

Abstract
ITF2357 (givinostat) is a histone deacetylase inhibitor with antiinflammatory properties at low nanomolar concentrations. We report here a phase I safety and pharmacokinetics trial in healthy males administered 50, 100, 200, 400 or 600 mg orally. After 50 mg, mean maximal plasma concentrations reached 104 nmol/L 2 h after dosing, with a half-life of 6.9 h. After 100 mg, maximal concentration reached 199 nmol/L at 2.1 h with a half-life of 6.0 h. Repeat doses for 7 consecutive days of 50, 100 or 200 mg resulted in nearly the same kinetics. There were no serious adverse effects (AEs) and no organ toxicities. However, there was a dose-dependent but transient fall in platelets. After 7 daily doses of 50 or 100 mg, the mean decrease in platelets of 17 and 25% was not statistically significant and returned to baseline within 14 d. Blood removed from the subjects after oral dosing was cultured ex vivo with endotoxin, and the release of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-1Ra, interferon (IFN)-γ and IL-10 was determined. Maximal reduction in IL-1β, TNFα, IL-6 and IFNγ was observed 4 h after dosing but returned to baseline at 12 h. There was no significant reduction in IL-1Ra or IL-10. With daily dosing, the fall in cytokine production in blood cultures observed on day 7 was nearly the same as that of the first day. We conclude that dosing of 50 or 100 mg ITF2357 is safe in healthy humans and transiently but repeatedly reduces the production of proinflammatory cytokines without affecting production of antiinflammatory cytokines.
INTRODUCTION
Acetylation and deacetylation of histones help to regulate gene expression with remodeling of chromatin, allowing the binding of transcription factors. The acetylation of histones is regulated by two classes of enzymes: histone acetyl-transferases and histone deacetylases (HDACs) (1,2). Whereas the base pair sequence of DNA provides the fundamental code for proteins, posttranslational modification of proteins plays a major role in the control of gene transcription. The amino acid tails of the core nucleosomal histones are subject to posttranslational modifications by acetylation of lysines, methylation of lysines and arginines, phosphorylation of serines and ubiquitination of lysines. Because the key acetylation takes place on lysine residues, the term “lysine deacetylases” (KDAC) can be used, particularly since inhibitors of lysine deacetylases can hyperacetylate cytoplasmic proteins.
Butyrate is a naturally occurring inhibitor of HDACs that is thought to play a role in the gastrointestinal tract. Butyrate compounds are also used to increase hemoglobin levels in patients with certain anemias due to genetic mutations. Valproic acid is also an HDAC inhibitor and is used widely to treat epilepsy and certain psychiatric problems. However, new synthetic HDAC inhibitors were developed primarily to treat cancer, by modulating the expression of pro-apoptotic genes that have been suppressed in malignant cells (2,3).
Orally active HDAC inhibitors appear to lack organ toxicities and are well tolerated. Presently, HDAC inhibitors are used to treat patients with advanced solid and hematological tumors (3–5). Suberoylanilide hydroxamic acid was approved for the treatment of cutaneous T-cell lymphoma. In general, HDAC inhibition selectively alters the transcription of few of the expressed genes (approximately 2% to 10% of expressed genes are increased or decreased in their rate of transcription) (6–9).
At concentrations lower than those used for antitumor effects, HDAC inhibitors can modulate inflammation primarily by reducing cytokine production as well as immune responses (10–14). Suberoylanilide hydroxamic acid can suppress acute graft versus host disease after allergenic bone marrow transplantation, in part, by reducing proinflammatory cytokine production (13,15). HDAC inhibitors have also been shown to reduce the severity of disease in mouse models of inflammatory bowel disease (16,17). In other inflammatory and immune-mediated diseases such as models of lupus (12,14,18), inhibitors of HDAC represent a new class of therapeutic options. In the present study, we investigated the HDAC inhibitor ITF2357 for safety and effects on ex vivo cytokine production in a phase I trial of healthy subjects.
MATERIALS AND METHODS
Human Subjects and Eligibility
The studies were approved by the Ethical Commission of the Bavarian Physician’s Chamber (Munich, Germany). All subjects provided written informed consent in accordance with the Declaration of Helsinki before enrollment. Subjects were deemed healthy on the basis of a clinical history, physical examination, electrocardiogram (ECG) and laboratory tests of blood and urine. Body weight was between 60 and 95 kg and body mass index was between 18 and 28 kg/ m2. Subjects were excluded because of evidence of hepatitis B and C and HIV-1 infection. In addition, drug or alcohol abuse, a donation of >400 mL blood during the previous 3 mo and the use of prescribed medication during the previous 30 d were exclusion criteria. Over-the-counter medicine in the previous 14 d or a history of severe allergic disease also served as exclusion criteria. Subjects were allowed to withdraw from the study at any time. Subjects were hospitalized from 24 h before dosing until 48 h after dosing and were followed for 7 d after dosing. Table 1 summarizes the subjects and the treatment schedules.
Table 1.
Pharmacokinetics studies design and volunteers characteristics.
| Study design | Treatment doses | Number of subjects | Age range (y) | M/F | Duration of treatment (and follow-up) |
|---|---|---|---|---|---|
| Single rising oral doses, double-blind, placebo-controlled, randomized | 50, 100, 200, 400, 600 mg | 40 | 21–39 | M | Single dose (1-wk follow-up) |
| Repeat rising oral doses, double blind, placebo-controlled, randomized | 50 mg every day, 100 mg every day, 200 mg every day, 100 mg twice a day | 41 | 21–40 | M | 7 d (4 wks follow-up) |
| Single dose, food interaction study; open-label randomized | 100 mg every day | 24 | 25–39 | M | Single-dose crossover |
Study Design and Treatment Plan
The doses of ITF2357 were administered as capsules in the morning after a 12-h fast. Placebo capsules were administered in equal numbers. Three studies were performed. The first was a single-dose escalation study involving 40 subjects treated with either a single dose of 50, 100, 200, 400 or 600 mg or placebo (six actives and two placebos per group) and 1 wk of follow-up. The second study was a double-blind, randomized, placebo-controlled, multiple-dose study in which 41 subjects received ITF2357 each morning for 7 consecutive days at the doses of 50, 100 and 200 mg once a day, or 100 mg twice a day (eight actives and two placebos per group; one subject did not complete the study for personal reasons). The subjects were treated for 1 wk, with a follow-up period of 4 wks. The third study was a single-dose open-label crossover randomized food interaction study where 24 subjects were treated with 100 mg once a day in the fed and fasted state and followed for 1 wk.
Pharmacokinetics
Pharmacokinetics was determined for single or repeat doses of ITF2357. Venous blood samples (5 mL) were collected into heparinized tubes according to a predetermined schedule at the following time points: 0, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 16, 24 and 48 h after dosing. After repeat doses, blood samples were collected on the first and the last day of treatment. Intermediate blood sampling was done before each morning dose on day 1 to day 7 and 2 h after dosing on days 3 and 5 to assess steady-state achievement. Plasma concentrations of ITF2357 and its main metabolites, ITF2374 and ITF2375, were assayed using a specific and validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method. The analyses were performed in two steps: the first one for the determination of ITF2357 and ITF2374 and the other for the determination of ITF2375.
Plasma samples (250 μL), spiked with the internal standards, were processed by liquid–liquid extraction with diethyl ether. After centrifugation, the organic layer was separated and evaporated under a nitrogen stream. The residue was treated with 150 μL reconstitution solvent (75% H2O/25% CH3CN, 0.1% tri-fluoracetate [TFA]) and applied to the LC-MS/MS system. The chromatographic analysis was performed using an end-capped Purospher STAR RP-18 column and mass spectrometry detection with an API3000 (ABSciex) with a Turbo Ion Spray source in positive mode. The lower limit of quantitation for the three compounds was 1 ng/mL. Pharmacokinetic parameters were calculated by non-compartmental analysis.
Safety and Tolerability
A total of 105 subjects were enrolled: 64 subjects involved in single-dose studies and 41 subjects in the repeated-dose study (Table 1). ITF2357 was studied at daily doses ranging from 50 to 600 mg in single-dose study and 50 to 200 mg in repeat-dose study, with administration frequencies of once or twice daily. All clinical adverse events (AEs) and laboratory abnormalities were evaluated by the investigators for a potential relationship to ITF2357. Those considered possibly, probably or definitely related to ITF2357 were classified as drug-related. Tolerability was monitored throughout the study periods and at follow-ups. Blood pressure and heart rate were monitored before dosing and at the times of blood sampling for pharmacokinetics. ECG was recorded before, 24 and 48 h and 7 d after dosing. Complete blood counts, blood chemistries and urinalysis were performed before and 12 h (only hematology), 24 h, 48 h and 7 d after the last dosing.
Ex Vivo Whole Blood Cultures for Cytokine Production
Blood samples were drawn at various times during the studies after oral dosing to measure ex vivo unstimulated and lipopolysaccharide (LPS)-induced cytokine production. One mL of blood was directly collected in sterile heparinized culture tubes containing a premeasured (1 mL) amount of RPMI 1640 (Cellgro Mediatech, Manassas, VA, USA) tissue culture fluid or 1 mL RPMI 1640 containing 20 ng LPS (Escherichia coli serotype 0127: B8; Sigma, St. Louis, MO, USA). Therefore, the final concentration of LPS was 10 ng/mL, since the blood was diluted one part in two. After 2, 4, 8, 12 and 24 h at 37°C, the supernatant was removed and frozen at −80°C until assayed for cytokines. The white blood cell (WBC) counts and the monocyte and lymphocyte counts were obtained for each experimental condition. The relative concentrations of cytokines were calculated per million cells. Mature IL-1β, tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-10, IL-1Ra and interferon (IFN)-γ were assayed by the electro-chemiluminescence (ECL) method (BioVeris, Gaithersburg, MD, USA). Antibodies for these assays were purchased (R&D Systems, Minneapolis, MN, USA) and biotinylated and ruthenylated according to the manufacturer’s instructions (BioVeris). The cytokine standards were obtained from Peprotech (Rocky Hill, NJ, USA). The lowest limit of detection of the ECL assay was 20 pg/mL for each cytokine.
RESULTS
Safety and Tolerability of ITF2357
A total of 104 subjects gave informed consent. In the single-dose study, nine moderate adverse effects (AEs) were reported by six (15%) subjects, but these occurred only in the groups treated with ≥200 mg. Seven of the nine moderate AEs were judged as possibly related to the study drug by the investigators. The most frequently reported AE was headache (n = 4) of mild-to-moderate severity.
In the repeat-dose study, there were 40 AEs. AEs were classified as mild (31/40, 77%) or moderate (9/40, 22%). No AEs were classified as severe. AEs were reported by 12 (29%) subjects out of 41 treated: 10 subjects treated with ITF2357 and 2 with placebo. No AEs were reported by subjects receiving 50 or 100 mg ITF2357. The most frequently reported AEs were headache (n = 6 by four subjects), palpitations (n = 8 by two subjects), dysuria (n = 5 by one subject), nausea (n = 2 by two subjects with one in placebo) and abdominal pain (n = 3 by two subjects with one in placebo). A total of 26 AEs (26/40, 65%) were assessed as possibly related to the study drug by the investigators.
Hematologic Changes
Nonspecific changes in WBC counts were observed at doses up to 200 mg/ day. A reduction in platelet counts was observed across all ITF2357-treated groups. The magnitude of the decrease was dose dependent. The effect was evident from day 5 onward, reaching the lowest value on day 9, that is, 48 h after last dosing. On day 9, the mean platelet counts decreased by 17%, 25% and 35% in groups receiving 50, 100 and 200 mg ITF2357 each day, respectively (Figure 1). For the most part, the effect on platelets recovered on day 14 (7 d after last dosing) and was fully reversed 4 wks after the last dosing (see Figure 1). The pattern of platelet decrease in the group receiving 100 mg ITF2357 twice a day was comparable to that observed in the group receiving 200 mg each day. Additional hematological evaluation performed in the 100-mg twice-a-day group revealed that the effect was more pronounced on day 11; the trend reversed toward normality on day 14 and full recovery was observed on day 21 onwards.
Figure 1.

Platelet counts in healthy volunteers receiving 7-day oral doses of ITF2357 or placebo. Mean platelet counts in ITF2357-treated group receiving 50, 100 and 200 mg ITF2357 each day, respectively, are shown.
Routine Laboratory Findings
There were no changes in liver or renal function tests at any dose or dosing schedule, using standard hospital clinical laboratory–based methods.
Pharmacokinetics of Single Oral Dose ITF2357
The mean plasma concentrations of ITF2357 after single oral administration are shown in Figure 2. They reached peak levels approximately 2 h after dosing for all dose levels and displayed biphasic elimination profiles with a mean apparent terminal elimination phase (half-life) ranging from 5.3 to 6.9 h. Dose-proportionality of the drug was assessed over the 50- to 600-mg dose range. A proportional increase in area under the curve (AUC) and Cmax of the drug with increasing dose levels of ITF2357 was observed (Table 2).
Figure 2.

Pharmacokinetics of ITF2357 in healthy male subjects after administration of a single oral dose. Mean (± SD) plasma concentrations of ITF2357 are shown. n = 9 in each group.
Table 2.
Pharmacokinetics parameters after single rising dose to healthy volunteers.
| Dose | 50 mg | 100 mg | 200 mg | 400 mg | 600 mg | |
|---|---|---|---|---|---|---|
| Cmax | ng/mL | 53 ± 5 | 99 ± 13 | 236 ± 25 | 526.3 ± 67.3 | 542 ± 93 |
| nmol/L | 111 ± 10 | 207 ± 28 | 495 ± 53 | 1,106 ± 141 | 1,139 ± 195 | |
| tmax | (h) | 2.0 ± 0.44 | 2.09 ± 0.45 | 2.17 ± 0.40 | 2.17 ± 0.40 | 3.67 ± 0.61 |
| AUC | ng • h/mL | 359 ± 26 | 671 ± 36 | 1,346 ± 158 | 2,503 ± 312 | 3,543 ± 486 |
| nmol/L • h | 754 ± 55 | 1,410 ± 76 | 2,828 ± 332 | 5,259 ± 656 | 7,444 ± 1,021 | |
| Half-life | (h) | 6.9 ± 0.43 | 6.0 ± 0.15 | 6.0 ± 0.40 | 5.7 ± 0.32 | 5.3 ± 0.32 |
| CL/Fa | (L/min) | 2.38 ± 0.17 | 2.52 ± 0.14 | 2.64 ± 0.28 | 2.96 ± 0.49 | 3.15 ± 0.51 |
Clearance fluid compartment.
Repeated Oral Doses
Doses of 50, 100 and 200 mg once a day and 100 mg twice a day were considered safe on the basis of toxicokinetic studies in various animal species and the single-dose study in volunteers. Therefore, a 7-d repeat-dose study in healthy volunteers was performed. In the once-a-day treatments, maximal concentration of the drug was reached within 2.1–2.6 h of administration. A slightly longer but statistically not significantly different tmax was observed with the twice-a-day treatment (Table 3 and Figure 3). The peak levels (Cmax) of the 100-mg ITF2357 twice-a-day regimen observed on day 7 are statistically different (P < 0.05) from those seen on day 1, whereas there was no statistical difference at the other dose regimens. The apparent elimination half-life of ITF2357 was fairly comparable between the 50-, 100- and 200-mg doses, ranging between 5.73 and 6.15 h on day 1 and 6.47 and 7.31 h on day 7. On the basis of the above data and the predose concentration values of ITF2357 (not shown), steady state was achieved within 5–7 d for the single-dose regimens. In contrast, steady state for the twice-a-day dose was reached after 7 d from the first treatment.
Table 3.
Pharmacokinetics parameters after a repeat (7-day) rising dose in healthy volunteers.
| 50 mg every day
|
100 mg every day
|
200 mg every day
|
100 mg twice a day
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Dose | Day 1 | Day 7 | Day 1 | Day 7 | Day 1 | Day 7 | Day 1a | Day 7a | |
| Cmax | (ng/mL) | 56 ± 5 | 67 ± 5 | 124 ± 9 | 135 ± 10 | 322 ± 26 | 283 ± 16 | 116 ± 15 | 182b ± 17 |
| Cmin | (ng/mL) | 2.4 ± 0.2 | 3.5 ± 0.2 | 4.1 ± 0.3 | 5.5 ± 0.3 | 10.2 ± 1.3 | 12.0 ± 1.4 | 17.7 ± 1.6 | 30.9 ± 2.6 |
| tmax | (h) | 2.5 ± 0.5 | 2.1 ± 0.4 | 2.6 ± 0.4 | 2.8 ± 0.34 | 2.4 ± 0.4 | 2.6 ± 0.4 | 2.8 ± 0.4 | 3.1 ± 0.6 |
| AUC0-t | (ng • h/mL) | 369 ± 23 | 452 ± 20 | 771 ± 27 | 891 ± 32 | 1875 ± 102 | 1,907 ± 130 | 1,326 ± 148 | 1,084 ± 84 |
| Half-life | (h) | — | 7.31 ± 0.16 | — | 6.47 ± 0.22 | — | 6.54 ± 0.16 | — | — |
First daily dose.
Statistically significant difference (P < 0.05) from day 1 Cmax.
Figure 3.

Pharmacokinetics of ITF2357 in healthy male subjects during seven daily doses. (A) Mean (± SD) plasma concentrations of ITF2357 after the first single oral dose are shown. (B) Mean (± SD) plasma ITF2357 on day 7 after seven daily doses. n = 8 in each group.
ITF2357 gives two main metabolites in vivo deriving from the biotransformation of the hydroxamate moiety into a carboxylate (ITF2375) or into an amide group (ITF2374). The metabolites inhibit HDAC enzymes at concentrations in the high micromolar–low millimolar range—therefore, 3 to 5 orders of magnitude larger than the effect of ITF2357 on these enzymes. In preclinical models, they do not contribute to the efficacy of active doses of ITF2357. These two metabolites were seen in both the single-and repeat-dose studies. ITF2375 was the more abundant one, with its metabolic ratio to the parent compound being approximately 4. Peak plasma concentrations of ITF2375 were observed 3 to 5 h after administration. The appearance of ITF2374 was delayed, with its concentrations peaking between 6.7 and 8 h. The metabolic ratio of ITF2374 to ITF2357 was <1. The AUC at 0 time (AUC0-t) and Cmax of ITF2357 and ITF2375 increased proportionally with the dose, whereas the increase of ITF2374 was more than proportional.
Cytokine Production in Ex Vivo Whole Blood Cultures
Whole heparinized blood taken at various time points after oral dosing was cultured for 24 h with endotoxin (10 ng/mL), and the supernatants were assayed for IL-1β, TNFα, IL-6, IL-1Ra, IL-10 and IFNγ. As shown in Figure 4A, the maximal decrease in TNFα production occurred at 4 h at all doses, and the 4-h time point was also the time of maximal decrease in IL-1β (Figure 4B). At a dose of 100 mg, the mean maximal concentration of ITF2357 of approximately 100 ng/mL (200 nmol/L) occurs at 2 h. However, the mean decrease in TNFα of about 37% (P = NS) of baseline occurs at 4 h. After an oral dose of 50 mg, the maximal concentration at 2 h is about 100 nmol/L. At 4 h after dosing, the mean reduction in LPS-induced TNFα was 51% (P = 0.02). The concentration of ITF2357 carried over in the plasma of the subjects into the culture is slightly lower (see Figure 2); however, since the blood is diluted in the whole blood culture one part in two, the concentration is 50% lower. For example, using a single dose of 50 mg, the concentration of ITF2357 at 4 h is approximately 40 nmol/L and the concentration carried over into the culture would be 20 nmol/L. At 20 nmol/L, LPS-induced cytokines in peripheral blood mononuclear cells (PBMCs) is significantly reduced (19). A similar reduction in IL-1β was also observed (see Figure 4B), particularly at the 100-mg dose (−77%, P = 0.001).
Figure 4.

LPS-induced TNFα and IL-1β in whole blood cultures. The percent change in cytokine levels shown on the y axis are derived from 24-h cultures of whole blood obtained at each time point shown under the horizontal axis. (A) Mean ± SEM percent change of TNFs per mL from basal production (mean for group 815 ± 205 pg/mL) just before a single oral dose. (B) IL-1β in the same samples shown in A. Mean ± SEM basal IL-1β for the group was 1,020 ± 525 pg/mL. n = 9 for each group.
By 12 and 24 h after a single dose of ITF2357, there is no significant reduction of either TNFα or IL-1β in whole blood cultures from subjects treated with 50 or 100 mg, although in the 200- and 400-mg dose group, reductions at 12 h but not 24 h were observed. We next examined the production of TNFα and IL-1β at 12 and 24 h, calculating the cytokines per million WBCs. As shown in Figure 5A and B, there is no suppression at 12 and 24 h when the amount of cytokine is examined per million WBCs in the 50- and 100-mg dose group, although suppression was present in the 200- and 400-mg group. By 24 h, all dosing groups produced cytokines comparable to that of the baseline level. From these data, we conclude that the ITF2357-associated reduction of LPS-induced TNFα and IL-1β production in whole blood cultures is transient and returns to predrug levels by 12 h after dosing.
Figure 5.
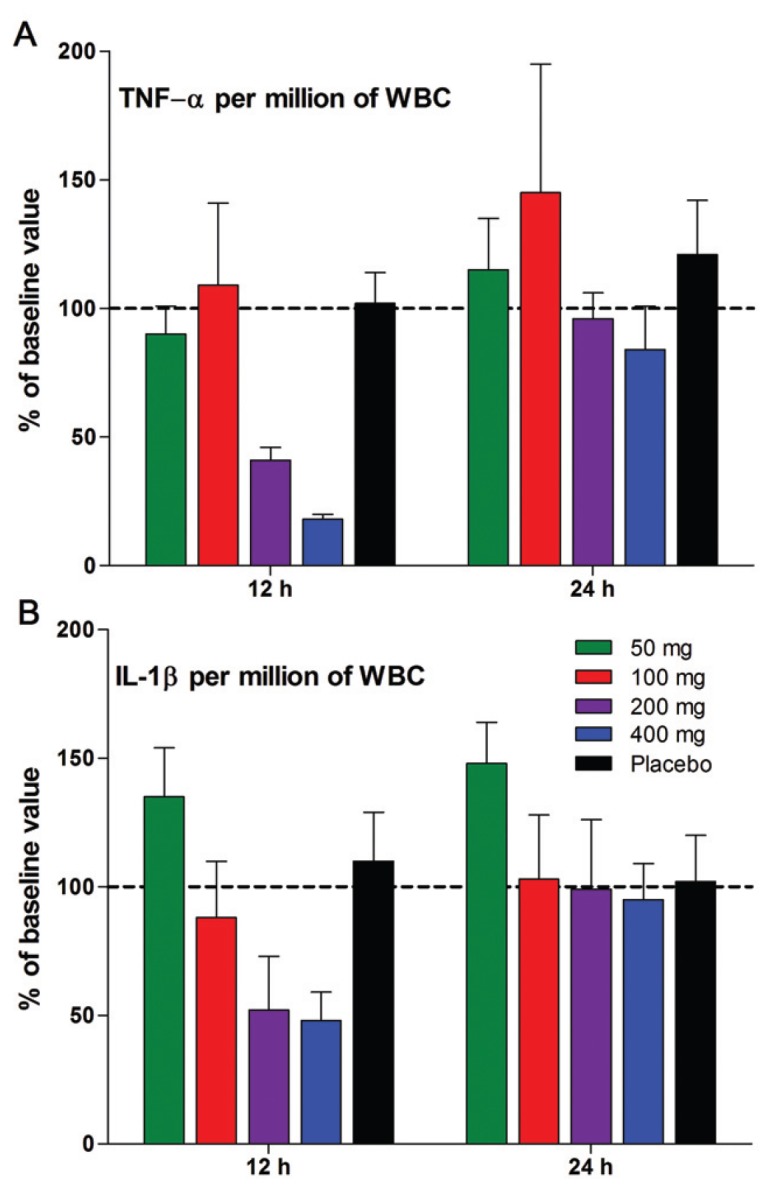
Production of LPS-induced TNFα and IL-1β per WBC. (A) Mean ± SEM of TNFα expressed as percent change from baseline. The percent change in cytokine levels shown on the y axis are derived from 24-h cultures of whole blood obtained at 12- and 24-h time points. (B) IL-1β in the same samples shown in A. n = 9 in each group.
Effect of Seven Daily Doses of ITF2357 on Cytokine Production
A daily dosing regimen was carried out to ascertain whether a sustained course of ITF2357 would affect cytokine production different from that observed after a single dose (Figure 6A, B). As shown in Figure 6B, the reduction in IL-1β after seven daily doses of 200 mg is nearly the same as that observed after the first day. Nearly the same pattern as shown in Figure 6 was observed on day 7 for TNFα, IL-6 and IFNγ (data not shown).
Figure 6.

Comparison of a single dose day 1 and day 7 of ITF2357 on LPS-induced IL-1β. The percent change in cytokine levels shown on the y axis are derived from 24-h cultures of whole blood obtained at each time point shown under the horizontal axis. (A) Mean ± SEM of IL-1β per mL expressed as percent change of basal production (980 ± 325 pg/mL) just before the single oral administration. (B) Mean ± SEM on day 7 of IL-1β after the 7th daily dose of 200 mg. Basal production was 1,144 ± 625 pg/mL. n = 9 for each group.
IL-6 and IFNγ Production
In the same samples of data shown in Figure 4, we measured levels of IL-6 and IFNγ induced by LPS. These data are shown in Figure 7A and B. A single dose of 50 mg resulted in a statistically significant (P = 0.01) reduction in IFNγ of 63% compared with that of the placebo group at the 4-h time point (see Figure 7B). The reduction was 77% at a dose of 100 mg. IL-6 was also reduced at the 4-h time point (−45%, P = 0.05). However, 12 h after dosing, production of IFNγ returns and exceeds that of basal levels (not significant). A similar pattern was observed for IL-6 (see Figure 7A).
Figure 7.

LPS-induced IL-6 and IFNγ in whole blood cultures. The percent change in cytokine levels shown on the y axis are derived from 24-h cultures of whole blood obtained at each time point shown under the horizontal axis. (A) Mean ± SEM percent change of IL-6 per mL from basal production (860 ± 356 pg/mL) just before a single oral dose. (B) IFNγ in the same samples shown in A. Mean ± SEM basal IFNγ was 485 ± 137 pg/mL. n = 9 for each group.
The amount of IFNγ was also calculated per million lymphocytes, since in the LPS model used in the present study, LPS induces IL-12 and IL-18, which in turn, induce T-cells to produce IFNγ. As shown in Figure 8, the production of IFNγ at 12 and 24 h based on per million lymphocytes is no different from that observed per million WBCs, except that the elevated production 12 h (Figure 9B) after 50 or 100 mg is no longer observed per million lymphocytes (see Figure 8).
Figure 8.
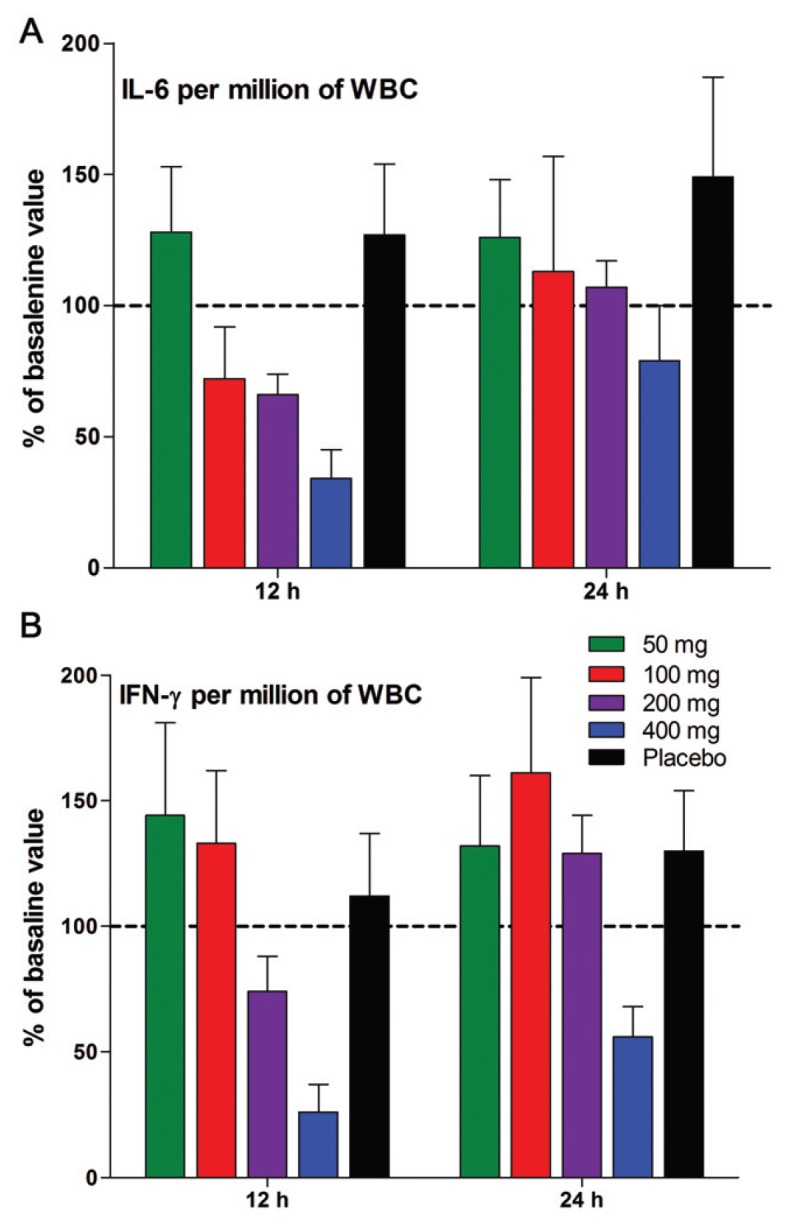
LPS-induced IL-6 and IFNγ per WBC. (A) Mean ± SEM of IL-6 per million WBC expressed as percent change of basal production just before the oral dosing. The hours after the dose are indicated under the horizontal axis. (B) IFNγ in the same sample shown in A. n = 9 for each group.
Figure 9.
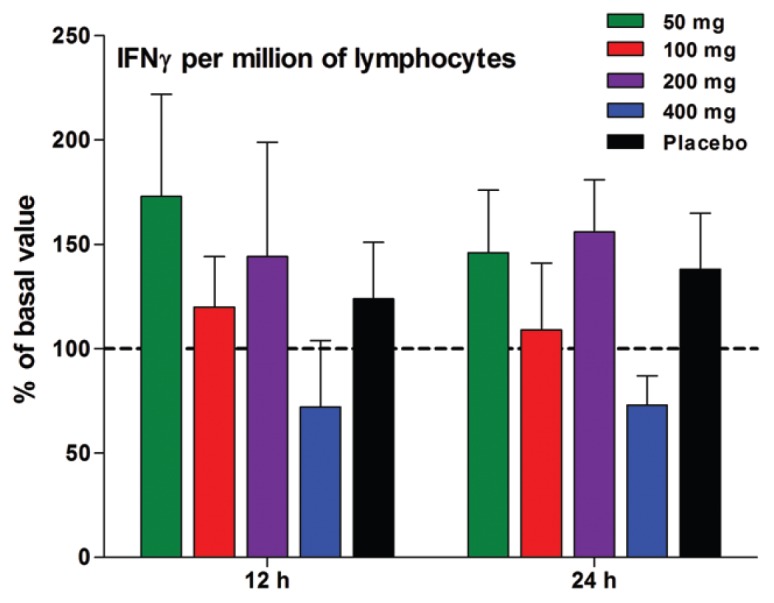
Production of LPS-induced IFNγ per lymphocyte. Mean ± SEM of IFNγ expressed as percent change from baseline per million lymphocytes 12 and 24 h after a single dose. n = 9 in each group.
No Decrease in IL-1Ra
As shown in Figure 10, there were no significant decreases at 4 h at all doses, including the placebo. Of note, at the 50-mg dose, there was an increased production of IL-1Ra at 4 h, and it was sustained after 8 h before returning to baseline levels after 24 h; however, these levels were not statistically significant. These samples in which IL-1Ra was determined are the same as those used to measure changes in TNFα and IL-1β production (see Figure 4A, B). There were no statistically significant changes in IL-10 production at doses of 50 or 100 mg each day (data not shown).
Figure 10.

LPS-induced IL-1Ra in while blood cultures. Mean ± SEM of IL-1Ra per mL expressed as percent change of basal production (1,212 ± 102 pg/mL) just before a single oral dose. The hours after the dose are indicated under the horizontal axis. The percent change in cytokine levels shown on the y axis are derived from the 24-h cultures of whole blood obtained at each time point shown under the horizontal axis. n = 9 for each group.
Effect of ITF2357 on Platelet Counts
Subjects were treated with daily dosing for 7 d and platelet counts were determined. A reduction in platelet counts was observed across all ITF2357-treated groups. As shown in Figure 1, the magnitude of the decrease was clearly dose dependent. The effect was evident from day 5 onward, reaching the lowest value on day 9 (that is, 48 h after the last dosing). On day 9, the mean platelet counts decreased by 17, 25 and 35% in groups receiving 50, 100 and 200 mg ITF2357 each day, respectively (see Figure 1). For the most part, the effect on platelets recovered on day 14 (7 d after the last dosing) and was fully reversed 4 wks after the last dosing (see Figure 1). The pattern of platelet decrease in the group receiving 100 mg ITF2357 twice a day was comparable to that observed in the group receiving 200 mg each day. Additional hematological evaluation performed in the 100 mg twice a day group revealed that the effect was more pronounced on day 11; the trend reversed toward normality on day 14, and full recovery was observed on day 21 onwards.
DISCUSSION
These studies described the first evaluation of the pharmacokinetics, safety and tolerability of the oral histone deacetylase inhibitor ITF2357 (givinostat) in humans after the administration of increasing single and multiple doses. Similar to the short-term use, there were no serious adverse effects of ITF2357 in the long-term studies (data not shown). In addition, food intake did not affect the pharmacokinetics of ITF2357 (data not shown). ITF2357 was found to be orally available, eliminated slowly and displayed dose-proportional pharmacokinetics up to a total dose of 600 mg/day. No further increases in concentrations in plasma were observed with the multiple doses after 7 days, suggesting that there was no accumulation of the drug. This result was confirmed by the ratio of the AUC on day 7 to the AUC on day 1, which was close to unity in all cases (Table 3). After the administration of all dose levels in both studies, ITF2357 was quickly metabolized with a half-life ranging from approximately 5 to 7 hours, depending on the dose regimen. This result implies that twice-daily dosing would be an appropriate clinical dosage regimen for treating inflammatory conditions. In fact, twice-daily dosing was used to treat children with systemic-onset juvenile idopathic arthritis with ITF2357 for 12 weeks at a 1.5 mg/kg total daily dose (20).
The results from these studies of the pharmacokinetics of ITF2357 in humans corroborate findings from preclinical investigations with animals, in which dose-related increases in exposure were observed in others species. In rats, dogs, rabbits and monkeys, the drug is eliminated by biotransformation and the metabolites are produced by several enzymes and are eliminated by biliary and renal excretion; only traces of unchanged ITF2357 were found in urine. In repeat oral toxicity studies in rats, the drug was administered consecutively up to 26 weeks. A dose of 50 mg/kg/day administered for 4 consecutive weeks was determined to be safe. In other two studies (of 13 and 26 weeks), there was no adverse effect level of the drug at 10 mg/ kg/day. The data in the 36-week study in monkeys showed no adverse effects at the dose of 12 mg/kg/day.
In humans, ITF2357 was well tolerated in both the single-dose and multiple-dose trials. The most commonly reported AEs were mild to moderate and nonspecific and included headache, nausea, palpitations and dysuria. There also was no suggestion that ITF2357 caused cardiac AEs, including clinically significant ECG findings or QT prolongation. There were clearly reduced platelets in subjects receiving the higher doses of ITF2357. Inhibition of histone deacetylase enzymes and reducing synthesis and release of several cytokines can explain the effect on platelet reduction. Thrombocytopenia is also reported for other HDAC inhibitors. The platelet count never dropped to critical values, even with higher doses, and consistently showed a rapid recovery after the discontinuation of the drug.
Safety is always a fundamental consideration when evaluating a drug, particularly when used in the treatment of diseases with no immediate danger to the patient. Although the single doses of 200 and 600 mg resulted in a marked decrease in cytokines, these doses were also associated with a greater lowering of platelets as well as gastrointestinal side effects. Therefore, the likely therapeutic dosing will be 50 or 100 mg/day in twice divided doses. A dose of 1.5 mg/kg divided in two doses was used to treat children with systemic-onset juvenile idiopathic arthritis. In that study, ITF2357 was safe and effective after 12 weeks of therapy (21). The equivalent dose in adult humans is approximately 100 mg total per day.
These studies also established that ITF2357 is an antiinflammatory agent in ex vivo cultures of whole blood. The present work expands the finding that HDAC inhibition reduces cytokine production from PBMCs in vitro, particularly cytokines relevant to autoimmune/ inflammatory diseases. The implications of these studies are that inhibition of HDAC by low oral doses of ITF2357 may be effective in the treatment of certain autoimmune and autoinflammatory diseases, particularly those currently treated with TNFα or IL-1β blockade. Inhibitors of HDAC increase as well as decrease an equal number of genes in the same cells (22). Although the general property of inhibitors of HDAC is one of increasing gene expression of proapoptotic genes in cancer cells (23), this property requires micromolar concentrations in vitro and comparable concentrations in vivo. At nanomolar concentrations, ITF2357 suppresses cytokines in LPS-stimulated PBMCs (19). Indeed, the concentrations of ITF2357, which are carried over into the whole blood cultures, were in the low nanomolar range, particularly at the dose of 100 mg at the 4-hour time point.
The precise mechanisms of cytokine inhibition by ITF2357 remain unclear. The acetylation state of the subunit p65 plays a critical role in the regulation of nuclear factor (NF)-κB (24). Such studies may explain the anti-cytokine effect of ITF2357, especially on TNFα production (19). Thus, an inhibition of NFκB- dependent transcription by the HDAC inhibitors could possibly contribute to suppression of inflammation. Whatever the mechanism of ITF2357 suppression of LPS-induced cytokines in the whole blood culture, the maximal reduction was consistently observed 4 hours after the 50- and 100-mg doses, not 2 hours after dosing (this latter time point being the maximal plasma concentration). These data suggest that the circulating cytokine-producing cells have been affected by exposure to ITF2357 in vivo and exhibit a reduced ability to produce LPS-induced cytokine hours later. Thus, the data do not support the concept that the suppression was due to the carryover of ITF2357 into the culture; rather, suppression appears to be via a mechanism that required at least 2 hours after maximal exposure to ITF2357.
The reduction in secretion of LPS-induced IL-1β is not inconsequential from a clinical perspective. Indeed, severe systemic inflammatory diseases are due to greater release of active IL-1β from cultured PBMCs in vitro compared with PBMCs from healthy subjects. These diseases include systemic-onset juvenile idiopathic arthritis (25), Muckle-Wells syndrome (26,27), familial cold- induced autoinflammatory syndrome (28), neonatal-onset multisystem inflammatory disease (29), hyper IgD syndrome (30) and Schnitzler’s syndrome (31). The increased secretion of IL-1β in these diseases is due to a single-point mutation in the NOD-like receptor family pryin domain containing 3 (NLRP3) gene, which controls the activation of procaspase-1 (27,32). Alternatively, ITF2357 may affect the ability to secrete IL-1β via the specialized secretory lysosomal pathway and activation of phosphatidyl-choline-specific phospholipase C (33). Another pathway in the secretion of IL-1β is the P2X7 receptor (34,35), which may also be affected by mechanisms of hyperacetylation.
Secretion of active IL-1β appears to be the likely mechanism by which there is a reduction in IL-1β activity. In the Leoni studies with suberoylanilide hydroxamic acid (10) as well as ITF2357 (19), there was no decrease in steady-state mRNA coding for IL-1β and no decrease in the amount of the IL-1β precursor. Thus, the likely mechanism for reduced IL-1β activity appears to be at the posttranslational level and specifically at processing the precursor to the active mature form. However, we have no evidence that ITF2357 inhibits caspase-1 activity. In a study by Carta et al. (36), suberoylanilide hydroxamic acid or ITF2357 prevented the exocytosis of IL-1β-containing secretory lysosomes. At nanomolar concentrations, ITF2357 reduced the secretion of IL-1β after ATP activation of the P2X7 receptor. Whereas the inhibition of HDACs results in hyperacetylation of tubulin, acetylation of HSP90 was unaffected. The reduction in IL-1β secretion appears to be due to disruption of microtubules impairing lyso-some exocytosis (36).
Although this study was a phase I trial on the safety and tolerability of ITF2357, the pharmacokinetic data are helpful in future trials, particularly concerning the twice-daily dosing schedules. The transient reduction in inducible cytokines at 50 and 100 mg in whole blood cultures can be viewed as both a mechanism of action at clinically achievable doses as well as a safety margin. Cytokine blockade such as the widespread use of anti-TNFα monoclonal antibodies are associated with increased opportunistic infections and possibly lymphoma. There appears to be a greater incidence of infections associated with long-lasting monoclonal antibodies such as infliximab and adalimumab compared with etanercept (37). One explanation for these differences affecting host responses to infection is the differences in half-lives of these agents. Infliximab has the longest duration in the circulation (weeks), whereas etanercept has the shortest (days). Therefore, the short duration of cytokine inhibition by ITF2357 may provide a greater parameter of safety compared with long-acting cytokine inhibitors.
ACKNOWLEDGMENTS
This study was supported by Italfarmaco, S.p.A., and by National Institutes of Health Grant AI-15614 (to CA Dinarello).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
CA Dinarello served as a consultant for Italfarmaco.
REFERENCES
- 1.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–87. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 2.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 3.Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3:344–51. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- 4.Kelly WK, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly WK, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–88. [PubMed] [Google Scholar]
- 6.Butler LM, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99:11700–5. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Della Ragione F, et al. Genes modulated by histone acetylation as new effectors of butyrate activity. FEBS Lett. 2001;499:199–204. doi: 10.1016/s0014-5793(01)02539-x. [DOI] [PubMed] [Google Scholar]
- 8.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–53. [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki H, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–9. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 10.Leoni F, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skov S, et al. Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood. 2003;101:1430–8. doi: 10.1182/blood-2002-07-2073. [DOI] [PubMed] [Google Scholar]
- 12.Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111:539–52. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy P, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101:3921–6. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly CM, et al. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. 2004;173:4171–8. doi: 10.4049/jimmunol.173.6.4171. [DOI] [PubMed] [Google Scholar]
- 15.Leng C, et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34:776–87. doi: 10.1016/j.exphem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Glauben R, et al. Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 2008;57:613–22. doi: 10.1136/gut.2007.134650. [DOI] [PubMed] [Google Scholar]
- 17.Glauben R, et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–22. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 18.Mishra N, Brown DR, Olorenshaw IM, Kammer GM. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc Natl Acad Sci U S A. 2001;98:2628–33. doi: 10.1073/pnas.051507098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leoni F, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med. 2011;17:397–403. doi: 10.2119/molmed.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vojinovic J, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:1452–8. doi: 10.1002/art.30238. [DOI] [PubMed] [Google Scholar]
- 22.Wakabayashi K, et al. Gene expression associated with the decrease in malignant phenotype of human liver cancer cells following stimulation with a histone deacetylase inhibitor. Int J Oncol. 2005;26:233–9. [PubMed] [Google Scholar]
- 23.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–6. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–7. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 25.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–12. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 27.Agostini L, et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman HM, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–85. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovell DJ, Bowyer SL, Solinger AM. Interleukin-1 blockade by anakinra improves clinical symptoms in patients with neonatal-onset multi-system inflammatory disease. Arthritis Rheum. 2005;52:1283–6. doi: 10.1002/art.20953. [DOI] [PubMed] [Google Scholar]
- 30.Drenth JP, Goertz J, Daha MR, van der Meer JW. Immunoglobulin D enhances the release of tumor necrosis factor-alpha, and interleukin-1 beta as well as interleukin-1 receptor antagonist from human mononuclear cells. Immunology. 1996;88:355–62. doi: 10.1046/j.1365-2567.1996.d01-672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Koning HD, et al. Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann Rheum Dis. 2006;65:542–4. doi: 10.1136/ard.2005.045245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffman HM, Wright FA, Broide DH, Wanderer AA, Kolodner RD. Identification of a locus on chromosome 1q44 for familial cold urticaria. Am J Hum Genet. 2000;66:1693–98. doi: 10.1086/302874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrei C, et al. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A. 2004;101:9745–50. doi: 10.1073/pnas.0308558101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solle M, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–32. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 35.Laliberte RE, Eggler J, Gabel CA. ATP treatment of human monocytes promotes caspase-1 maturation and externalization. J Biol Chem. 1999;274:36944–51. doi: 10.1074/jbc.274.52.36944. [DOI] [PubMed] [Google Scholar]
- 36.Carta S, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of micro-tubules. Blood. 2006;108:1618–26. doi: 10.1182/blood-2006-03-014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinarello CA. Differences between anti-tumor necrosis factor-alpha monoclonal antibodies and soluble TNF receptors in host defense impairment. J Rheumatol Suppl. 2005;74:40–47. [PubMed] [Google Scholar]