

Abstract
Cocaine binds and inhibits dopamine transporter (DAT), norepinephrine transporter (NET) and serotonin transporter. The residues forming cocaine binding sites are unknown. RTI-113, a cocaine analog, is 100x more potent at inhibiting DAT than inhibiting NET. Here we show that removing the hydroxyl group from residue Tyr151 in NET by replacing it with Phe, the corresponding residue in DAT, increased the sensitivity of NET to RTI-113, while the reverse mutation in DAT decreased the sensitivity of DAT to RTI-113. In contrast, RTI-31, another cocaine analog having the same structure as RTI-113 but with the phenyl group at the 2β position replaced by a methyl group, inhibits the transporter mutants equally well whether a hydroxyl group is present at the residue or not. The data suggest that this residue contributes to cocaine binding site and is close to the 2β position of cocaine analogs. These results are consistent with our previously proposed cocaine-DAT binding model where cocaine initially binds to a site that does not overlap with, but is close to, the dopamine-binding site. Computational modeling and molecular docking yielded a binding model that explains the observed changes in RTI-113 inhibition potencies.
1.0 Introduction
Cocaine inhibits the dopamine transporter (DAT), norepinephrine transporter (NET) and the serotonin transporter at similar concentrations and thus it is presumed that the cocaine binding sites are similar in the three transporters (Ritz et al., 1987), (Amara and Sonders, 1998), (Wu and Gu), (Han and Gu, 2006), (Beuming et al., 2006). Recently, the crystal structure of a leucine transporter (LeuTAa) from a bacterium, Aquifex aeolicus, has been solved (Yamashita et al., 2005), which is in the same superfamily of the monoamine transporters. It is believed that the LeuTAa structure provides the general overall structure for this family of proteins which is supported by the presence of the antidepressant binding site in LeuTAa and the mammalian monoamine transporters (Singh et al., 2007). However, the exact cocaine binding site is unknown. Some biochemistry and computer modeling studies propose that cocaine binds to a site overlapping with the dopamine binding site (Loland et al., 2008), (Beuming et al., 2008), (Schmitt et al., 2008), (Bisgaard et al., 2010). Our previous studies using molecular modeling and dynamics simulations suggest that cocaine binds initially to a site that does not overlap with, but is close to, the dopamine-binding site (Huang et al., 2009).
In contrast to cocaine, some cocaine analogs have very different affinities to DAT and NET (Carroll et al., 1995). Since DAT and NET share 67% sequence identity (Chen and Reith, 2002) and are believed to have similar structures and conformations (Singh, 2000), (Dutta et al., 2003), residues that are different between DAT and NET could be responsible for the analog affinity differences. It is possible that these residues are distal and can impact the packing within the core of the protein and the cocaine binding site. It is also possible that these residues are at the cocaine binding site and have direct interactions with specific functional groups on cocaine or cocaine analogs, allowing the identification of residues contributing to the cocaine binding site.
Many cocaine analogs have been generated and these compounds contain distinct chemical modifications to the cocaine structure (Carroll, 2003). In this study, we compared affinities to the cocaine analogs of DAT, NET and their mutants with reciprocal mutations. We identified interactions between certain residues in the transporters and a single group on the cocaine analogs.
2.0 Materials and Methods
2.1 Specific mutagenesis of mDAT and mNET
Plasmid DNA containing mouse DAT (mDAT) and mouse NET (mNET) were cloned into bluescript vector with a T7 promoter as described (Chen et al., 2005), (Wei et al., 2009). Using a ClustalW alignment screen (Thompson et al., 1994), the sequences of NET and DAT were aligned and the 12 transmembrane (TM) regions of mDAT and mNET were assigned (see below about the computational details). The starting and ending positions of the assigned TMs of NET were similar as that of structure-based alignment previously reported by Beuming et al. (Beuming et al., 2006). Residues in TM3, TM6, and TM8 that are different between mDAT and mNET were mutated using site-directed mutagenesis with mutagenic oligonucleotide primers. The mutants were then assayed for uptake activity and the sequences of the mutant constructs were determined by sequencing.
2.2 Substrate uptake into transiently transfected cells
HeLa cells (American Type Culture Collection, Rockville, MD) were grown in 96-well plates, infected with recombinant vTF-7 vaccinia virus, carrying the T7 polymerase gene, and transiently transfected with the plasmids bearing cDNAs using Lipofectin (Invitrogen Corp., Carlsbad, CA) as described previously (Chen et al., 2005).
About 20–24 h after transfection, HeLa cells were assayed for substrate uptake in 96-well plates using the PBS/Ca/Mg buffer as described (Hill et al., 2009). For the determination of Km and Vmax values, cells were incubated in PBS/Ca/Mg buffer containing 60 nM [3H]-labeled dopamine or norepinephrine in the presence of increasing concentrations of unlabeled monoamine substrates (0.1–20 μM) for 10 min at room temperature. For determination of IC50 values, transfected cells were incubated in the PBS/Ca/Mg buffer containing added 60 nM [3H]-labeled monoamine substrates and increasing concentrations of an inhibitor (e.g., cocaine, RTI-31, or RTI-113) for 10 min at room temperature. Substrate uptakes were terminated by two successive washes with PBS/Ca/Mg. Amounts of [3H]-labeled substrates accumulated in the cells were quantitated by liquid-scintillation counting. Protein concentrations were determined in triplicate using Bio-Rad dye and bovine serum albumin (gamma V) as the standard. Cells transfected with vehicle were used as controls and radioactivity associated with these cells were considered the background. This background was subtracted from the total scintillation counts of the wells.
The WT mNET and mDAT cDNAs were described previously (Han and Gu, 2006). [3H] labeled dopamine and norepinephrine were purchased from PerkinElmer (Boston, MA). Cold dopamine and norepinephrine were from Sigma–Aldridge (St. Louis, MO). Cocaine, RTI-31 and RTI-113 were synthesized at the Research Triangle Institute or provided by NIDA drug supply program.
2.3 Random mutagenesis of mDAT and mNET
To generate random mutations at mNET Tyr151/mDAT Phe155 position, PCR primers were used with nucleotides NNS (N being A, T, G, or C; and S being G or C) as the desired mutation codon. Nucleotides NNS encode for all amino acids but reduce the number of stop codons and increase the relative abundance of rare codons for Met and Trp. When necessary, additional primers were designed with specific nucleotides codon at the desired mutation site to encode for generating a specific mutant. The random mutants were then assayed for uptake activity and functional mutants were selected for further characterization. The sequences of the mutant constructs were determined by sequencing.
2.4 Data analysis
The Km, Vmax, and IC50 values were determined by a nonlinear regression analyses of one-site binding model concentration-response experimental data using GraphPad Prism 3.0 (San Diego, CA). The Km, Vmax, and IC50 values presented are averages ± standard error of means (SEM) calculated from 3 independent uptake experiments. Statistical analyses for the differences between the IC50 values between mDAT and mNET or between the wild type transporter and a mutant transporter were performed with one-way ANOVA followed by Dunnett’s post-hoc analysis using GraphPad Prism 5 (La Jolla, CA).
2.5 Computational details
2.5.1 Homology Modeling of NET and Molecular Docking
As well known, NET has a similar physiological function as DAT, i.e. transporting the neurotransmitter from the synaptic cleft to pre-synapse in the central nervous system (Torres et al., 2003). DAT and NET share 67% sequence identity (Chen and Reith, 2002) and the two transporters both co-transport Na+, Cl−, and the monoamine substrates dopamine and norepinephrine (Gu et al., 1994). It is reasonable to assume that these two transporters have high homology in structure. Instead of using the bacterial homolog LeuTAa as the template (Yamashita et al., 2005), the DAT model developed in our previous studies based on the LeuTAa structure (PDB entry of 2A65 at 1.65Å) (Huang and Zhan, 2007), (Huang et al., 2009) was used as the template to construct the structure of NET (more rationales in Discussion). The process of homology modeling for the structure of NET was performed in a similar way as that used for construction of DAT in our previous studies (Huang and Zhan, 2007), (Huang et al., 2009). Briefly, the amino acid sequence of NET was directly extracted from the NCBI databank (access No. CAA62566). Sequence alignment between NET and DAT was performed by using ClustalW with Blosum scoring function (Thompson et al., 1994), (Henikoff and Henikoff, 1992). The best alignment was selected according to both the alignment score and the reciprocal positions of conserved residues between NET and DAT, e.g. the residues around the Na+ binding site. The sequence identity is as high as 66.0% and the whole sequence homology became 87.4%. The TM regions of NET were assigned based on the aligned sequences and the corresponding positions of conserved residues in the TM regions of DAT, e.g. aromatic residues FYY (#150 to #152) at TM3 of NET correspond to aromatic residues FFY (#154 to #156) of TM3 of DAT. The starting and ending positions of the assigned 12 TMs of NET in the present study were similar with that of the structure-based alignment for neurotransmitter/Na+ symporters (Beuming et al., 2006). The first 51 residues at the N-terminal and the last 32 residues at the C-terminal of NET were omitted because there is no corresponding homolog sequence in the X-ray crystal structure of LeuTAa (Yamashita et al., 2005) and our previous DAT model (Huang and Zhan, 2007), (Huang et al., 2009). The coordinates of the conserved regions were directly transformed from the template structure, while the nonequivalent residues were mutated from the template to the corresponding ones of NET. The side chains of those nonconserved residues were relaxed by using the Homology module of InsightII (Version 2000.2, Accelrys, San Diego, CA) to remove the possible steric overlap or hindrance with the neighboring conserved residues.
After the initial NET structure was built, it was subject to energy minimization in the environment of a lipid bilayer and solvent water. The NET molecule was inserted into a preequilibrated palmitoyloleoyl phosphatidylcholine (POPC) phospholipid bilayer structure and then solvated by two layers of water molecules at each side of the phospholipid bilayer. Atomic charges and force field parameters for POPC molecules were directly adopted from those in our previous study (Huang and Zhan, 2007). The final system size was ~146 Å × 146 Å × 109 Å and composed of 141,208 atoms, including 351 POPC molecules and 28,536 water molecules. The energy minimization was carried out by using the Sander module of Amber 9 program suit (Case et al., 2006) with a nonbonded cutoff of 10 Å and a conjugate gradient energy-minimization method. The first 2000 steps were performed on the backbone of NET while the side chains were fixed, and then the next 50,000 steps on the side chain atoms, lipid bilayer molecules and water molecules. Finally the whole system was energy-minimized for 5000 steps on all atoms and a convergence criterion of 0.001 kcal*mol−1*A−1 was achieved.
Based on the constructed molecular model of NET, the binding mode of NET with its substrate norepinephrine was explored through molecular docking by using the AutoDock3.0.5 program (Morris et al., 1998). The atomic charges of the protonated norepinephrine were determined as the restrained electrostatic potential (RESP)-fitted charges determined by using the standard RESP procedure implemented in the Antechamber module of the Amber 9 program (Case et al., 2006) after the ab initio electronic structure and electrostatic potential calculations at the HF/6-31G* level. The similar RESP-fitting calculations based on the ab initio electronic structure method were used in our previous computational studies of other protein-ligand systems and led to the identification of reasonable binding structures that explained experimental data (Zhan et al., 2003), (Huang et al., 2005). During the AutoDock docking process, a conformational search was performed using the Solis and Wets local search method (Solis and Wets, 1981) and the Lamarckian genetic algorithm (Morris et al., 1998) was applied to deal with the NET- norepinephrine interactions. Among a series of docking parameters, the grid size was set to be 60 × 60 × 60 and the grid space was the default value of 0.375 Å. The possible binding site in NET was first roughly defined as a similar site as that for dopamine in the DAT structure (Huang et al., 2009), i.e., the cavity around transmembrane helices 1, 3, 6, and 8. All the obtained possible complex structures were evaluated and ranked in terms of geometric matching quality and binding affinity by using the standard scoring function implemented in the AutoDock 3.0.5 program (Morris et al., 1998). By visualizing the docking structures, the microscopic binding structure of NET-norepinephrine that had the best score and best geometric matching quality was selected. The NET-norepinephrine complex obtained from docking operations was further relaxed by energy minimization using the Sander module of the Amber9 program (Case et al., 2006). The energy minimization of NET-norepinephrine binding structure was performed in the same way as that of the NET as described above.
In order to explore how the norepinephrine-bound NET (here denoted by NET-NE) was inhibited by cocaine, RTI-31 and RTI-113, the possible binding modes of NET-NE with these three inhibitors were investigated by performing molecular docking and energy minimization. The RESP charges for all atoms of the protonated species of these inhibitors were determined in the same way as that for the RESP charges of norepinephrine. Our previous study suggested that cocaine was initially bound at a site which is near the extracellular end of substrate-entry pathway (Huang et al., 2009). This new cocaine-binding site did not overlap, but was close to the dopamine-binding site. After DAT made some conformational changes and expanded the binding site cavity, a cocaine molecule could move to and bind to DAT at a site overlapping the dopamine-binding site. Such a molecular mechanism of cocaine binding had reasonably explained all the experimental observations about cocaine blocking the transport of dopamine (Huang et al., 2009). When cocaine was bound at the new binding site we have identified, it interacted directly with residue F155 of DAT. In contrast, there was no direct molecular contact between residue F155 and cocaine bound to the overlap binding site model reported by Beuming et al (Beuming et al., 2008). Based on the structural similarity among the inhibitors cocaine, RTI-31 and RTI-113, the binding site in NET-NE for each of these inhibitors was first roughly assigned the similar site as the new cocaine binding site proposed in our previous study (Huang et al., 2009). The search area for the binding site in the NET-NE for each of these inhibitors was increased by changing the center and the size. The docking process for each of these inhibitors was performed in the same way as that of the docking with norepinephrine. The selected complex structure of NET-NE with each of these three inhibitors from docked candidates was energy-minimized by using the Amber 9 program (Case et al., 2006), and then visually checked for the geometric matching quality. In order to compare the difference in binding mode caused by different transporters for the same inhibitor, RTI-31 and RTI-113 molecules were also docked into the possible binding site at DAT-DA, and their complex structures with DAT-DA were also constructed and energy- minimized.
2.5.2 Mutational Modeling of NET and DAT
For NET-NE system, Y151F mutational modeling was performed in order to explore the role of residue Y151 on the inhibitory selectivity of each of these inhibitors as cocaine, RTI-31 and RTI-113 on NET-NE. As revealed by sequence alignment, residue F155 of DAT-DA is in the corresponding amino acid position as Y151 in NET-NE, and F155 of DAT-DA was involved in the hydrophobic interactions with cocaine as demonstrated in our previous studies (Huang et al., 2009). In order to explore whether F155 of DAT-DA plays an important role in the inhibitory selectivity of these three inhibitors, computational mutation was also performed on F155Y mutant of DAT-DA. The mutational modeling on F155Y of DAT-DA was also carried out to see whether the difference of amino acids at the same functional position between NET-NE and DAT-DA contributes to the observed difference in inhibitory effects for the same inhibitor (e.g. RTI-113) against these two different transporters, i.e. NET-NE and DAT-DA.
The modeling of Y151F mutation of NET-NE and F155Y mutation of DAT-DA was started from the modeled complex structures of NET-NE/inhibitor and DAT-DA/inhibitor, respectively. Each of the mutants was generated automatically by using a script developed in house at University of Kentucky and the X-leap module of Amber 9 program (Case et al., 2006). In order to more accurately reflect the structural perturbation from the mutation to the binding site for each of the NET-NE/inhibitor complexes, and for the convenience of rapid geometry refinement, we only needed to change the hydroxyl group on Y151 side chain of NET-NE into H atom for the Y151F mutation. For each of the DAT-DA/inhibitor structures, a hydroxyl group was added to replace the H atom on the side chain of F155 to generate F155Y mutant.
Once a mutant was generated, it was subject to energy minimization in order to optimize the interactions between the mutant and the inhibitor. The energy minimization was performed by using the Sander module of Amber 9 (Case et al., 2006), via a combined use of the steepest descent/conjugate gradient algorithms, with a convergence criterion of 0.01 kcal*mol−1*Å−1, and the non-bonded cutoff distance was set to 10.0 Å. The energy minimization was performed first on the mutated residue, and then on those residues which were within 5 Å distance around the mutated residue, and finally on the entire complex structure at the environment of lipid bilayer and solvent water. After energy minimization, we visually checked the entire mutant-type complex structures, in order to make sure that there was no any obvious structural distortion. Most of the modeling and MD simulations were performed on a supercomputer (e.g. IBM X-series Cluster with 340 nodes or 1360 processors) at the University of Kentucky Center for Computational Sciences. Some other modeling and computations were carried out on SGI Fuel workstations.
3.0 Results
Figure 1 highlights the similarities between chemical structures of cocaine and two cocaine analogs (RTI-31 and RTI-113). RTI-31 has the 3β ester removed from cocaine and a Chlorine added, resulting in a compound that is 100x more potent in inhibiting DAT and NET compared with cocaine (Carroll et al., 1995). When the methyl group on the 2β ester in RTI-31 is replaced with a phenyl group, the resulting compound (RTI-113) (Figure 1C) retains the same potency to DAT but is about 100 times less potent to NET (Carroll et al., 1995). Therefore, these cocaine analogs provide a unique tool to identify the critical residues that confer transporter specific affinity and can help identify the cocaine binding site.
Figure 1.
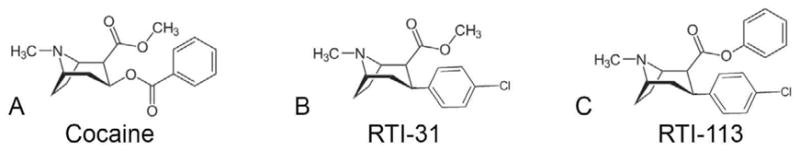
Chemical structures of monoamine transporter inhibitors. The chemical structures of cocaine (A), RTI-31 (B), and DAT-selective inhibitor RTI-113, (C).
Identifying residues interacting with the 2β position of a transporter selective cocaine analog involved three steps. First, reciprocal mutations were made at aqueous-facing residues that differ between mDAT and mNET. Next, the mutants were examined in uptake inhibition and kinetic assays. Individual mutants with altered analog specificities were selected and the residue position was randomly mutated to every amino acid to determine the basis of the interaction. Lastly, molecular simulations were performed to reveal the mechanism of the interaction.
3.1 Identifying the critical TM region
Identifying TM regions or residues in biogenic amine transporters that are critical in cocaine binding has been the subject of years of mutagenesis studies (Henry et al., 2006). The substituted-cysteine accessibility method (SCAM) pioneered by Kaback on lac permease was adapted to the biogenic amine transporters (Ferrer and Javitch, 1998). Extensive screening of the Serotonin transporter localized cocaine interaction with these transporters to sections of TM1 and TM3 (Chen et al., 1997), (Henry et al., 2003). These two TMs were also identified as having substrate interaction when the crystal structure of the Aquifex aeolicus leucine transporter (LeuTAa) was solved (Yamashita et al., 2005).
Figure 2 show that the WT mNET and mDAT expressed in cultured cells exhibited inhibition IC50 values similar to published results for cocaine and analogs (Carroll et al., 1995). The IC50 values for cocaine, RTI-31, and RTI-113 respectively were: for mNET, 398±66.8 nM, 2.04±0.96 nM, and 170±25.3 nM; and for mDAT, 318±126 nM, 0.91±0.52 nM, and 0.65±0.28 nM. We compared between the potencies of each drug in inhibiting mDAT versus mNET. One-way ANOVA followed by Dunnett’s post-hoc analysis was performed which showed that the IC50 values were significantly different for RTI-113 in inhibiting mDAT versus mNET (n=6, p<0.001) but not for cocaine and RTI-31.
Figure 2.

Inhibition of monoamine uptake. Wild type mNET (A) or mDAT (B) were transiently expressed in cultured cells. [3H] labeled norepinephrine or dopamine added to the cells in the presence of increasing concentration of cocaine, RTI-31 and RTI-113. The amount of substrate uptake was determined by scintillation counting. RTI-113 is significantly more potent at inhibiting substrate uptake for mDAT than mNET (p < 0.001, n = 6).
The crystal structure of the Aquifex aeolicus leucine transporter (LeuTAa) shares ~25% homology to mDAT and mNET and is proposed to be a bacterial homolog to the mammalian neurotransmitter transporters (Yamashita et al., 2005). The LeuTAa crystal structure indicates that TM1, TM3, TM6 and TM8 form the pocket or channel that likely contribute to the substrate and/or inhibitor binding sites (Yamashita et al., 2005). Based on this and the SCAM evidence, aqueous facing residues of these four transmembrane domains are more likely to interact with cocaine analogs and thus contribute to the transporter selectivity that some cocaine analogs exhibit.
First, we determined which TM region(s) is responsible for the different RTI-113 potencies observed between mDAT and mNET. Replacing multiple residues in TM6 or TM8 of mNET with the corresponding residues from mDAT slightly but significantly decreased the potencies of uptake inhibition by RTI-113 (Figure 3 and Table 1, p<0.05, n=3–6). However, when residues in TM3 of mNET were replaced by those from mDAT, the resulting mutant was 14-fold more sensitive to RTI-113 (Figure 3 and Table 1, p<0.001, n=6), suggesting that TM3 of mDAT has one or more residues that is at least partially responsible for the higher affinity of mDAT for RTI-113.
Figure 3.

Inhibition by RTI-113 of uptake by TM substituted mNET mutants. Mutations were made by replacing multiple residues in mNET TM3, 6, or 8 with the corresponding residues in mDAT. mNET-TM3m: Y151F/S159A/Y161H; mNET-TM6m: T306S/I315V/F316C/A321; and mNET-TM8m: A415T/L418I/S421A/A427S/A433V/D435E. Only mNET-TM3m showed a significant increase compared to mNET in RTI-113 inhibition potency of substrate uptake (p<0.001, n=3–6).
Table 1.
RTI-113 IC50 values for selected transporter mutants. One-way ANOVA was performed followed by Dunnett’s post-hoc analysis to see if the mNET mutants were significantly different from the wild type mNET.
| Transporter | RTI-113 IC50 [nM] uptake inhibition | Dunnett’s post-hoc versus mNET (n=3–6), p value |
|---|---|---|
| mNET | 170±25.3 | |
| mNET-TM8m | 269±35.8 | <0.05 |
| mNET-TM6m | 341±77.2 | <0.05 |
| mNET-TM3m | 1.89±1.29 | <0.001 |
|
| ||
| mDAT | 0.65±0.28 | <0.001 |
| mDAT F155Y | 13.4±1.9 | <0.001 (versus mDAT) |
|
| ||
| mNET Y151F | 12.1±0.8 | <0.001 |
| mNET Y151A | 130±15.1 | n/s |
| mNET Y151C | 211±23.0 | n/s |
| mNET Y151D | 306±105 | n/s |
| mNET Y151E | 770±55.3 | 0.001 |
| mNET Y151G | 165±28.4 | n/s |
| mNET Y151H | Low uptake | |
| mNET Y151I | 199±65.1 | n/s |
| mNET Y151K | 453±71.0 | <0.01 |
| mNET Y151L | 342±110 | n/s |
| mNET Y151M | 679±174 | <0.01 |
| mNET Y151N | 291±51.5 | n/s |
| mNET Y151P | Low uptake | |
| mNET Y151Q | 831±69.0 | <0.01 |
| mNET Y151R | Low uptake | |
| mNET Y151S | 99.3±43.8 | n/s |
| mNET Y151T | 115±33.2 | n/s |
| mNET Y151V | 181±81.1 | n/s |
| mNET Y151W | 256±66.9 | n/s |
3.2 Identifying the critical mNET151/mDAT155 residue
The individual residues different in TM3 of mNET and mDAT were further investigated by mutagenesis. Replacing Y151 in mNET with Phe, the corresponding residue in mDAT (mNET-Y151F) resulted in 14-fold increase in the potency for inhibition of substrate uptake by RTI-113, the cocaine analog with a phenyl group at the 2β position (Figure 4A, p<0.001, n=6). However, reciprocal mutations at the residues have no significant effect on the inhibition potency of RTI-31, the cocaine analog with a methyl group at the 2β position (Figure 4B. IC50 = 1.44±0.67 nM for Y151F, 2.04±0.96 nM for WT; p>0.05, n=3). The mNET-Y151F mutation has minimal effect on transporter function as compared to WT. The transport kinetic Km and Vmax values respectively were, 1.01±0.2 μM and 11.5±2.5 pmol/mg/min for WT mNET and 1.73±0.6 μM and 8.14±1.8 pmol/mg/min for mNET-Y151F.
Figure 4. Inhibition of mNET-Y151F and mDAT-F155Y by cocaine analogs.

The removal of a hydroxyl group at mNET residue 151 (Y151F) significantly increased the potency of RTI-113 (p<0.001, n=6) in inhibiting uptake, while the addition of a hydroxyl group at the corresponding residue in mDAT (F155Y) had significant opposite effect (p<0.001, n=6) (A). In contrast, these mutations did not significantly alter the inhibition potencies of RTI-31, (p>0.05, n=3–6) (B).
To further confirm that the presence of a hydroxyl group on this residue is responsible for the change in potency for RTI-113, the reverse mutation was performed in mDAT. As expected, adding the hydroxyl group to the residue in mDAT by F to Y substitution at corresponding position #155 decreased the potency for RTI-113 by 20-fold (Figure 4A, IC50s: 13.4±1.9 nM for mDAT-F155Y, 0.65±0.28 nM for WT; p<0.001, n=3). Similar to the mNET-Y151F mutation, the mDAT-F155Y mutation also has little effect on the binding of RTI-31 (Figure 4B, IC50s: 1.65±1.19 nM for mDAT-F155Y, 0.91±0.52 nM for WT; p>0.05, n=3). These data show that the presence of a tyrosine hydroxyl group at the mNET Y151 or mDAT F155 position is affecting the binding of cocaine analog with a phenyl group at the 2β position but has no effect if this phenyl group is replaced by a methyl group.
Next, to determine if other amino acid residues at this position could also affect this 2β-specific interaction, random mutagenesis was performed. Mutant mNET-Y151X transporters were created using degenerate primers at the #151 position. RTI-113 IC50 values for 17 NET-Y151X mutations were determined. None of these other mutations tested produced the large increases in IC50 values for RTI-113 that were observed with the Y151F mutation (Table 1). The decreased RTI-113 affinity results for some large and charged residues corresponds to previously observed decrease in cocaine inhibition of substrate potency with the same large residues at the Y151 position (Wei et al., 2009).
3.3 Binding mode of NET-NE with cocaine, RTI-31 and RTI-113
To elucidate the interaction of the tyrosine hydroxyl with RTI-113, molecular modeling and molecular docking were performed to gain insight on how the transporter residues might interact with cocaine analogs. As depicted in Figure 5, the overall structure of NET in each of the energy-minimized NET-NE/inhibitor complexes appears to be a shallow glasslike shape opening toward the extracellular side. The positively charged R81 from helix 1 forms a stable salt bridge with the negatively charged side chain of D473 from helix 10. Figure 5 shows each of the three inhibitors is oriented perpendicularly inside the vertical vestibule formed by helices 1, 3, 6, 8 and 10 of NET-NE, with the cationic head group of each of these inhibitors pointing to the extracellular end of the vestibule. The cationic head group of these inhibitors is located nearby the aromatic side chain of Y84 from helix 1, and packed with other surrounding residues as W80, V387, F388, Y467 and L469, and partly covered by and directly interact with the turning point of the “v-shaped” structure of extracellular loop 4 (EL-4) at residues G383 and A384 (typical GAGL motif). The cationic head group of these inhibitors has no direct cation-π interactions with the aromatic side chain of Y84 or Y467. Besides one intra-molecular hydrogen bonding with the carbonyl oxygen on the methyl ester group of cocaine or RTI-31 (or the 2β-carbophenoxy group of RTI-113), the proton at the cationic head group likely can form a hydrogen bond with a water molecule from the surrounding solvent as viewed from our energy-minimized structures. As observed in each of the NET-NE/inhibitor complex structures, the 3β-position (benzoyl ester group of cocaine or the 4-chlorophenyl group of RTI-31 and RTI-113) are all in close contacts with the aromatic side chains of Y151, Y152 and F317, and interacts tightly with the hydrophobic side chains of L76, A77 and I155. The aromatic side chain of Y152 packs perpendicularly with the 3β group. Meanwhile, the aromatic side chain of Y152 also packs in parallel with the phenyl group of the substrate norepinephrine. Such an interacting mode of Y152 with both the above inhibitor and the beneath substrate makes it impossible for the kinetic turnover of NET-NE complex, thus the substrate-transporting is blocked by the bound inhibitor.
Figure 5.
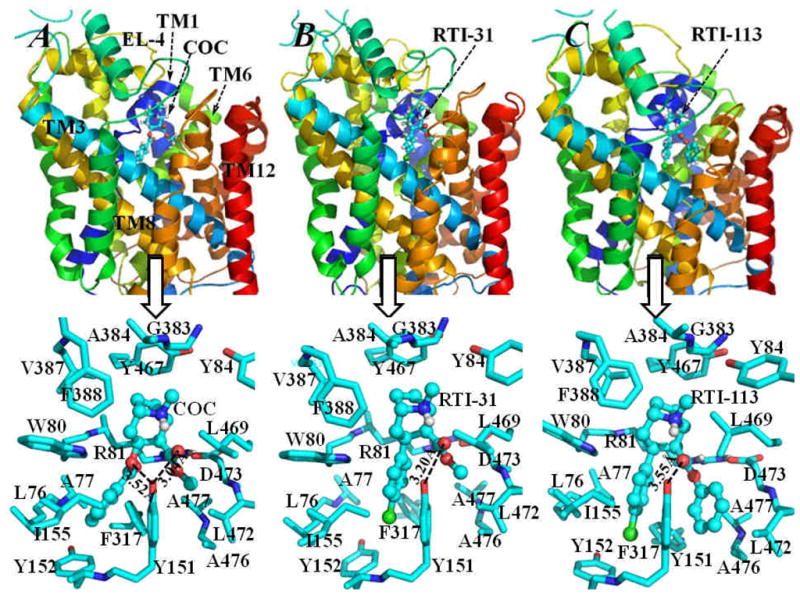
Energy-minimized structures of NET-NE/inhibitor complexes: (A) NET-NE/cocaine; (B) NET-NE/RTI-31; (C) NET-NE/RTI-113. The NET-NE protein is represented as colored ribbons and the inhibitors, i.e. cocaine (COC), RTI-31 and RTI-113, are shown in ball-and-stick style. The transmembrane helices 1, 3, 6, 8 and12, and extracellular loop 4 (EL-4) are also labeled. The low panel of this figure shows more details of inter-molecular interactions of NET-NE with each of the three inhibitors, residues of NET-NE within 5 Å around the inhibitor are shown as stick and colored by atom types. The distances between the hydroxyl oxygen on the side chain of Y151 of NET-NE and the carbonyl oxygen on the methyl ester group of both cocaine and RTI-31 are labeled. Also labeled is the distance between the hydroxyl oxygen of Y151 and the carbonyl oxygen on the benzoyl ester group of cocaine. For comparison, the distance between the hydroxyl oxygen of Y151 of NET-NE and the carbonyl oxygen on the 2β-carbophenoxy group of RTI-113 is also labeled.
Besides these common inter-molecular interactions for the NET-NE/inhibitor complexes, the largest differences in the binding mode come from the 2β subsite for the methyl ester group of cocaine and RTI-31, and the 3β subsite for the 4-chlorophenyl group of RTI-113. In the NET-NE/cocaine or the NET-NE/RTI-31 complex, the methyl ester group interacts with the side chains of several hydrophobic residues (such as Y151, L469, L472, and A477) and the backbone of D473. The aromatic side chain of Y151 packs at its left-side with the benzoyl ester group of cocaine or the 4-chlorophenyl group of RTI-31, and contacts on its right-side with the methyl ester group of cocaine or RTI-31. Although the hydroxyl group on the side chain of Y151 of NET-NE is close to the carbonyl oxygen of the methyl ester group of cocaine or RTI-31, and is also close to the carbonyl oxygen of the benzoyl group of cocaine, there is no direct hydrogen bonding interaction with each other. As observed in the NET-NE/RTI-113 complex structure, the 2β-carbophenoxy group of RTI-113 is surrounded by residues Y151, L418, L421, A425 and A426 of NET-NE. Besides the hydrophobic residue A425 of NET-NE is close to the 2β-carbophenoxy group of RTI-113, the aromatic side chain of Y151 packs on its left-side in parallel with the 3β subsite 4-chlorophenyl group of RTI-113, and on its right-side with the 2β-carbophenoxy group of RTI-113. The hydroxyl group on the side chain of Y151 points away from the carbonyl oxygen of the 2β-carbophenoxy group of RTI-113, i.e. no direct hydrogen bonding with each other. The binding mode of DAT-DA with RTI-31 is quite similar to that of DAT-DA/cocaine binding (data not shown). The RTI-113 also interacts with DAT-DA in a similar way as it interacts with NET-NE (data not shown). The significant difference in inter-molecular interactions between DAT-DA/RTI-113 and NET-NE/RTI-113 complexes exists in participating residue forming the π-π stacking with the 2β-carbophenoxy group of RTI-113. In DAT-DA/RTI-113 structure, the participating residue for the π-π stacking is F155, whereas this residue changes to Y151 in NET-NE/RTI-113 structure.
3.4 Mutational effects on the inhibitory selectivity of RTI-113
According to our mutational modeling resutls, the Y151F mutation of NET-NE still kept the π-π stacking between the Y151F mutant and the 2β-carbophenoxy group of RTI-113, and there is no significant change for the orientation of RTI-113 in the binding site (Figure 6). The F155Y mutation of DAT-DA also did not destroyed the π-π stacking between the F155Y mutant and the 2β-carbophenoxy group of RTI-113 (data not shown). At first glance, these modeling results are not helpful to understand our experimental observations that the Y151F mutation of NET-NE increased the inhibitory potency of RTI-113, and the F155Y mutation of DAT-DA decreased the inhibitory potency of RTI-113. However, the mutation from a residue of Y to a residue of F at the position #151 of NET-NE must result in the change of the strength for this particular π-π stacking interaction. Generally speaking, the higher electronic density, the stronger the π-π stacking will be (Churchill and Wetmore, 2009), (Geiss et al., 2009). Compared to Y151 residue in the wild-type, the Y151F mutant has an enhanced electronic density on the aromatic side chain of F151, because the electron-drawing group (i.e. hydroxyl group) is removed from the side chain of residue #151 after the Y151F mutation. Thus, F151 residue in the mutant helps to enhance the local binding between the NET-NE and the 2β-carbophenoxy group of RTI-113. This enhanced inter-molecular interaction becomes the structural determinant for the observed increase in the inhibitory potency of RTI-113 against the substrate-transport function of NET. On the other hand, the F155Y mutation of DAT-DA must decrease the electronic density on the side chain of residue #155. Thus the local π-π stacking interactions between the residue #155 of DAT-DA and the 2β-carbophenoxy group of RTI-113 are weakened by the F155Y mutation. So, the F155Y mutation on the binding affinity results in a decrease in the inhibitory potency of RTI-113 against the substrate-transport of DAT. It has been shown before that pi-pi interactions are improved changing a residue from F to Y (Geiss et al., 2009).
Figure 6. Impact of Y151F mutation on RTI-113 π-π stacking interactions.
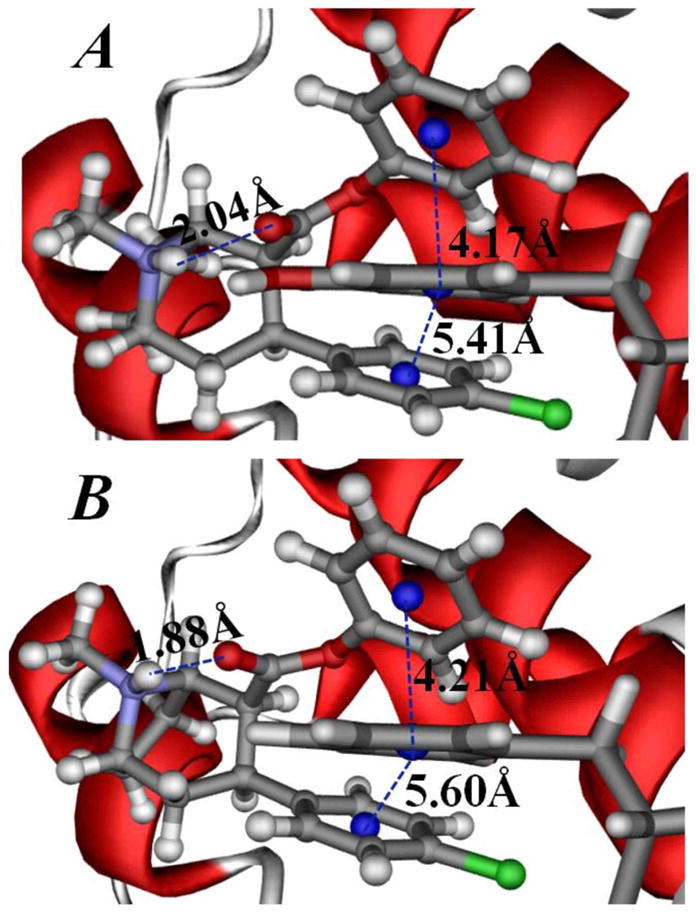
Representative π-π stacking interactions between the 2β-carbophenoxy group of RTI-113 and the aromatic side chain of residue #151 in the binding structures of wild-type NE-NE/RTI-113 complex (A) and Y151F mutant-type NET-NE/RTI-113 complex (B). The RTI-113 molecule is shown in ball-and-stick style. The NET-NE protein is represented by the colored ribbon and residue #151 is shown in stick style. Critical distances from the aromatic center of the 2β-carbophenoxy group and aromatic center of the 3β 4-chlorophenyl group of RTI-113 to the aromatic center of the side chain of residue #151 of NET-NE are labeled. Also labeled is the distance for the intra-molecular hydrogen bonding between the cationic head and the carbonyl oxygen at the 2β-carbophenoxy group of RTI-113.
4.0 Discussion
Before the LeuTAa structure was solved, years of SCAM analyses and species-scanning mutagenesis, random mutagenesis, and other approaches were used to identify putative aqueous facing residues of the biogenic amine transporters (Ferrer and Javitch, 1998), (Chen and Reith, 2002), (Henry et al., 2003). LeuTAa is a bacterial leucine transporter belonging to the neurotransmitter transporter family with 20–25% sequence identities to transporters such as DAT, NET, SERT etc. (Yamashita et al., 2005). The crystal structure of LeuTAa provides a structural template for other members of the neurotransmitter transporters. Structure and function studies and additonal SCAM analyses confirmed that LeuTAa and neurotransmitter transporters share structural features, but there are clear differences between the prokaryotic and eukaryotic transporters, including the absence of the cocaine binding site in LeuTAa (Rudnick, 2007), (Tao et al., 2009).
According to the LeuTAa structure, TM1, TM3, TM6, and TM8 form the pocket or channel that likely contribute to the substrate and/or inhibitor binding sites (Yamashita et al., 2005). Reciprocal mutations between mDAT and mNET in TM6 and TM8 did not have significant impact on the potency of RTI-113 in inhibiting uptake while reciprocal mutations in TM3 did (Figure 3). It is expected that most reciprocal mutations do not have an impact because cocaine and RTI-31 have the same potencies in inhibiting mDAT and mNET. We narrowed this TM3 potency change down to a single residue in TM3 (155 in mDAT and 151 in mNET) that is mostly responsible for the difference in RTI-113 sensitivity between mDAT and mNET. Removing the hydroxyl group from residue Tyr151 in mNET by replacing it with Phe, the corresponding residue in DAT, increased the sensitivity of NET to RTI-113, while the reciprocal mutation (DAT- F155Y) decreased the sensitivity of DAT to RTI-113. In contrast, RTI-31, another cocaine analog having the same structure as RTI-113 but with the phenyl group at the 2β position replaced by a methyl group, inhibits the transporter mutants equally well whether a hydroxyl group is present or absent at the residue. The data suggest that F155 in DAT (or Y151 in NET) contribute to cocaine binding site and is close to the 2β position of cocaine analogs.
There has been abundant evidence indicating the involvement in substrate and drug binding by residues in TM1, TM3, TM6 and TM8 of the monoamine transporters. The TM6 in human SERT was shown to contribute to an aqueously accessible binding pocket for serotonin and psychostimulants (Field et al., 2010). Y95 in TM1 and I172 (corresponding to V152 in DAT) in TM3 of human SERT has been shown to interact to establish high affinity recognition of antidepressants (Henry et al., 2006). Residue Y156 in human DAT, next to the key residue F155, was mutated to Ala and Cys and the resulting mutants exhibited no uptake function or cocaine analog binding (Beuming et at., 2008). Mutations of V152, the residue just below F155 in the α–helix model of TM3, to Ala, Ile, or Met resulted in DAT mutants with decreased affinity for DA uptake and cocaine analogs (Beuming et at., 2008). These studies are consistent with our current findings indicating the important roles of residues in this region.
We then attempted to understand how these mutations affect cocaine analog binding. We selected a DAT model we previously proposed (Huang et al., 2009) as the template to build the NET structural model because a DAT model is a much better starting point than the LeuTAa structure for building the NET model. As well known (Norregaard et al., 1998), (Torres et al., 2003), (Beuming et al., 2006), (Chen et al., 2007), NET is much closer to DAT than to LeuTAa in terms of sequence identity, physiological functions, ion dependence, substrate structure, inhibitor specificity, etc. Much of the information learned from structural studies of DAT can be applied to NET and vice versa, while many important structural features shared by DAT and NET are absent in LeuT. We built the DAT model based on the crystal structure of LeuT structure conserving the pseudo-two-fold axis between TMs 1–5 and TMs 6–10. In addition, we incorporated into the DAT model as much structural information learned from abundant structural studies of DAT that can be applied to NET. DAT has a unique Zn2+-binding site composed of residues H193, H375 and E396, and this Zn2+-binding site was located near the extracellular end of TM7, TM8 and extracellular loop 2 (EL-2) (Norregaard et al., 1998), (Loland et al., 2003). The corresponding conserved residues in NET are H372 and E393. Residue K189 of NET is at the corresponding position of residue H193 of DAT. Although the wild-type NET could not bind with Zn2+, the K189H mutant of NET was found to be able to bind with Zn2+ (Norregaard et al., 1998). Such results of Zn2+- probing (Norregaard et al., 1998), (Loland et al., 2003) indicated that the structure surrounding the Zn2+-binding site is conserved in DAT and NET. Our recent study (Chen et al., 2007) found that there is a disulfide bound (residues C180–C189) in the EL-2 of DAT. Such disulfide bond is conserved in NET (C176–C185), suggesting the conformation of EL-2 of NET is similar to the conformation of EL-2 of DAT. LeuTAa (Yamashita et al., 2005) does not have a corresponding Zn2+-binding site. The crystal structure of LeuTAa (Yamashita et al., 2005) does not have a disulfide bond in EL-2. The substrate transporting of LeuTAa is Cl−-independent, and therefore, LeuTAa does not have a Cl−-binding site. In contrast, both experimental and computational studies have confirmed the substrate-transporting of DAT or NET is Na+/Cl−-dependent, and the Cl−-binding site has been confirmed to be conserved from DAT to NET (Forrest et al., 2007), (Zomot et al., 2007). As shown in our previous studies (Huang and Zhan, 2007), (Huang et al., 2009), the refined DAT structure corresponds to the conformational state opening toward extracellular side, and has conserved the pseudo-two-fold axis between TMs 1–5 and TMs 6–10 as found in the LeuTAa crystal structure (Yamashita et al., 2005). However, our previous refined DAT model is not a suitable template to be used to model the other conformation states of NET (e.g. the intracellularly-open state). For the purpose to study cocaine-binding, our refined DAT model (Huang and Zhan, 2007), (Huang et al., 2009) is a reasonable selection as the template for the homology modeling of NET.
According to our published structural model of cocaine-bound DAT-DA, the 2β position of cocaine is within 3 Ǻ of residue Phe155 (Huang et al., 2009). The corresponding residue in NET is Tyr151. Replacing Tyr with Phe in NET (NET-Y151F) and thus removing the hydroxyl group at this position resulted in a large increase in the potency for RTI-113 (the 2β phenyl substituted cocaine analog), suggesting specific interactions or removal of steric inhibition. Introducing a hydroxyl group by replacing F with Y in DAT has the opposite effect (Figure 4A). Interestingly, this residue change has no effect on the potency of RTI-31, the cocaine analog with a methyl group at the 2β position, (Figure 4B) and only minimal impact on transporter function. Reciprocal mutations at the selected positions between mDAT and mNET did not affect the potencies of cocaine and other nonspecific monoamine uptake inhibitors (data not shown).
We utilized molecular modeling and molecular docking to determine binding structures after energy minimization of DAT-DA/inhibitor and NET-NE/inhibitor complexes constructed from docking operations. As described above, there are only general hydrophobic interactions between the side chain of residue #151 of NET-NE and the methyl ester group of cocaine or RTI-31. Hence, it can be expected that the Y151F mutation will not significantly change the inhibitory potency of cocaine or RTI-31. The same rationale can be applied to the case of the F155Y mutant-type DAT-DA/inhibitor complex formed by either cocaine or RTI-31 as residue #155 of DAT-DA has the same role in the inter-molecular interactions.
In order to quantitatively understand the experimental observations on the changes of inhibitory potency of RTI-113, we estimated the binding free energy difference, ΔΔGbind (the mutation-caused shift of the binding free energy), between the RTI-113 binding with Y151F mutant-type NET-NE and RTI-113 binding with the wild-type NET-NE by using the same molecular mechanics-Poison-Boltzmann surface area (MM-PBSA) method as used in our previous studies (Huang and Zhan, 2007), (Huang et al., 2009). On the basis of the binding free energy calculations, we obtained ΔΔGbind = ~−1.8 kcal/mol, indicating that the RTI-113 binding affinity is increased by ~1.8 kcal/mol after Y151F mutation of the transporter NET-NE. Using our experimentally measured IC50 values of RTI-113 before and after the Y151F mutation, the ΔΔGbind value for RTI-113 caused by the mutation is estimated to be ~−1.6 kcal/mol according to the relationship ΔΔGbind = RTln(IC50mut/IC50wt) at room temperature T = 298.15 K. Using the same method for the binding free energy calculations, the ΔΔGbind between the RTI-113 binding with F155Y mutant-type DAT-DA and the wild-type DAT-DA was calculated to be ~1.5 kcal/mol. Correspondingly, the experimentally derived ΔΔGbind value is ~1.8 kcal/mol based on the measured IC50 values of RTI-113. The agreement between the calculated ΔΔGbind value of ~1.5 kcal/mol and the experimentally derived ΔΔGbind value of 1.8 kcal/mol strongly supports the computational insights into the protein-ligand binding associated with the observed effects of the Y151F mutation of NET-NE and the F155Y mutation of DAT-DA on the inhibitory potency of RTI-113. Taken together with our results of calculated binding free energy differences between the wild type and the mutant-type, the data provide further support that the side chain hydroxyl group of Y151 in NET determines the inhibitory selectivity of RTI-113 between NET and DAT.
In conclusion, the data here suggest an interaction or close proximity of NET-151Y/DAT-155F with the 2β ester group on cocaine backbone-based monoamine uptake inhibitors. Interaction between the hydroxyl group of the tyrosine residue and the 2β phenyl group on the cocaine analog compound RTI-113 prevents tighter binding observed when the hydroxyl group is not present. Supporting evidence for this hydroxyl interaction theory comes from the serotonin transporter which has a tyrosine residue at the equivalent Y151/F155 position and has an RTI-113 IC50 that is similar to NET (Carroll, 2003). Due to the strong structural similarity between RTI-113 and cocaine, the data here strongly support that an initial cocaine binding site exists, which is separate and not overlapping the substrate binding site.
Abbreviations
- LeuTAa
Aquifex aeolicus leucine transporter
- DA
dopamine
- DAT
dopamine transporter
- IC50
half maximal inhibition constant
- NE
norepinephrine
- NET
norepinephrine transporter
- POPC
palmitoyloleoyl phosphatidylcholine
- RESP
restrained electrostatic potential
- TM
transmembrane domain
- WT
wild-type protein
Footnotes
Financial Support Disclosure: This work was supported by National Institutes on Drug Abuse grant #R01DA014610 (HHG), R01DA020124 (HHG), R01DA013930 (CGZ), and R01DA025100 (CGZ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. doi: 10.1016/s0376-8716(98)00068-4. [DOI] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- Bisgaard H, Larsen MA, Mazier S, Beuming T, Newman AH, Weinstein H, Shi L, Loland CJ, Gether U. The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology. 2010 doi: 10.1016/j.neuropharm.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI. 2002 Medicinal Chemistry Division Award address: monoamine transporters and opioid receptors. Targets for addiction therapy. J Med Chem. 2003;46:1775–1794. doi: 10.1021/jm030092d. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Kotian P, Dehghani A, Gray JL, Kuzemko MA, Parham KA, Abraham P, Lewin AH, Boja JW, Kuhar MJ. Cocaine and 3 beta-(4′-substituted phenyl)tropane-2 beta-carboxylic acid ester and amide analogues. New high-affinity and selective compounds for the dopamine transporter. J Med Chem. 1995;38:379–388. doi: 10.1021/jm00002a020. [DOI] [PubMed] [Google Scholar]
- Case D, Darden T, Cheatham, Simmerling C, Wang J, Duke R, Luo R, Merz K, Pearlman D, Crowley M, Walker R, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong K, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews D, Schafmeister C, Ross W, Kollman P. Amber. 2006:9. [Google Scholar]
- Chen JG, Sachpatzidis A, Rudnick G. The third transmembrane domain of the serotonin transporter contains residues associated with substrate and cocaine binding. J Biol Chem. 1997;272:28321–28327. doi: 10.1074/jbc.272.45.28321. [DOI] [PubMed] [Google Scholar]
- Chen N, Ferrer JV, Javitch JA, Justice JB., Jr Transport-dependent accessibility of a cytoplasmic loop cysteine in the human dopamine transporter. J Biol Chem. 2000;275(3):1608–14. doi: 10.1074/jbc.275.3.1608. [DOI] [PubMed] [Google Scholar]
- Chen N, Reith ME. Structure-function relationships for biogenic amine neurotransmitter transporters. In: Reith MEA, editor. Neurotransmitter Transporters: Structure, Function, and Regulation. Humana Press; Totowa, NJ: 2002. pp. 53–109. [Google Scholar]
- Chen R, Han DD, Gu HH. A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate. J Neurochem. 2005;94:352–359. doi: 10.1111/j.1471-4159.2005.03199.x. [DOI] [PubMed] [Google Scholar]
- Chen R, Wei H, Hill ER, Chen L, Jiang L, Han DD, Gu HH. Direct evidence that two cysteines in the dopamine transporter form a disulfide bond. Mol Cell Biochem. 2007;298:41–48. doi: 10.1007/s11010-006-9348-7. [DOI] [PubMed] [Google Scholar]
- Churchill CD, Wetmore SD. Noncovalent interactions involving histidine: the effect of charge on pi-pi stacking and T-shaped interactions with the DNA nucleobases. J Phys Chem B. 2009;113:16046–16058. doi: 10.1021/jp907887y. [DOI] [PubMed] [Google Scholar]
- Dutta AK, Zhang S, Kolhatkar R, Reith ME. Dopamine transporter as target for drug development of cocaine dependence medications. Eur J Pharmacol. 2003;479:93–106. doi: 10.1016/j.ejphar.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Ferrer JV, Javitch JA. Cocaine alters the accessibility of endogenous cysteines in putative extracellular and intracellular loops of the human dopamine transporter. Proc Natl Acad Sci U S A. 1998;95:9238–9243. doi: 10.1073/pnas.95.16.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field JR, Henry LK, Blakely RD. Transmembrane domain 6 of the human serotonin transporter contributes to an aqueously accessible binding pocket for serotonin and the psychostimulant 3,4-methylene dioxymethamphetamine. J Biol Chem. 2010;285(15):11270–80. doi: 10.1074/jbc.M109.093658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B. Identification of a chloride ion binding site in Na+/Cl -dependent transporters. Proc Natl Acad Sci U S A. 2007;104:12761–12766. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci U S A. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss BJ, Thompson AA, Andrews AJ, Sons RL, Gari HH, Keenan SM, Peersen OB. Analysis of flavivirus NS5 methyltransferase cap binding. J Mol Biol. 2009;385:1643–1654. doi: 10.1016/j.jmb.2008.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–7130. [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LK, Adkins EM, Han Q, Blakely RD. Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J Biol Chem. 2003;278:37052–37063. doi: 10.1074/jbc.M305514200. [DOI] [PubMed] [Google Scholar]
- Henry LK, Defelice LJ, Blakely RD. Getting the message across: a recent transporter structure shows the way. Neuron. 2006;49:791–796. doi: 10.1016/j.neuron.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hill ER, Tian J, Tilley MR, Zhu MX, Gu HH. Potencies of cocaine methiodide on major cocaine targets in mice. PLoS One. 2009;4:e7578. doi: 10.1371/journal.pone.0007578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong WC, Amara SG. Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J Biol Chem. 2010;285:32616–32626. doi: 10.1074/jbc.M110.150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Gu HH, Zhan CG. Mechanism for cocaine blocking the transport of dopamine: insights from molecular modeling and dynamics simulations. J Phys Chem B. 2009;113:15057–15066. doi: 10.1021/jp900963n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhan CG. How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophys J. 2007;93:3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zheng F, Crooks PA, Dwoskin LP, Zhan CG. Modeling multiple species of nicotine and deschloroepibatidine interacting with alpha4beta2 nicotinic acetylcholine receptor: from microscopic binding to phenomenological binding affinity. J Am Chem Soc. 2005;127:14401–14414. doi: 10.1021/ja052681+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol Pharmacol. 2008;73:813–823. doi: 10.1124/mol.107.039800. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Norgaard-Nielsen K, Gether U. Probing dopamine transporter structure and function by Zn2+-site engineering. Eur J Pharmacol. 2003;479:187–197. doi: 10.1016/j.ejphar.2003.08.068. [DOI] [PubMed] [Google Scholar]
- Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson A. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- Norregaard L, Frederiksen D, Nielsen EO, Gether U. Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 1998;17:4266–4273. doi: 10.1093/emboj/17.15.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Rudnick G. What is an antidepressant binding site doing in a bacterial transporter? ACS Chem Biol. 2007;2:606–609. doi: 10.1021/cb7001818. [DOI] [PubMed] [Google Scholar]
- Schmitt KC, Zhen J, Kharkar P, Mishra M, Chen N, Dutta AK, Reith ME. Interaction of cocaine-, benztropine-, and GBR12909-like compounds with wild-type and mutant human dopamine transporters: molecular features that differentially determine antagonist-binding properties. J Neurochem. 2008;107:928–940. doi: 10.1111/j.1471-4159.2008.05667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. Chemistry, design, and structure-activity relationship of cocaine antagonists. Chem Rev. 2000;100:925–1024. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Solis FJ, Wets RJB. Minimization by Random Search Techniques. Mathematics of Operations Research. 1981;6:19–30. [Google Scholar]
- Tao Z, Zhang YW, Agyiri A, Rudnick G. Ligand effects on cross-linking support a conformational mechanism for serotonin transport. J Biol Chem. 2009;284:33807–33814. doi: 10.1074/jbc.M109.071977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Wei H, Hill ER, Gu HH. Functional mutations in mouse norepinephrine transporter reduce sensitivity to cocaine inhibition. Neuropharmacology. 2009;56:399–404. doi: 10.1016/j.neuropharm.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Gu HH. Cocaine affinity decreased by mutations of aromatic residue phenylalanine 105 in the transmembrane domain 2 of dopamine transporter. Mol Pharmacol. 2003;63:653–658. doi: 10.1124/mol.63.3.653. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhan CG, Zheng F, Landry DW. Fundamental reaction mechanism for cocaine hydrolysis in human butyrylcholinesterase. J Am Chem Soc. 2003;125:2462–2474. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Rudnick G. The cytoplasmic substrate permeation pathway of serotonin transporter. J Biol Chem. 2006;281:36213–36220. doi: 10.1074/jbc.M605468200. [DOI] [PubMed] [Google Scholar]
- Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI. Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature. 2007;449:726–730. doi: 10.1038/nature06133. [DOI] [PubMed] [Google Scholar]