gel (Invitrogen, Carlsbad, CA, USA) and transferred to PVDF membranes. The membranes were saturated with blocking buffer (10 mmol/L Tris, 150 mmol/L NaCl, and 0.05% Tween-20 supplemented with 5% dry milk) for 1 h at room temperature and incubated with the respective primary antibodies: Bnip3 (Abcam Inc, Cambridge, MA, USA), Pgc1-α (Cell Singling Technology, Beverly, CA, USA), or β-actin (Abcam). The membranes then were washed 5× with TBST (Tris-buffered saline supplemented with 0.05% Tween-20) followed by incubation with an appropriate secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) conjugated with horse-radish peroxidase for 1 h at room temperature. The membranes were again washed 5× with TBST and probed using ECL (Amersham, Piscataway, NJ, USA), and autoradiographed.
All supplementary materials are available online at www.molmed.org.
RESULTS
Cardiac Function Decreases with Aging and following Trauma-Hemorrhage Injury
Blood loss following hemorrhage procedure was confirmed by the blood hematocrit level (data not shown) following reperfusion. Additionally, T-H resulted in increased plasma lactate and decreased mean arterial pressure as compared with sham controls in both age groups (data not shown). Two h following T-H and resuscitation, heart performance was tested by measuring +dP/dt and −dP/dt. As seen in Figure 1, there was a significant decrease in left ventricular function (±dP/dt) in the 22-month-old sham- operated rats when compared with 6-month-old rats demonstrating an age-dependent decline in cardiac performance. Furthermore, there was a marked decrease in cardiac performance of the aged animals (Figure 1A) subjected to T-H as compared with the younger ones, demonstrating the age-dependent effect of hemorrhagic injury on heart performance.
Figure 1.
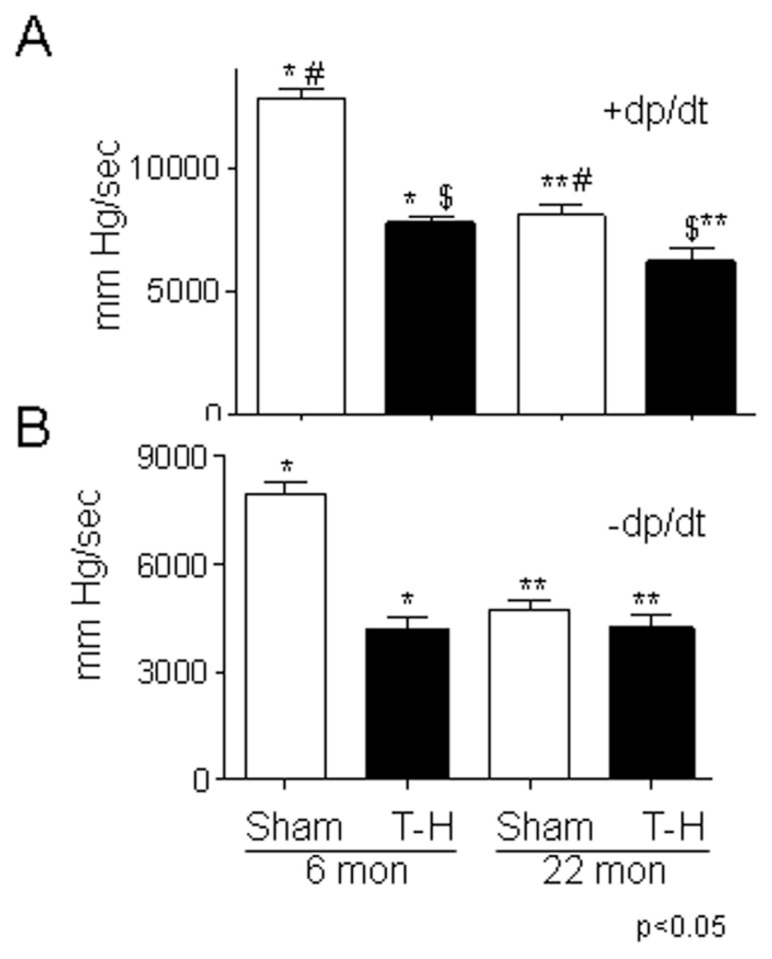
Left ventricular performance decreases with aging and T-H. Six- and 22-month-old rats were subjected to sham or T-H procedure and left ventricular performance (+ and −dP/dt) was measured. dP/dt values expressed as mean > SE. Panel A, >dP/dt and panel B, −dP/dt. See “Materials and Methods” section for experimental details.
Mitochondrial Gene Expression in Cardiomyocytes following Hypoxia
Mitochondrial energetics is critical to cardiac function and, to investigate molecular changes that may have contributed to altered mitochondrial function following T-H and in the aged, our objective was to profile the mitochondrial transcriptome in the left ventricular tissue of 6- and 22-month-old rats. The RoMitochip was designed to study mitochondrial gene expression in the rats and mice, and we previously tested the mouse probe sets using an in vitro hypoxia model (11). In this study, prior to using the RoMitochip in the in vivo T-H model, we tested the rat probe sets using RNA isolated from hypoxic or nomoxic neonatal rats. The objective of this study was mainly to test the mitochondrial gene chip. This experiment was not intended to infer hypoxic molecular pathways in vivo. The isolated neonatal cardiomyocytes were subjected to 24-h hypoxia (1% oxygen) and microarray experiments were performed in quadruplicate. Of the 419 rat mitochondrial probe sets on RoMitochip, 208 demonstrated significant alteration as defined by P < 0.05 and 163 probe sets demonstrated more than 1.2-fold with significance (P < 0.05). Among the probe sets that demonstrated most upregulation were Bnip3 (10-fold), Egln3 (7.5-fold), HK2, Myc (1.9-fold), Mdm2 (1.8-fold) and Vdac1 (1.5-fold) (Supplementary Table 1A). All these genes (except Egln3) also were found to be upregulated in the mouse cardiomyocytes in the previous study (11). The expression of Pla2g2a (phospholipase A2, group IIA) was the most down-regulated (29-fold). Among the other genes that were downregulated following hypoxia were Bcl2 (2.7-fold), Mtch2 (2.2-fold) Casp7 (2.1-fold), Casp3 (2.9-fold) and Bid (1.7-fold) (Supplementary Table 1B). Among the 163 probe sets that demonstrated significant change beyond 1.2-fold, 100 were downregulated and 63 were upregulated following 24-h hypoxia (Supplementary Table S1).
Influence of Age on Gene Expression Following T-H
Following T-H, there were more downregulated genes than upregulated in both 6-month as well as in 22-month groups (Supplementary Table 2). More strikingly, the total number of genes altered (upregulated or downregulated) was markedly less in the aged group and it was more prominent in the case of downregulated genes. Overall, when expression of 142 mitochondrial probe sets demonstrated significant change following T-H in the 6-month group, the expression of only 66 were altered in the aged group (Supplementary Table 2). Among the upregulated genes, the majority of them were upregulated uniquely in each age group—in contrast to the downregulated genes, among which most of the downregulated genes in the 22-month group also were changed similarly in the 6-month group, demonstrating a clear difference in mitochondrial gene expression profile following injury in relation to aging.
Age-Independent Expression of Mitochondrial Genes following T-H
There were 11 genes upregulated and 25 downregulated following T-H, common to both age groups (Supplementary Table 3). Myc, Jak2, glutaredoxin (Glrx1), hexokinase 2 (Hk2), HIF-1α and Sod2 were among the genes significantly upregulated in both 6- and 22-month-old animals. Among these, Myc was the most upregulated (6-month: 6.2-fold; 22-month: 3.8-fold) followed by Jak2 (6-month: 3.9-fold; 22-month: 2.3-fold) (Figure 2). Whereas, the expression of genes carnitine palmitoyltransferase 1 (Cpta), glycerol 3-phosphate acyltransferase (Gpam), uncoupling protein (Ucp) 3 and isocitrate dehydrogenase 1 (Idh1) were downregulated in both age groups (see Figure 2). Though expression of these genes were altered following T-H irrespective of the age of the animal, the amplitude of expression was less in the aged than in the younger (see Figure 2, Supplementary Table 3).
Figure 2.
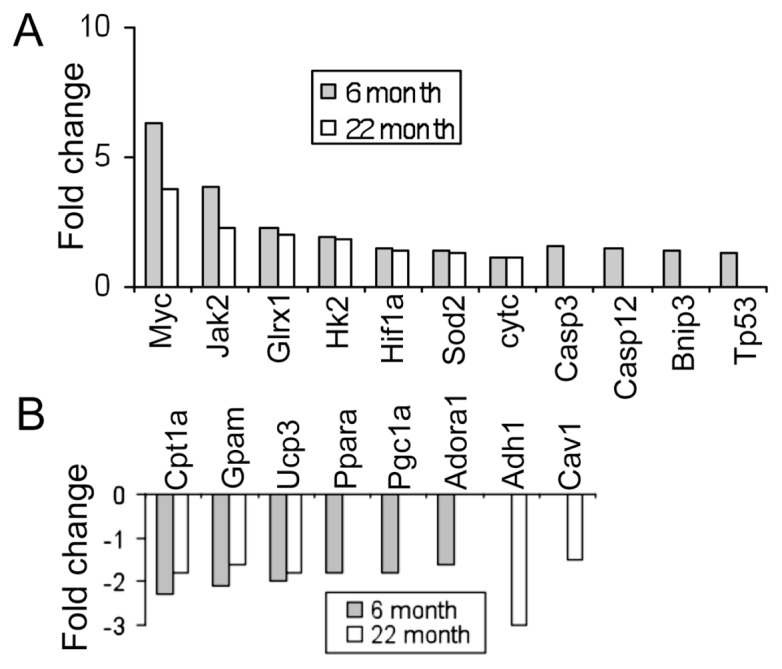
Mitochondrial gene expression changes following T-H. Changes in expression of a subset of mitochondrial genes following T-H in two different age groups (6- and 22- months). Upregulated (A) and downregulated (B) genes in each group are shown as bars. Bars represent mean fold change with a P < 0.05.
Mitochondrial Gene Expression in 6-Month-Old Animals following T-H
The expression of a total of 36 probe sets were increased and that of 103 probe sets were decreased in 6-month-old rats, after T-H and resuscitation. When the subset of genes that were similarly altered in the aged rats was excluded, the uniquely increased or decreased probe sets were 28 and 78 respectively (Supplementary Table S2). Among the genes that demonstrated a significant upregulation following T-H only in the 6-month age group were: tissue-type transglutaminase (Tgm2), caspase 3 (Casp3), Mdm2, caspase 12 (casp12), Bnip3, Tp53 and Tfam. Ppar-α, Pgc-α, Casp9 and Ogt expression were significantly downregulated following T-H in the 6-month-old rats. The Western blot experiments confirmed that the increased gene expression of Bnip3 and decreased expression of Pgc1-α were consistent with their protein expression (Figure 3A, B).
Figure 3.
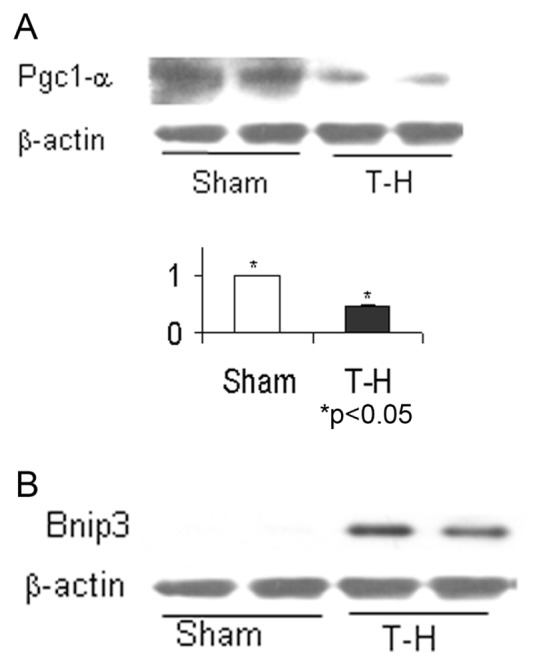
Pgc-1α and Bnip3 protein expression changes following T-H. Left ventricular tissues from 6-month-old rats subjected to T-H or sham operation were tested for Pgc-1α (A) and Bnip3 (B) by Western blot. A representative result from two sham and two T-H rats are given. The bar graph represents mean and standard error of three experiments using tissues from four sham and four T-H rats.
Mitochondrial Gene Expression in 22-Month-Old Animals following T-H
The gene expression profile of 22-month-old rats demonstrated 30 probe sets to be upregulated and 36 downregulated following T-H. However, 11 of the 30 upregulated and 25 of the 36 downregulated genes also were altered in the 6-month-old rats. Therefore expression of only a smaller number of genes was observed as unique to the aged group (Supplementary Table 4). Among the uniquely upregulated transcripts in the 22-month age group, 12 of the 19 probe sets represented mtRNA transcripts. The 11 uniquely downregulated genes included, alcohol dehydrogenase (aldh) 1 and 2, caveolin 1 (Cav1) α and nitric oxide synthase (Nos) 1.
mtDNA Transcript Alteration following T-H
The RoMitochip had 48 probe sets representing mtDNA transcripts, none of these were found to be significantly altered in 6-month-old rats. However, 13 of these probe sets were upregulated significantly in the 22-month-old rats. Twelve of the 13 probe sets represented tRNA transcripts: t-RNA to threonine, aspartate, isoleucine, phenylalanine, alanine, glycine, arginine, glutamate and serine (Figure 4). We had previously reported similar increased expression of tRNA transcripts following hypoxia in cardiomyocytes (11).
Figure 4.

Alteration of tRNA transcripts on mtDNA following T-H. Transcripts of tRNAs that demonstrated a significant upregulation in 22-month-old rats following T-H. Expression changes in each animal are represented as a heat map (see “Materials and Methods” for details).
Myc Expression following T-H in 6- and 22-Month-Old Rats
As shown in Figure 2, the most upregulated gene following T-H in both age groups was c-Myc. The increase in mRNA levels of Myc was consistent with its protein expression (Figure 5A–C). The increase in c-Myc expression was associated with decreased expression of Pgc-1α gene in the 6-month-old rats, which is also confirmed by Western blot (Figure 3A).
Figure 5.

Upregulation of c-Myc following T-H. Left ventricular tissues from rats subjected to T-H or sham operation were tested by Western blot (A and B) or real time PCR (C) for c-Myc expression. The bands were quantified by densitometry (C).
DISCUSSION
A prolonged depression of cardiovascular function occurs following hemorrhagic shock despite fluid resuscitation. Aging also is found to be a contributing factor to posttraumatic complications and mortality (21). Impaired mitochondrial function and decreased mitochondrial ATP have been observed following T-H (22).
Hemorrhagic shock causes a whole body hypoxia/reperfusion (H/R) injury, leading to dysregulation of biochemical pathways and multiple organ dysfunction syndrome (6,23–27). Cardiac output, stroke volume, and cardiac contractility decrease significantly following T-H (7,28–30). Experimental evidences indicate that reactive oxygen species (ROS) play significant role in cardiac ischemia/reper-fusion (I/R) injury and mtDNA has been shown to be a major target of ROS- mediated cellular injury (31,32). Also, mitochondrial calcium overload plays an important part in I/R injury (33,34). During I/R, ischemia induced by the deprivation of coronary blood flow and subsequent reoxygenation by reperfusion results in a dramatic elevation of mitochondrial ROS generation (35,36). This may lead to either programmed cell death or a preconditioning response that may prevent cell death after a subsequent, more severe ischemia. The studies from our group and others have demonstrated hepatic and cardiac dysfunction following T-H (37–39). T-H causes tissue hypoxia and the oxygen deficiency has been demonstrated to have direct effects on sarcolemma and on mitochondria (40). Furthermore, when an overwhelming amount of evidence links mitochondrial dysfunction with cellular senescence and aging, it has been observed that subsarcolemmal and interfibrillar mitochondria experience functional decline during aging with a selective reduction in oxidative phosphorylation (41). These functional correlates might explain age- dependent effect of T-H on contractility and relaxation. Additionally, our experiments demonstrated enhanced endoplasmic reticulum (ER) stress in metabolically active organs following T-H (20,42). Previous studies have successfully demonstrated the usefulness of focused microarrays in profiling mitochondrial gene expression (11,43,44). One of the factors involved in T-H injury that causes ER stress is hypoxia, and our recent in vitro studies have identified over 400 signature mitochondrial genes that were altered following hypoxia in cardiomyocytes (11). Along with decreased ATP levels, we also observed augmented expression of genes corresponding to glycolysis (for example, hexokinase) as well as those involved in preventing the entry of pyruvate to mitochondria (for example, Pdk1) (11), consistent with the well-known Warburg effect. One of the most upregulated genes in mouse cardiomyocytes subjected to hypoxia was Bnip3. Similarly, when rat cardiomyocytes were subjected to hypoxia and gene expression profiled using RoMitochip, as reported in this communication, Bnip3 was the most profoundly expressed gene (Supplementary Table 1A). In addition to this, the results from rat experiments shared altered expression of several genes with the mouse cardiomyocytes that were subjected to 24-h hypoxia. Interestingly, as shown in this paper, many of these genes (such as Bnip3, caspase 3 and hexokinase) also were altered following T-H in an in vivo model. Apart from identifying augmentation of the cardiac death promoting gene in this in vivo injury model, the study also unraveled the increased expression of a gene less known in injury models, c-Myc.
The results of the study using 6- and 22-month-old rats indicate that there were fewer genes altered following T-H in the aged (22-month) group as compared the younger (6-month). In addition, the extent of change for individual genes that demonstrated altered expression was less in the aged group as compared with the 6-month-old animals (see Figure 2). These observations are important in the context of cellular senescence induced by the aging process (Supplementary Table 1)
Interestingly, there was an upregulation of 10 tRNAs located in mtDNA in the old (22-month) rats (see Figure 4). This increased level of tRNAs was consistent with the observation in in vitro hypoxia experiments using cardiomyocytes (11). This might imply that there is a transcriptional-to-translational switch in the aged-to-conserved energy. As seen in Figure 2, we observed a significant downregulation of Pgc1-α, Ppar-α and the purine receptor Adora1 and upregulation of Bnip3, caspase 3, caspase 12 and p53 in 6-month-old rats. However, these genes remained unaltered in 22-month-old rats indicating an age-dependent decline or inability in their expression. Additionally, among the most upregulated genes following T-H in both age groups were c-Myc (Figure 5), Janus kinase (Jak) 2, hexokinase (Hk) 2 and Hif-1α; and among the most downregulated genes in both age groups included carnitine palmitoyltransferase I (Cpt1) and glycerol 3-phosphate acyltransferase (Gpam). Among these, c-Myc mRNA expression was the most augmented. These observations confirm a reduced utilization of fatty acid oxidation products in the mitochondria (downregulation of Cpt1) and a switch to glycolysis following T-H, akin to hypoxic condition.
Our results are quite consistent with the previous reports that c-Myc activation leads to downregulation of Pgc-1α as well as the downstream target Cpt-1 implicated in fatty acid oxidation (45), though we observed an age-related difference in the expression pattern of these genes. Using Western blot, we confirmed that the increase in gene expression of c-Myc was followed by marked increase in its protein expression (see Figure 5). Pgc-1α coactivates the coordinated expression of genes involved in mitochondrial biogenesis, energy production and defense systems against reactive oxygen species (ROS), such as the genes encoding Tfam, ATP synthase, and uncoupling proteins. Pgc-1α acts with transcription factors such as Ppar-γ, retinoid receptor (RXR)-α and nuclear respiratory factor-1 (Nrf-1). It has been shown previously that Ppar expression after T-H decreases, and its augmented expression reduces liver apoptosis following T-H (46). Upon events such as oxidative or cold stress, expression of Pgc-1α is upregulated by transcriptional activities and downstream target genes are activated (47). Pgc-1 family of transcriptional coactivator control fatty acid oxidation and mitochondrial biogenesis in the adult heart. In ischemic and hypoxic conditions, Pgc-1 is downregulated to promote glucose metabolism and suppress fatty acid oxidation (48). However, c-Myc, a pleiotropic transcription factor that enhances the glycolytic pathway, also is reported to enhance mitochondrial activity by producing biosynthetic substrates. c-Myc was increased significantly in both age groups (6.3- and 3.8-fold), though more at 6 months (see Figure 5). HIF, the transcript of which was also increased significantly at both age groups (1.5- and 1.4-fold, at 6 and 22 months respectively) (see Figure 2), specifically blocks access of glycolytic end products to mitochondria. Hif-1 is an oxygen-sensing transcription factor that initiates transcription of an array of genes during hypoxic insult. Decreased cellular oxygen results in decreased breakdown and stable cytoplasmic level of Hif-1α. This allows the dimerization of Hif-1α with Hif-1β and binding of the dimer to hypoxia response element (HRE) to recruit transcriptional coactivators (49). The effect of hypoxia in promoting glycolysis while reducing ATP production through decreased mitochondrial oxidation in cardiomyocytes subjected to hypoxia was observed previously in in vitro experiments (11). The enhancement of c-Myc following T-H is suggestive of its role in promoting glycolytic process in injury. Furthermore, c-Myc activates p53—promoting apoptosis. The increased expression of c-Myc is also important in T-cell activation and in proinflammatory mediators, as it instructs a complex inflammatory program, including induction of IL-1β (50,51). HIF and c-Myc act on multiple targets to regulate carbon metabolism and act in concert to fine tune adaptive responses to hypoxic environment (52). In pathological stress, c-Myc regulates the increased metabolic energy demand, then mitochondrial biogenesis (45). Additionally, the c-Myc protein level has been shown to increase markedly after hypoxia and ischemia (53). The upregulation of c-Myc was consistent with the observed decreased expression of genes negatively regulated by c-Myc (Cpt1 [−2.3-fold] and Gpam [−2.1-fold]). The critical function of c-Myc in regulating cardiac energy metabolism during stress and its marked upregulation after T-H in both age groups give substantial credence to its significance in energy balance in response to T-H injury and ensuing cell death (20).
The reason for the lack of change in Pgc-1α or Bnip3 transcripts in the old age (22 months), to be consistent with an increase in c-Myc and Hif-1α, remains to be elucidated and we speculate that this is characteristic of cellular senescence concomitant with aging. In this study, we examined the mitochondrial gene expression changes at 2 hours following T-H. In further studies, we also may have to test at 24 hours and 72 hours following T-H to determine the influence of aging and injury on temporal gene expression changes. It also remains to be explored whether, in aging, c-Myc-induced mitochondrial biogenesis involves pathways independent of Pgc-1α and its downstream effectors. Therefore, the genes induced by c-Myc following T-H in the different age groups remain to be identified, and this is expected to bring attention to the observed age-specific gene expression profile.
Aging is associated with a reduced aerobic activity in humans and declined mitochondrial function (54). A reduction in maximal mitochondrial ATP production rate and mitochondrial DNA abundance occurs with age in association with muscle weakness and reduced endurance in elderly people (55,56). A progressive decline in muscle mitochondrial DNA abundance and protein synthesis with age also has been reported by others (57,58). The identification of c-Myc as a potential target gene in injury and aging and its relationship to mitochondrial function may unravel novel pathways in injury and its resolution in relation to aging (Figure 6). Our future studies using clinical specimens will further ascertain the significance of c-Myc-induced mitochondrial functional alteration and its role in energy balance.
Figure 6.
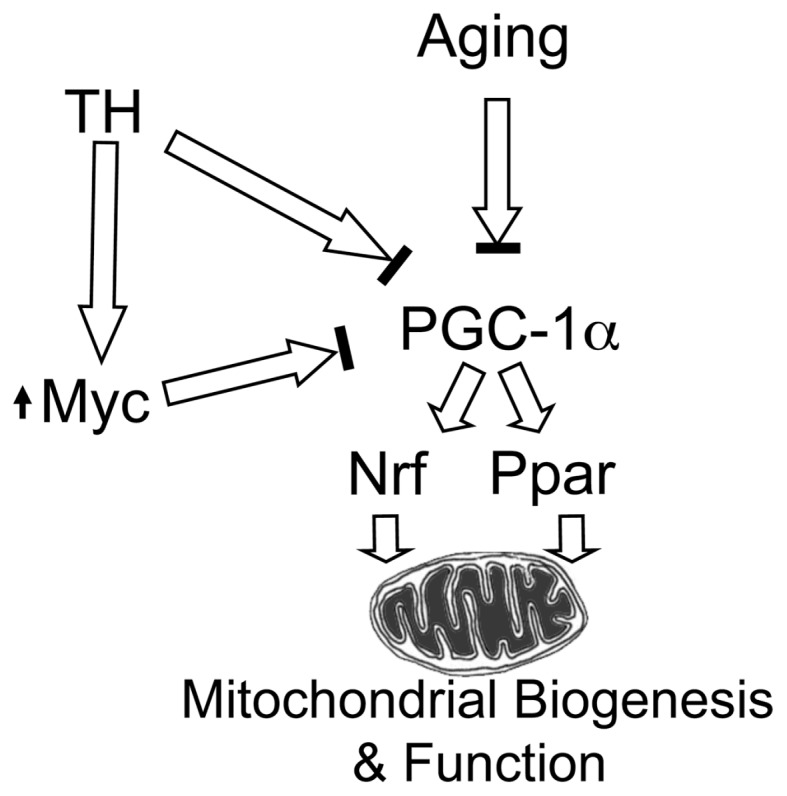
Regulation of Pgc-1α and mitochondrial function by c-Myc, T-H and aging. Aging and T-H are known to reduce Pgc-1α activity. Our results indicate that c-Myc expression increases following T-H in both age groups and this transcription factor might be the mediator for decreased Pgc-1α following injury, resulting in diminished mitochondrial biogenesis and function.
Supplemental Data
ACKNOWLEDGMENTS
The study was supported by NIH grants AG 031440 (R Raju), GM 39519 (I Chaudry), HL101192, HL079364 and HL67464 (JC Chatham) and the UAB HSF GEF Scholar Award (R Raju). The microarray experiments were carried out in the Heflin Center for Genomic Science by Michael Crowley and supported by the UAB Comprehensive Cancer Center Core Grant 5P30 CA13148-37.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
RoMitochip is a custom chip made with funding from the National Institute of Aging. None of the authors had financial profit from this chip in this study.
REFERENCES
- 1.Terzioglu M, Larsson NG. Mitochondrial dysfunction in mammalian ageing. Novartis Found Symp. 2007;287:197–208. doi: 10.1002/9780470725207.ch14. [DOI] [PubMed] [Google Scholar]
- 2.Wang P, et al. Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res. 2010;106:911–9. doi: 10.1161/CIRCRESAHA.109.206185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ladiges W, Wanagat J, Preston B, Loeb L, Rabinovitch P. A mitochondrial view of aging, reactive oxygen species and metastatic cancer. Aging Cell. 2010;9:462–5. doi: 10.1111/j.1474-9726.2010.00579.x. [DOI] [PubMed] [Google Scholar]
- 4.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–55. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh YC, et al. Inhibition of cardiac PGC-1alpha expression abolishes ERbeta agonist-mediated cardioprotection following trauma-hemorrhage. FASEB J. 2006;20:1109–17. doi: 10.1096/fj.05-5549com. [DOI] [PubMed] [Google Scholar]
- 6.Ba ZF, et al. Alterations in tissue oxygen consumption and extraction after trauma and hemorrhagic shock. Crit Care Med. 2000;28:2837–42. doi: 10.1097/00003246-200008000-00026. [DOI] [PubMed] [Google Scholar]
- 7.Mizushima Y, et al. Estradiol administration after trauma-hemorrhage improves cardiovascular and hepatocellular functions in male animals. Ann Surg. 2000;232:673–9. doi: 10.1097/00000658-200011000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochanek PM, Bayir H. Titrating oxygen during and after cardiopulmonary resuscitation. JAMA. 2010;303:2190–1. doi: 10.1001/jama.2010.715. [DOI] [PubMed] [Google Scholar]
- 9.Yang S, et al. Mechanism of cardiac depression after trauma-hemorrhage: increased cardiomyocyte IL-6 and effect of sex steroids on IL-6 regulation and cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H2183–91. doi: 10.1152/ajpheart.00624.2003. [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Chaudry IH. Crystalloid resuscitation restores but does not maintain cardiac output following severe hemorrhage. J Surg Res. 1991;50:163–9. doi: 10.1016/0022-4804(91)90241-d. [DOI] [PubMed] [Google Scholar]
- 11.Jian B, et al. Hypoxia induced alteration of mitochondrial genes in cardiomyocytes- role of Bnip3 and Pdk1. Shock. 2010;34:169–75. doi: 10.1097/SHK.0b013e3181cffe7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou L, et al. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am J Physiol Heart Circ Physiol. 2009;296:H515–23. doi: 10.1152/ajpheart.01025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capasso JM, Palackal T, Olivetti G, Anversa P. Left ventricular failure induced by long-term hypertension in rats. Circ Res. 1990;66:1400–12. doi: 10.1161/01.res.66.5.1400. [DOI] [PubMed] [Google Scholar]
- 14.Forman DE, Cittadini A, Azhar G, Douglas PS, Wei JY. Cardiac morphology and function in senescent rats: gender-related differences. J Am Coll Cardiol. 1997;30:1872–7. doi: 10.1016/s0735-1097(97)00411-7. [DOI] [PubMed] [Google Scholar]
- 15.Hacker TA, McKiernan SH, Douglas PS, Wanagat J, Aiken JM. Age-related changes in cardiac structure and function in Fischer 344 x Brown Norway hybrid rats. Am J Physiol Heart Circ Physiol. 2006;290:H304–11. doi: 10.1152/ajpheart.00290.2005. [DOI] [PubMed] [Google Scholar]
- 16.Reddy PH. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromolecular Med. 2008;10:291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlick NE, et al. Sequence analysis of the complete mitochondrial DNA in 10 commonly used inbred rat strains. Am J Physiol Cell Physiol. 2006;291:C1183–92. doi: 10.1152/ajpcell.00234.2006. [DOI] [PubMed] [Google Scholar]
- 18.Raju R, Dalakas MC. Gene expression profile in the muscles of patients with inflammatory myopathies: effect of therapy with IVIg and biological validation of clinically relevant genes. Brain. 2005;128(Pt 8):1887–96. doi: 10.1093/brain/awh518. [DOI] [PubMed] [Google Scholar]
- 19.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 20.Jian B, et al. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim Biophys Acta. 2008;1782:621–6. doi: 10.1016/j.bbadis.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nomellini V, Gomez CR, Gamelli RL, Kovacs EJ. Aging and animal models of systemic insult: trauma, burn, and sepsis. Shock. 2009;31:11–20. doi: 10.1097/SHK.0b013e318180f508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh YC, et al. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol. 2006;41:511–21. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Ayala A, Ertel W, Chaudry IH. Trauma- induced suppression of antigen presentation and expression of major histocompatibility class II antigen complex in leukocytes. Shock. 1996;5:79–90. doi: 10.1097/00024382-199602000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Bulger EM, Cuschieri J, Warner K, Maier RV. Hypertonic resuscitation modulates the inflammatory response in patients with traumatic hemorrhagic shock. Ann Surg. 2007;245:635–41. doi: 10.1097/01.sla.0000251367.44890.ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. 2009;40:912–18. doi: 10.1016/j.injury.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 26.Raju R, Bland KI, Chaudry IH. Estrogen: a novel therapeutic adjunct for the treatment of trauma-hemorrhage-induced immunological alterations. Mol Med. 2008;14:213–21. doi: 10.2119/2008-00001.Raju. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rushing GD, Britt LD. Reperfusion injury after hemorrhage: a collective review. Ann Surg. 2008;247:929–37. doi: 10.1097/SLA.0b013e31816757f7. [DOI] [PubMed] [Google Scholar]
- 28.Yarlagadda S, et al. Cardiovascular predictors of in-patient mortality after subarachnoid hemorrhage. Neurocrit Care. 2006;5:102–7. doi: 10.1385/NCC:5:2:102. [DOI] [PubMed] [Google Scholar]
- 29.Yu HP, et al. The PI3K/Akt pathway mediates the nongenomic cardioprotective effects of estrogen following trauma-hemorrhage. Ann Surg. 2007;245:971–7. doi: 10.1097/01.sla.0000254417.15591.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sayeed MM, Zhu M, Maitra SR. Alterations in cellular calcium and magnesium during circulatory/septic shock. Magnesium. 1989;8:179–89. [PubMed] [Google Scholar]
- 31.Ambrosio G, Flaherty JT. Effects of the superoxide radical scavenger superoxide dismutase, and of the hydroxyl radical scavenger mannitol, on reperfusion injury in isolated rabbit hearts. Cardiovasc Drugs Ther. 1992;6:623–32. doi: 10.1007/BF00052564. [DOI] [PubMed] [Google Scholar]
- 32.Chen Q, Lesnefsky EJ. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med. 2006;40:976–82. doi: 10.1016/j.freeradbiomed.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 33.Delcamp TJ, Dales C, Ralenkotter L, Cole PS, Hadley RW. Intramitochondrial [Ca2+] and membrane potential in ventricular myocytes exposed to anoxia-reoxygenation. Am. J. Physiol. 1998;275(2 Pt 2):H484–94. doi: 10.1152/ajpheart.1998.275.2.H484. [DOI] [PubMed] [Google Scholar]
- 34.Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of postis-chemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am. J. Physiol. 1996;271(5 Pt 2):H2145–53. doi: 10.1152/ajpheart.1996.271.5.H2145. [DOI] [PubMed] [Google Scholar]
- 35.Baue AE. MOF, MODS, and SIRS: what is in a name or an acronym. Shock. 2006;26:438–49. doi: 10.1097/01.shk.0000228172.32587.7a. [DOI] [PubMed] [Google Scholar]
- 36.Meldrum DR, et al. Intracellular signaling mechanisms of sex hormones in acute myocardial inflammation and injury. Front Biosci. 2005;10:1835–67. doi: 10.2741/1665. [DOI] [PubMed] [Google Scholar]
- 37.Hsu JT, et al. Role of p38 mitogen-activated protein kinase pathway in estrogen-mediated car-dioprotection following trauma-hemorrhage. Am. J. Physiol. Heart Circ. Physiol. 2007;292(6):H2982–7. doi: 10.1152/ajpheart.01303.2006. [DOI] [PubMed] [Google Scholar]
- 38.Not LG, Brocks CA, Vamhidy L, Marchase RB, Chatham JC. Increased O-linked beta-N-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 hours after trauma-hemorrhage in rats. Crit Care Med. 2010;38:562–71. doi: 10.1097/CCM.0b013e3181cb10b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambol JT, et al. Mesenteric lymph duct ligation prevents trauma/hemorrhage shock-induced cardiac contractile dysfunction. J Appl Physiol. 2009;106:57–65. doi: 10.1152/japplphysiol.90937.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walshe TE, D’Amore PA. The role of hypoxia in vascular injury and repair. Annu Rev Pathol. 2008;3:615–43. doi: 10.1146/annurev.pathmechdis.3.121806.151501. [DOI] [PubMed] [Google Scholar]
- 41.Ruiz-Meana M, Fernandez-Sanz C, Garcia-Dorado D. The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res. 2010;88:30–9. doi: 10.1093/cvr/cvq225. [DOI] [PubMed] [Google Scholar]
- 42.Kozlov AV, et al. Effect of estrogen on mitochondrial function and intracellular stress markers in rat liver and kidney following trauma-hemorrhagic shock and prolonged hypotension. Mol Med. 2010;16:254–61. doi: 10.2119/molmed.2009.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnston DS, Su YA, Alesci S. Mitochondrial gene profiling: translational perspectives. Pharmacogenomics. 2009;10:1645–55. doi: 10.2217/pgs.09.112. [DOI] [PubMed] [Google Scholar]
- 44.Voss JG, et al. A focused microarray to study human mitochondrial and nuclear gene expression. Biol Res Nurs. 2008;9:272–9. doi: 10.1177/1099800408315160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahuja P, et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest. 2010;120:1494–505. doi: 10.1172/JCI38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zingarelli B, et al. Liver apoptosis is age-dependent and is reduced by activation of peroxisome proliferator activated receptor-{gamma} in hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2009;298:G133–41. doi: 10.1152/ajpgi.00262.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross CA, Thompson LM. Transcription meets metabolism in neurodegeneration. Nat Med. 2006;12:1239–41. doi: 10.1038/nm1106-1239. [DOI] [PubMed] [Google Scholar]
- 48.Lehman JJ, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jian B, et al. Anosmin-1 involved in neuronal migration is hypoxia inducible and cancer regulated. Cell Cycle. 2009;8:3770–6. doi: 10.4161/cc.8.22.10066. [DOI] [PubMed] [Google Scholar]
- 50.Grumont R, et al. The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity. 2004;21:19–30. doi: 10.1016/j.immuni.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Borrello MG, Degl’Innocenti D, Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 2008;267:262–70. doi: 10.1016/j.canlet.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 52.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–13. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosano GM, Fini M, Caminiti G, Barbaro G. Cardiac metabolism in myocardial ischemia. Curr Pharm Des. 2008;14:2551–62. doi: 10.2174/138161208786071317. [DOI] [PubMed] [Google Scholar]
- 54.Rogers MA, Hagberg JM, Martin WH, III, Ehsani AA, Holloszy JO. Decline in VO2max with aging in master athletes and sedentary men. J Appl Physiol. 1990;68:2195–9. doi: 10.1152/jappl.1990.68.5.2195. [DOI] [PubMed] [Google Scholar]
- 55.Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A. 1996;93:15364–9. doi: 10.1073/pnas.93.26.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tatpati LL, et al. The effect of branched chain amino acids on skeletal muscle mitochondrial function in young and elderly adults. J Clin Endocrinol Metab. 2010;95:894–902. doi: 10.1210/jc.2009-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bua E, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–80. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zahn JM, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS. Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.