


Abstract
The SET- and MYND-domain containing (Smyd) proteins constitute a special subfamily of the SET-containing lysine methyltransferases. Here we present the structure of full-length human Smyd3 in complex with S-adenosyl-l-homocysteine at 2.8 Å resolution. Smyd3 affords the first example that other region(s) besides the SET domain and its flanking regions participate in the formation of the active site. Structural analysis shows that the previously uncharacterized C-terminal domain of Smyd3 contains a tetratrico-peptide repeat (TPR) domain which together with the SET and post-SET domains forms a deep, narrow substrate binding pocket. Our data demonstrate the important roles of both TPR and post-SET domains in the histone lysine methyltransferase (HKMT) activity of Smyd3, and show that the hydroxyl group of Tyr239 is critical for the enzymatic activity. The characteristic MYND domain is located nearby to the substrate binding pocket and exhibits a largely positively charged surface. Further biochemical assays show that DNA binding of Smyd3 can stimulate its HKMT activity and the process may be mediated via the MYND domain through direct DNA binding.
INTRODUCTION
It has been well established that covalent modifications of histones are involved in the regulation of chromatin structure and function and hence play critical roles during development and disease pathogenesis (1). Histone methylation, one of the major forms of histone modification, has been shown to exert important functions in various biological processes such as heterochromatin formation, X-chromosome inactivation and transcriptional regulation (2). Methylation of histones can occur at different lysine or arginine residues and is correspondingly catalyzed by various methyltransferases. Since about a decade ago when it was discovered that the SET domain-containing proteins are able to selectively methylate lysine residues of histones (3,4), the SET family histone lysine methyltransferases (HKMTs) have been found to be responsible for methylation of all (H3K4, H3K9, H3K27, H3K36 and H4K20) but one lysine residue (H3K79) of histones (5).
In contrast to the vast majority of the SET-containing lysine methyltransferase, five proteins share a special characteristic with the SET domain being split by a myeloid-Nervy-DEAF-1 (MYND) insertion, and thus cluster to a subfamily named SET- and MYND-domain containing proteins (Smyd) (6,7). In the Smyd subfamily, three proteins have been proven to possess methyltransferase activities: Smyd1 is able to specifically methylate histone H3-Lys4 (H3K4) (8); Smyd2 harbors a methyltransferase activity towards histone H3-Lys36 (H3K36) (7,9), p53-Lys370 (10), and probably H3K4 as well (9); and Smyd3 specifically catalyzes di- and tri- methylation of H3K4 (6) and methylation of Lys831 of vascular endothelial growth factor receptor 1 (VEGFR1) (11).
Consistent with the HKMT function of Smyd3 for methylation of H3K4 which is a hallmark of active gene transcription (12–14), Smyd3 has been demonstrated to bind with RNA helicase HELZ and hence is associated with RNA polymerase II for transcription elongation (6). The other substrate of Smyd3, VEGFR1, plays an important role in regulation of angiogenesis and has been shown to be involved in inflammatory responses, tumor growth and atherosclerosis (15). Given the functional roles of Smyd3 in gene transcription and the VEGFR1 signaling pathway, it is not surprising that dysregulation of Smyd3 is involved in disease pathogenesis. Smyd3 was originally identified as a gene overexpressed in hepatocellular carcinoma (HCC) and colorectal carcinoma (CRC) cells (6). Upregulation of Smyd3 promoted the growth of HCC and CRC cells (6), and the presence of three tandem repeats of an E2F-1-binding element in the Smyd3 promoter region is a risk factor for HCC, CRC and breast cancer (16).
Despite the important functional roles of Smyd3 and its association with cancers, the knowledge about the molecular mechanism of its methyltransferase activity and the regulation of the enzymatic activity is quite limited. It has been suggested that the MYND domain might bind specific DNA elements, and a chaperon, namely heatshock protein 90 α (Hsp90α) has been demonstrated to interact with Smyd3 and enhance its HKMT activity in a dose-dependent manner (6). However, the potential function and structural basis of the DNA binding by the MYND domain of Smyd3, and the mechanism underlying the regulation of the HKMT activity of Smyd3 by Hsp90α remain unclear. In addition, more fundamental questions about Smyd3 and the other Smyd proteins are in queue to be answered. For example, it has been well known that the ‘pre-SET’ and ‘post-SET’ domains are necessary for the activity of SUV39H1 (3); however, for the Smyd subfamily members, there is no ‘pre-SET’ domain, and the exact function of the ‘post-SET’ domain has not been investigated yet. In addition, all Smyd proteins except Smyd5 have a large C-terminal region, but the structure and function of the region are still unknown.
We carried out structural and functional studies of human Smyd3, and present here the crystal structure of Smyd3 in complex with the cofactor product, S-adenosyl-l-homocysteine (AdoHcy) at 2.8 Å resolution. Comparison of the Smyd3–AdoHcy complex with the reported structures of other SET enzymes (17–29) reveals unique structural characteristics of Smyd3. Based on the structural information, we further performed mutagenesis analyses and biochemical assays. Together, our results reveal the unique features of Smyd3 compared with the other SET methyltransferases, and provide insights into the structural basis and regulatory mechanism of the HKMT activity of Smyd3.
MATERIALS AND METHODS
Protein expression and purification
The full-length human Smyd3 gene was amplified with PCR from HEK293 cDNA and was subcloned into the pET-28a and pET-28b vectors (Novagen) with a His6 tag at the N-terminus or the C-terminus. The constructed plasmids were transformed into Escherichia coli BL21 (DE3) Codon Plus strain. The bacterial cells were grown in LB medium at 37°C to OD600 of 0.6, and induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside at 16°C for 24 h. The cells were collected by centrifugation at 6000 g, suspended in a lysis buffer [50 mM Tris–HCl (pH 8.0), 300 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol and 1 mM phenylmethylsulfonyl fluoride], and lysed on ice by sonication. The cell lysate was precipitated by centrifugation at 18 000 g for 30 min, and the supernatant was used for protein purification.
The human Smyd3 protein was purified by affinity chromatography using a Ni2+-NTA column (Qiagen) equilibrated with a binding buffer [20 mM Tris–HCl (pH 8.0), 300 mM NaCl and 5 mM β-mercaptoethanol]. The column was washed with the binding buffer supplemented with 30 mM imidazole, and then the target protein was eluted with the binding buffer supplemented with 200 mM imidazole. The protein sample was further purified by gel filtration using Superdex 200 16/60 column (Amersham Biosciences). Finally, half of the protein was stored in storage buffer A containing 20 mM Tris–HCl (pH 8.0), 100 mM NaCl and 1 mM DTT, while the rest in buffer B containing 20 mM Tris–HCl (pH 8.0), 50 mM Li2SO4 and 1 mM DTT.
The Smyd3 mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene) and verified by sequencing. Expression and purification of the mutant proteins were performed following the same procedure as for the wild-type protein.
Crystallization, diffraction data collection and structure determination
The purified C-terminally tagged Smyd3 protein was concentrated to 5–10 mg/ml in storage buffers (A and B) and then incubated with 600 μM AdoHcy. Crystallization was performed using the hanging drop vapor diffusion method. Crystals belonging to space group P21 (form I) were obtained at 4°C with an equal volume of the protein in storage buffer A and the reservoir solution containing 80 mM sodium cacodylate (pH 6.5), 160 mM calcium acetate, 14.4% polyethylene glycol 8000 and 20% glycerol. Crystals belonging to space group P212121 (form II) were obtained with the protein in storage buffer B and the reservoir solution containing 2.8 M sodium acetate (pH 7.0).
The purified N-terminally tagged Smyd3 protein was concentrated to 7.5–15 mg/ml in storage buffer A, and then incubated with 600 μM AdoHcy. Crystals belonging to space group P61 (form III) were obtained using the sitting drop vapor diffusion method with the reservoir solution containing 0.1 M bicine (pH 9.0), 0.1 M NaCl and 20% polyethylene glycol monomethyl ether 550.
The diffraction data were collected from flash-cooled crystals at 100 K at beamline 17U of Shanghai Synchrotron Radiation Facility, China. The diffraction data were processed, integrated and scaled together with HKL2000 (30). The statistics of the diffraction data are summarized in Table 1. The structure of Smyd3 in complex with AdoHcy was solved by the molecular replacement method using CNS (31) with the structure of Smyd3 in complex with S-adenosylmethionine (AdoMet) as the search model (PDB code 3MEK). The model building was performed using Coot (32), and the structure refinement was carried out using CNS (31) and REFMAC5 (33). The stereochemical geometry of the structures was analyzed using Procheck (34). The figures were generated using Pymol (http://www.pymol.org). The statistics of the structure refinement and the quality of the final structure models are also summarized in Table 1. The structural model derived from form I crystals has the highest resolution and best quality. For this crystal form, there are two molecules in an asymmetric unit, and the NCS restraint was applied during the initial refinement but released in the later stage of refinement. The two molecules assume almost identical overall structures [superposition of all Cα atoms yields a root-mean-square deviation (RMSD) of 0.27 Å], and the molecule with more detectable residues and better electron density was chosen for structural analyses and discussion in this article.
Table 1.
Summary of diffraction data and structure refinement statistics
| Form I | Form II | Form III | |
|---|---|---|---|
| Diffraction data | |||
| Wavelength (Å) | 0.99985 | 0.97908 | 0.97908 |
| Space group | P21 | P212121 | P61 |
| Cell parameters | |||
| a, b, c (Å) | 58.0, 118.0, 82.8 | 55.0, 101.0, 117.3 | 103.4, 103.4, 112.2 |
| α, β, γ (°) | 90.0, 91.8, 90.0 | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 50.0–2.8 (2.90–2.80)a | 50–3.6 (3.73–3.60)a | 50–3.4 (3.52–3.40)a |
| Observed reflections | 86 956 | 29 147 | 69 514 |
| Unique reflections (I/σ(I) > 0) | 26 338 | 6662 | 9395 |
| Average redundancy | 3.3 (3.0) | 4.4 (3.0) | 7.4 (5.9) |
| Average I/σ(I) | 9.8 (2.9) | 10.1 (2.1) | 16.6 (2.5) |
| Completeness (%) | 97.9 (94.4) | 82.8 (68.4) | 99.9 (100) |
| Rmerge (%)b | 12.2 (33.9) | 12.6 (33.3) | 13.0 (64.9) |
| Refinement and structure model | |||
| Reflections (Fo ≥ 0σ(Fo)) | |||
| Working set | 24 970 | 6299 | 8866 |
| Test set | 1323 | 340 | 487 |
| R factor/free R factor (%)c | 21.1/26.1 | 24.1/28.3 | 22.4/24.9 |
| Number of non-H atoms | 6896 | 3443 | 3426 |
| Number of amino acid residues | 851 | 426 | 424 |
| Number of water molecules | 24 | – | – |
| Average B factor (Å2) | |||
| All atoms | 40.3 | 103.4 | 97.4 |
| Protein | 40.3 | 103.4 | 97.8 |
| Ligand/ion | 39.8/39.4 | 108.1/104.4 | 54.7/58.6 |
| Water | 24.6 | – | – |
| RMSD | |||
| Bond lengths (Å) | 0.007 | 0.009 | 0.008 |
| Bond angles (°) | 1.1 | 1.2 | 1.1 |
| Ramachandran plot (%) | |||
| Most favoured regions | 92.1 | 83.6 | 86.2 |
| Allowed regions | 7.7 | 15.9 | 12.5 |
| Generously allowed regions | 0.3 | 0.5 | 1.3 |
aNumbers in parentheses represent the highest resolution shell.
b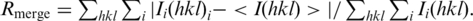
c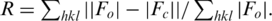
Gel shift assay
Different amounts of the wild-type and mutant Smyd3 proteins were incubated with 1 μg 6 bp DNA (5′-CCCTCC-3′) in a buffer containing 50 mM Tris–HCl (pH 7.4), 100 mM NaCl and 1 mM DTT. After incubation at 30°C for 2 h, the samples were loaded to a 2% agarose gel and visualized under UV with Gel Green staining.
In vitro HKMT activity assay
For the HKMT activity assay, 10 μg each of the wild-type and mutant Smyd3 proteins were incubated with 40 μg histone mixture extracted from calf thymus (Sigma) along with 0.5 μCi [methyl-3H]-S-adenosylmethionine (PerkinElmer Life Sciences) as the methyl donor for 2 h at 30°C, in a total volume of 40 μl with a buffer containing 50 mM Tris–HCl, 100 mM NaCl and 1 mM DTT. The HKMT activity was analyzed by liquid scintillation counting. As a negative control, the background reading was measured from the assay system containing the wild-type Smyd3 and the labeled cofactor but no histone mixture, which was subtracted from the readings of the other assay experiments. For some Smyd3 mutants, the experimental readings were slightly lower than the background reading, resulting in the apparent ‘negative activities’ after subtraction of the background reading. This also indicates that these mutants had no detectable enzymatic activity towards H3K4. The HKMT activity of Smyd3 increases substantially with the increase in pH (Supplementary Figure S1). For analyses of the effect of mutations of residues at the active site, the assays were performed at pH 8.0. To examine the stimulatory effect by DNA, the HKMT activities of the wild-type Smyd3 and the R66E mutant were measured at the physiological pH 7.4 with and without the 6 bp potential target DNA (5′-CCCTCC-3′).
RESULTS AND DISCUSSION
Overall structure
Crystallization of the full-length human Smyd3 in complex with the cofactor product AdoHcy was carried out, and three different forms of crystals belonging to three different space groups (P21, P212121 and P61) have been obtained (Table 1). The structures derived from the three forms of crystals are all similar with AdoHcy and three Zn2+ ions bound at similar positions, and the one (form I) refined to the highest resolution (2.8 Å) was used for further structural analysis and discussion. As shown in Figure 1A and B, the Smyd3–AdoHcy complex assumes a compact globular structure. The N-terminal region including the SET domain (residues 1–43 and 94–242), the MYND domain (residues 44–93) and the post-SET domain (residues 243–270) has a mixed structure consisting of α helices (α1–α7), β-strands (β1–β12) and long extended loops, while the C-terminal region (residues 271–428) is comprised of mainly α helices (α8–α15) (Figure 1A and Supplementary Figure S2). Our Smyd3–AdoHcy structure is very similar to the Smyd3 structure in complex with AdoMet (PDB code 3MEK) which was derived from a crystal of space group P212121 with an RMSD of 0.9 Å for all Cα atoms. In addition, the structures of the active site are almost identical in the two complexes with AdoHcy and AdoMet bound in a similar mode.
Figure 1.

Structure of the Smyd3–AdoHcy complex. (A) Overall structure of the Smyd3–AdoHcy complex. Top: a schematic representation of the full-length Smyd3 with the N-terminal SET domain (residues 1–43 and 94–242), the MYND domain (residues 44–93), the post-SET domain (residues 243–270), and the C-terminal region (residues 271–428) colored in magenta, yellow, cyan and blue, respectively. Bottom: two views of the overall structure of the Smyd3–AdoHcy complex. The domains are colored accordingly and the secondary structure elements are marked. The cofactor product AdoHcy is shown with a ball-and-stick model and colored in cyan. (B) Structural comparison of the SET and post-SET domains of Smyd3 with the equivalent regions of SET7/9 (PDB code 1O9S). Superposition of the Smyd3 and Set7/9 structures was performed based on the core region of the SET domain. SET7/9 is colored in green, and the color coding for Smyd3 is the same as in Figure 1A. The cofactors are shown with ball-and-stick models and colored accordingly. (C) Comparison of the Zn2+-binding site in the catalytic core of Smyd3 with the equivalent regions of SET7/9 (left panel) and Dim 5 (PDB code 1PEG, right panel). The post-SET regions of Smyd3, SET7/9 and Dim5 are shown with ribbon representations and colored in cyan, green and wheat, respectively. The side chains of the involved Cys residues and the bound cofactors are shown with ball-and-stick models and colored accordingly. The Zn2+ ions are shown with sphere models and colored accordingly. The secondary structure elements and the involved Cys residues in Smyd3 are labeled. (D) Zinc-binding sites in the MYND domain. The MYND domain (yellow) is characterized by a C6HC zinc chelating motif. The side chains of the Cys and His residues chelating the two Zn2+ ions are shown with ball-and-stick models. The Zn2+ ions are shown with sphere models. The secondary structure elements and the involved residues are labeled.
The overall architecture of the SET domain of Smyd3 is similar to the other SET domains of HKMTs. The core region (residues 1–40 and 180–242) of the SET domain is comprised largely of three canonical β sheets (β1 and β2; β3, β10 and β11; and β4, β7 and β8) and extended loops. Structural comparison of the SET domain of Smyd3 (Smyd3-SET) with those of the other SET methyltransferases (17–21,23–29,35–37) especially the H3K4-specific enzymes SET7/9 (18,21,23,25) and MLL1 (29) shows that the SET domain of Smyd3 mostly resembles that of SET7/9 with an RMSD of 2.2 Å based on superposition of the core region of the SET domain (Figure 1B). In addition, in a surface pocket formed by the SET and post-SET domains, the cofactor product AdoHcy binds in the same position and configuration as those in the SET7/9–AdoHcy complex (Figure 1B), and the overall structure of the cofactor binding site is generally similar to that of SET7/9 (see details later). However, unlike SET7/9, the post-SET domain of Smyd3 is Cysteine-rich and a Zn2+ ion is bound and coordinated by residue Cys208 of the SET domain, and residues Cys261, Cys263 and Cys266 of the post-SET domain (Figure 1C, left panel). Comparison of Smyd3 with the other methyltransferases containing a Cysteine-rich post-SET domain such as MLL1 (29), Dim5 (22,38) and Suv39H2 (17) shows that the position and coordination of the bound Zn2+ in these enzymes are similar; however, the overall structures of the post-SET domains are quite different: the post-SET domain of Smyd3 is comprised of an N-terminal α helix (α6), a loop and a C-terminal α helix (α7), whereas that of the other enzymes mainly forms a long loop (Figure 1C, right panel). Intriguingly, although the post-SET region of Smyd3 shares little sequence similarity with that of SET7/9, the N-terminal part of the post-SET region of Smyd3 resembles that of SET7/9 by forming an α helix (α6) (Figure 1C, left panel). These results demonstrate that Smyd3 has a distinctive Zn2+-binding post-SET motif.
The characteristic MYND domain forms a relatively independent structure, comprised of mainly a long extended loop, a short β sheet (β5 and β6) and two α helices (α1 and α2) (Figure 1D). The MYND domain is characterized by a C6HC zinc chelating motif, and as expected, the MYND domain in our structure is bound with two Zn2+ ions (Figure 1D).
Tetratrico-peptide repeat domain
Search with the Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server) reveals that the C-terminal region is mainly comprised of a tetratrico-peptide repeat (TPR) domain (residues 280–428, α9–α15 and η5) consisting of three TPR motifs which are degenerate 34 amino acid sequences and assume helix-turn-helix structures (Supplementary Figure S3). TPR repeats have been found in many proteins to mediate protein–protein and sometimes, protein–lipid interactions. It has been reported that Hsp90α binds to Smyd3 to enhance its methyltransferase activity (6), and intriguingly, numerous TPR domain-containing proteins, including Hop, CyP40, FKBP51/52 and p23, have been shown to bind to Hsp90 (39–42). In the crystal structure of the C-terminal TPR2 domain of Hop in complex with a C-terminal pentapeptide (MEEVD) of Hsp90, the peptide binds to a helical groove and is anchored to the TPR domain of Hop mainly through interactions of a highly conserved two-carboxylate clamp of the peptide with five residues of TPR2 which are also conserved in the TPR1 domain of Hop (43). Although the overall configuration of the TPR domain of Smyd3 is similar to TPR2 of Hop (RMSD of 4.9 Å based on 128 Cα atoms), the C-terminal region of the second TPR motif (α12) forms a long distorted α helix and hence the α helices at the C-terminus are almost perpendicular to their corresponding regions in the structure of the Hop–Hsp90 peptide complex. The molecular mechanism underlying the Smyd3–Hsp90α interaction is under investigation.
Histone binding pocket
Our attempts to obtain a structure of Smyd3 in complex with the histone peptide were unsuccessful. The Smyd3–AdoHcy structure was superposed to the structure of SET7/9 in complex with a methylated histone peptide (PDB code 1O9S) (25) (Figures 1B and 2A). In SET7/9, a narrow lysine channel is detected connecting the cofactor binding site and the histone peptide binding site (25). In the Smyd3–AdoHcy complex, a similar channel is present (see details later) and at one end of the channel AdoHcy binds at an almost identical position as in the SET7/9 complex (Figure 1B). Thus, we reason that the histone substrate of Smyd3 should bind at the other end of the channel as observed in SET7/9. In the SET7/9 structures, the histone peptide binding site is quite open as the N-terminal pre-SET domain is distant from the active site (21,25). In the Smyd3 structure, however, the TPR domain encloses a large part of the substrate binding site, and together with the SET and post-SET domains forms a deep and narrow substrate binding pocket (Figure 2A and B). Detailed examination of the pocket shows that several acidic residues, including Glu192 and Asp241 of the SET domain and Asp332 of the TPR domain are located at the opening of the substrate binding pocket, and might be involved in histone binding (Figure 2B).
Figure 2.

Histone binding pocket. (A) Potential histone peptide binding site. Superposition of the Smyd3 and SET7/9 (PDB code 1O9S) structures was performed as in Figure 1B. Smyd3 is shown in a ribbon representation with the same color coding as in Figure 1A. For simplicity, for SET7/9 only the bound histone peptide is shown in a ribbon representation and colored in green and the side chain of the methyllysine of the histone peptide is shown with a ball-and-stick model and colored accordingly. The cofactor is also shown with a ball-and-stick model. (B) Electrostatic potential surface of the potential histone peptide binding pocket in Smyd3. The surface charge distribution is displayed as blue for positive, red for negative, and white for neutral. A close-up view of the pocket (middle panel) shows that several acidic patches are present at the opening of the binding pocket. Some of the acidic residues in these patches are labeled. The TPR domain forms part of the substrate binding pocket and removal of the TPR domain would leave an incomplete pocket (right panel). (C) HKMT activity assays of the wild-type Smyd3 and the mutants with truncation or mutations at the substrate binding pocket. The activities of the truncate with deletion of the C-terminal region (Δ277–428) and the mutants carrying one or two point mutations of the residues potentially involved in histone binding were determined. Activity is shown as the relative activity of the proteins normalized to that of the wild-type protein. The experiments were performed in triplicates and the error bars indicate the standard deviation.
The mutagenesis analyses clearly show that truncation of the TPR domain (Δ277–428) resulted in an approximately 4-fold decrease in the HKMT activity, indicating an important, but not critical, role of the TPR domain (Figure 2C). Furthermore, mutation of either Asp241 or Asp332 to Ala indeed had a negative impact with 60–75% reduction of the HKMT activity of Smyd3, and intriguingly, double mutation of Asp241 and Asp332 almost completely abrogated the enzymatic activity (Figure 2C). Mutation of Glu192 to Ala had a minor effect, implying that Glu192 might not be directly involved in histone binding (Figure 2C). Taken together, the structural and biochemical data demonstrate that the TPR domain and the acidic property of the opening of the substrate binding pocket are important for the HKMT activity of Smyd3. In particular, residues Asp241 of the SET domain and Asp332 of the TPR domain play key roles in the HKMT reaction, most likely through the binding of the histone substrate.
Cofactor binding pocket
At the active site of the Smyd3–AdoHcy structure, AdoHcy is bound in a pocket surrounded by the β1–β2 loop, the η1–η2 loop and β9 of the SET domain and the α6–α7 loop of the post-SET domain (Figure 1B) with well-defined electron density (Figure 3A). Similar to AdoHcy bound in the SET7/9 structures (18,25), the cofactor takes a U-shape conformation in the Smyd3 structure (Figure 3B). The adenine ring of AdoHcy makes a π–π stacking interaction with the side chain of Phe259, and the N6 atom of the adenine forms a hydrogen bond with the main-chain carbonyl of His206. For stabilization of the ribose ring, the O2′ atom forms a hydrogen bond with the side-chain amino group of Asn132, and the O3′ atom forms hydrogen bonds with the side-chain carbonyl of Asn132 and the main-chain carbonyl of Tyr257. The amide group of the homocysteine is hydrogen-bonded to the main-chain carbonyl groups of Arg14 and Asn16 and the side-chain carbonyl of Asn205. The carboxyl group forms hydrogen bonds with the main-chain amide of Asn16 and the phenolic hydroxyl of Tyr124.
Figure 3.

Cofactor binding pocket. (A) A representative simulated-annealing omit Fo–Fc electron density map (contoured at 2.0 σ level) for the cofactor product AdoHcy. (B) A detailed comparison of the cofactor binding mode of Smyd3 (left panel) with that of SET7/9 (right panel, PDB code 1O9S). The hydrogen bonds are indicated with black dotted lines. The residues contributing to the cofactor binding with their side chains are labeled in orange, and those with their backbones in black. Depending on the contributing moieties, the side chains or backbones of the residues involved in the AdoHcy binding are shown with ball-and-stick models. The color coding is the same as in Figure 1A. (C) HKMT activity assays of the wild-type Smyd3 and the mutants carrying point mutations at the cofactor binding pocket.
Besides the residues that interact directly with AdoHcy, residue Asp262 of the post-SET domain might contribute to the cofactor binding as it forms a salt bridge with Arg14 to stabilize the positions of Arg14 and the β1–β2 loop where Arg14 is located, and perhaps interacts with the N1 and N6 atoms of the adenine moiety via a water molecule not seen at this resolution of diffraction data (Figure 3B). In addition, similar to its equivalent (Tyr335) in SET7/9 (25), Tyr239 may also make van der Waals interactions with the O4′ atom of the ribose ring (Figure 4A).
Figure 4.

Structure of the lysine channel. (A) Comparison of the lysine channels of Smyd3 and SET7/9 (PDB code 1O9S). Superposition of the active sites of Smyd3 and SET7/9 reveals a narrow channel in Smyd3 which is similar to the lysine channel of SET7/9. The key residues forming the lysine channel are shown with ball-and-stick models. The color coding is the same as in Figure 1A. (B) Superposition of the active sites of Smyd3 and pea Rubisco LSMT (PDB code 1P0Y). The residues are shown with ball-and-stick models, and for clarity, the side chains of Asn181 of Smyd3 and Arg222 of Rubisco LSMT are hidden. The color coding for Smyd3 is the same as in Figure 1A, and Rubisco LSMT is colored in light gray. (C) HKMT activity of the wild-type Smyd3 and the mutants carrying point mutations at the lysine channel.
Although in Smyd3 the cofactor binds at an equivalent position and assumes a similar conformation as in SET7/9 (18,25), there are notable differences in the cofactor binding mode between Smyd3 and SET7/9. As shown in Figure 3B, in the SET7/9–AdoHcy structure, the adenine ring of AdoHcy makes a π–π stacking interaction with Trp352, and the N1 atom forms a hydrogen bond with Glu356 of the post-SET domain. The carboxylate of the homocysteine moiety is not stabilized by the equivalent residue of Tyr124, instead by the main-chain carbonyl and amide of Glu228. In addition, the ribose moiety of SET7/9 does not make interactions with the surrounding residues.
Validation of the functional roles of these residues with mutagenesis analyses has not been performed previously except for the equivalents of Asn205 (21) and Tyr239 (20–22,27). Since Tyr239 has other important functions besides the cofactor binding, the results of the mutagenesis analyses of Tyr239 will be discussed later. As shown in Figure 3C, mutations of Tyr124, Asn132, Phe259 and Asp262 to Ala substantially impaired the HKMT activity of Smyd3, indicating the importance of their functional roles. In particular, mutation of Phe259 which interacts with the adenine ring and mutation of Asn132 which is hydrogen-bonded to both O2′ and O3′ of the ribose almost completely abolished the enzymatic activity of Smyd3. Since Phe259 belongs to the post-SET domain, the results are consistent with a critical role of the post-SET domain in the cofactor binding and the HKMT activity of Smyd3.
Lysine channel and implications on the catalytic mechanism
In the SET7/9 structure, a lysine channel connecting the histone peptide binding site and the cofactor binding site is found to accommodate the side chain of the methylated H3K4 (25). In the Smyd3 structure, a narrow channel is also found at a similar site and thus is a potential binding site of the side chain of H3K4 (Figure 4A). However, a detailed comparison of the two enzymes reveals significant differences at this site: the lysine channel of SET7/9 is formed mainly by residues Leu267, Tyr245, Tyr305, Tyr335 and Tyr337, while the lysine channel of Smyd3 contains only two Tyr residues (Tyr257 and Tyr239) (Figure 4A). In Smyd3, the positions where Leu267 and Tyr305 of SET7/9 are located are occupied by Phe183 and Ile214, respectively, and those where the phenol rings of Tyr337 and Tyr245 reside are vacant. In addition, although the position of the hydroxyl of Tyr337 is occupied by the hydroxyl of Tyr257, the side chain of Tyr257 approaches this site from an opposite side and correspondingly the hydroxyl group points to a different direction. Surprisingly, further comparison of the structure of the active site of Smyd3 with those of other lysine methyltransferases shows that the lysine channel of Smyd3 most resembles that of a non-HKMT, namely pea Rubisco large subunit methyltransferase (LSMT) (20) which, like Smyd3, is able to catalyze tri-methylation of lysine (Figure 4B). The base of the lysine channel should be the binding site for the methyl groups of the lysine substrate. In Smyd3, the base of the lysine channel which is surrounded by Phe183, Asn181 and Tyr239 is slightly larger than that in Rubisco LSMT. Further examination and comparison of the Smyd3 structure with that of the Y245A mutant of SE7/9 in complex with a tri-methylated peptide of TAF10 (PDB code 3M5A) clearly show that the base of the lysine channel in Smyd3 is sufficient to accommodate three methyl groups (Supplementary Figure S4), which is required by its activity to tri-methylate the substrate.
In all of the three structures, the Tyr residues (Tyr239 of Smyd3, Tyr335 of SET7/9 and Tyr287 of Rubisco LSMT) which correspond to the absolutely conserved Tyr residue in all of the identified SET enzymes, adopt similar positions and configurations (Figure 4A and B). During the methyl transfer reaction, the methyl acceptor Lys needs to be deprotonated, and it has been proposed that Tyr residues at the active site of methyltransferases are responsible for deprotonating the Nε group of the Lys substrate (18,20,22,23,25). Specifically, Trievel et al. originally proposed that in pea Rubisco LSMT, the methyl transfer is catalyzed by Tyr287 (20). However, later this notion was put in doubt by the same group as in their ternary structure of Rubisco LSMT in complex with a free ε-N-methyllysine and AdoHcy, the Nε group of Lys forms a hydrogen bond with the main-chain carbonyl of Arg222 and is more than 3.3 Å away from the Tyr287 hydroxyl (44). In addition, a study of a viral histone H3K27 methyltransferase argued that the corresponding Tyr of this enzyme is unlikely to act as a general base as the Tyr to Phe mutant exhibited little activity at pH 8.0 or above pH 9.0 (27). However, recently a deprotonation role of SET7/9-Tyr335 is again supported by simulation studies of SET7/9 which indicate that prior to the binding of the cofactor, Tyr335 can be deprotonated by bulk water molecules and then acts as a general base to deprotonate the Lys amine group (45). Thus, the mechanism to achieve deprotonation of the Nε group of the Lys substrate is still inconclusive and deserves more investigation.
Although the active site of Smyd3 is similar to that of Rubisco LSMT, there are notable differences (Figure 4B). In particular, Asn181 of Smyd3, equivalent to Arg222 of Rubisco LSMT, is unlikely to interact with the Lys substrate as the main-chain carbonyl of Asn181 points away from the lysine channel and this conformation of Asn181 is stabilized by the surrounding residues. We examined the effect of mutations of the residues involved in the formation of the lysine channel on the HKMT activity of Smyd3, including Phe183, the only two Tyr residues (Tyr239 and Tyr257), and Ser202. Mutation of Phe183 to Ala completely abrogated the HKMT activity of Smyd3, exhibiting a critical role of Phe183 in the maintenance of the integrity of the lysine channel and in the interaction of Smyd3 with the Lys substrate (Figure 4C). The S202A and Y257F mutants retain more than half of the HKMT activity, indicating that they play less important roles in the catalysis (Figure 4C). In contrast, mutation of Tyr239 to Phe or Ala completely abolished the enzymatic activity, demonstrating a key role of this residue in the HKMT reaction (Figure 4C).
Mutation of Tyr239 may affect the HKMT activity of Smyd3 in three ways: change of the cofactor binding affinity, disruption of the lower part of the lysine channel and loss of the ability to accelerate deprotonation of the Nε group of the Lys substrate. The possibility of the involvement of Tyr239 in the cofactor binding is inferred from the observation that its equivalent of SET7/9 (Tyr335) makes van der Waals contacts with the ribose ring of AdoHcy (18). However, in Smyd3, the effect of the Tyr239 mutation on the cofactor binding should be trivial as the ribose ring of AdoHcy is stabilized by three hydrogen bonds with Asn132 and Tyr257 (Figure 3B) and the potential interactions of Tyr239 with the cofactor might be expected to contribute little to the cofactor binding. It is also noteworthy that the hydroxyl group of Tyr239 forms a hydrogen bond with the main-chain carbonyl of Leu204 of the η4-β9 loop which might stabilize the position of the side chain of Tyr239, and hence mutation of Tyr239 to Ala or Phe might lead to disruption of the lysine channel due to the loss of the large side chain in the Y239A mutant or the instability of the phenol ring in the Y239F mutant. Therefore, we further mutated Tyr239 to Lys or a neutral residue Gln both of which might retain the interaction with the carbonyl of Leu204 but are unable to deprotonate a Lys amine. Again, both the Y239K and Y239Q mutations led to complete loss of the enzymatic activity of Smyd3 (Figure 4C). However, the intactness of the lysine channel in these mutants needs further investigation. Taken together, the structural analyses and biochemical assays indicate that the hydroxyl group of Tyr239 is critical for the HKMT activity of Smyd3.
Modulation of the HKMT activity via DNA binding of the MYND domain
The MYND domain was originally named due to its presence in ETO (also named myeloid tumor gene 8) and Drosophila proteins Nervy and Deaf1 (46), and later has been found in numerous other proteins including the Smyd proteins (47), BS69 (48), mammalian programmed cell death proteins 2 (49), and AML1/ETO which is a fused protein resulted from the t(8;21) translocation in acute myeloid leukemia. Comparison of the structure of the MYND domain of Smyd3 with other reported MYND structures shows that it most resembles the MYND domain of AML1/ETO (50) with an RMSD of 0.57 Å. Despite a very similar overall structure, there are differences in certain regions. For example, a loop encompassing residues 55–59 in Smyd3 takes a conformation different from that of the equivalent loop in AML1/ETO. In addition, residues 86–93 of Smyd3 form helix α2 while the equivalent residues form a loop in AML1/ETO.
It has been indicated that the MYND domain of AML1/ETO binds the PPPL1 motif of retinoid and thyroid hormone receptor SMRT corepressor (50). In the structure of the MYND domain of AML1/ETO which is fused to the PPPL1 motif of SMRT, the conformation of the MYND domain is similar to that of the unbound form, and the three residues Ser675, Gln688 and Trp692 of the MYND domain located in a hydrophobic pocket interact with the PPPLI motif of the SMRT peptide (50). In the MYND domain of Smyd3, the equivalent residues (Ser63, Glu76 and Trp80, respectively) are conserved. However, the electrostatic properties of the residues surrounding the hydrophobic pocket exhibit substantial differences, with the replacement of acidic residues (Glu672 and Glu692) of AML1/ETO with hydrophobic residues (Met60 and Pro81) and substitution of residues Thr673 and His689 with highly basic residues (Arg61 and Lys77) (Figure 5A). With the presence of additional Lys and Arg residues, the surface of the MYND domain in Smyd3 is largely positively charged (Figure 5A), which is in agreement with its potential role in the binding of specific DNA sequences such as 5′-CCCTCC-3′ and further in transcriptional regulation of the targets including Nkx2.8 (6).
Figure 5.

Modulation of the HKMT activity of Smyd3 via DNA binding of the MYND Domain. (A) Electrostatic potential surface of the MYND domain with the negative charge in red and the positive charge in blue. All the positively charged residues are labeled. (B) Analysis of the interaction between the wild-type and mutant Smyd3 proteins and the potential target DNA with gel shift assays. The wild-type (WT) Smyd3 and the R66E and E192A mutants were loaded to 1 µg DNA with different molar ratios as indicated. DNA binding of the wild-type and mutant Smyd3 were analyzed with agarose gel electrophoresis. (C) HKMT assays of the wild-type and R66E mutant Smyd3 in the presence and absence of the potential target DNA. The HKMT activity of the wild-type but not the mutant Smyd3 is stimulated when the potential target DNA is supplemented in the HKMT reaction system. (D) Sequence alignment of the MYND domain among all human Smyd proteins. The sequence number and secondary structure elements of Smyd3 are marked. The invariant residues across these proteins are denoted with filled red boxes, and the highly conserved ones in open boxes. The residues conserved in Smyd1-4 but not Smyd5 are denoted with asterisks.
To examine the hypothesis that the MYND domain of Smyd3 may directly bind the DNA element, two Smyd3 mutants were generated: the R66E mutant in which an acidic residue is placed on the positively charged MYND surface and a negative control mutant E192A which has an intact MYND domain. The wild-type and mutant Smyd3 proteins were incubated with the potential target of 6-bp duplex DNA with the sequence of 5′-CCCTCC-3′. As shown in Figure 5B, the DNA exhibited a slower migration rate in the presence of the wild-type Smyd3 or the E192 mutant but not the R66E mutant. It was reported that pre-incubation of yeast methyltransferase Dot1p with DNA could stimulate its HKMT activity on histones (51); therefore, we further examined the possibility that the DNA-binding ability of the MYND domain might affect the HKMT activity of Smyd3. In the presence of the potential target DNA, the HKMT activity of the wild-type Smyd3 was indeed increased in a dose-dependent manner (Figure 5C). The enhancement of the HKMT activity by DNA, however, did not occur when Arg66 is mutated to Glu, indicating that the DNA-binding ability of the MYND domain is essential for stimulating the enzymatic activity of Smyd3 by DNA. Taken together, our data demonstrate for the first time that the DNA binding of Smyd3 stimulates its HKMT activity, and the MYND domain may mediate the process through direct binding with the target DNA. Intriguingly, comparison of the MYND domains of the Smyd proteins shows that some of the basic residues of Smyd3 (Arg61, Arg66, Lys77 and Lys84) are conserved in Smyd1 and Smyd2 (Figure 5D), implying that Smyd1 and Smyd2 may utilize a similar mechanism to regulate their HKMT activities.
PROTEIN DATA BANK ACCESSION CODES
The structures of Smyd3 in crystal forms I, II and III have been deposited with the RCSB Protein Data Bank under accession codes 3OXF, 3OXL and 3OXG, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology of China (grants 2007CB914302 and 2011CB966301); National Natural Science Foundation of China (grant 30730028); Chinese Academy of Sciences (grants KSCX2-YW-R-107 and SIBS2008002) and the Science and Technology Commission of Shanghai Municipality (grant 10JC1416500). Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the staff members at the Shanghai Synchrotron Radiation Facility of China for technical supports in diffraction data collection, and other members of their group for helpful discussion. C.Z. gratefully acknowledges the support of SA-SIBS scholarship program.
REFERENCES
- 1.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 2.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 3.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 4.Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R, Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004;6:731–740. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 7.Brown MA, Sims RJ, 3rd, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer. 2006;5:26. doi: 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan X, Rotllant J, Li H, De Deyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl Acad. Sci. USA. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell Proteomics. 2008;7:560–572. doi: 10.1074/mcp.M700271-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 11.Kunizaki M, Hamamoto R, Silva FP, Yamaguchi K, Nagayasu T, Shibuya M, Nakamura Y, Furukawa Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007;67:10759–10765. doi: 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- 12.Noma K, Allis CD, Grewal SI. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science. 2001;293:1150–1155. doi: 10.1126/science.1064150. [DOI] [PubMed] [Google Scholar]
- 13.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 14.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 15.Cao Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal. 2009;2:re1. doi: 10.1126/scisignal.259re1. [DOI] [PubMed] [Google Scholar]
- 16.Tsuge M, Hamamoto R, Silva FP, Ohnishi Y, Chayama K, Kamatani N, Furukawa Y, Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 2005;37:1104–1107. doi: 10.1038/ng1638. [DOI] [PubMed] [Google Scholar]
- 17.Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, et al. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs SA, Harp JM, Devarakonda S, Kim Y, Rastinejad F, Khorasanizadeh S. The active site of the SET domain is constructed on a knot. Nat. Struct. Biol. 2002;9:833–838. doi: 10.1038/nsb861. [DOI] [PubMed] [Google Scholar]
- 19.Min J, Zhang X, Cheng X, Grewal SI, Xu RM. Structure of the SET domain histone lysine methyltransferase Clr4. Nat. Struct. Biol. 2002;9:828–832. doi: 10.1038/nsb860. [DOI] [PubMed] [Google Scholar]
- 20.Trievel RC, Beach BM, Dirk LM, Houtz RL, Hurley JH. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell. 2002;111:91–103. doi: 10.1016/s0092-8674(02)01000-0. [DOI] [PubMed] [Google Scholar]
- 21.Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell. 2002;111:105–115. doi: 10.1016/s0092-8674(02)00964-9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–127. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon T, Chang JH, Kwak E, Lee CW, Joachimiak A, Kim YC, Lee J, Cho Y. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet. EMBO J. 2003;22:292–303. doi: 10.1093/emboj/cdg025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manzur KL, Farooq A, Zeng L, Plotnikova O, Koch AW, Sachchidanand, Zhou MM. A dimeric viral SET domain methyltransferase specific to Lys27 of histone H3. Nat. Struct. Biol. 2003;10:187–196. doi: 10.1038/nsb898. [DOI] [PubMed] [Google Scholar]
- 25.Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 26.Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 2005;19:1444–1454. doi: 10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian C, Wang X, Manzur K, Sachchidanand, Farooq A, Zeng L, Wang R, Zhou MM. Structural insights of the specificity and catalysis of a viral histone H3 lysine 27 methyltransferase. J. Mol. Biol. 2006;359:86–96. doi: 10.1016/j.jmb.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Briknarova K, Zhou X, Satterthwait A, Hoyt DW, Ely KR, Huang S. Structural studies of the SET domain from RIZ1 tumor suppressor. Biochem. Biophys. Res. Commun. 2008;366:807–813. doi: 10.1016/j.bbrc.2007.12.034. [DOI] [PubMed] [Google Scholar]
- 29.Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 35.Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, Zhang X, Bedford MT, Shinkai Y, Snyder JP, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005;19:1455–1465. doi: 10.1101/gad.1318405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S, Prapapanich V, Rimerman RA, Honore B, Smith DF. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol. Endocrinol. 1996;10:682–693. doi: 10.1210/mend.10.6.8776728. [DOI] [PubMed] [Google Scholar]
- 40.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 41.Dolinski KJ, Cardenas ME, Heitman J. CNS1 encodes an essential p60/Sti1 homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol. Cell. Biol. 1998;18:7344–7352. doi: 10.1128/mcb.18.12.7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marsh JA, Kalton HM, Gaber RF. Cns1 is an essential protein associated with the hsp90 chaperone complex in Saccharomyces cerevisiae that can restore cyclophilin 40-dependent functions in cpr7Delta cells. Mol. Cell. Biol. 1998;18:7353–7359. doi: 10.1128/mcb.18.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 44.Trievel RC, Flynn EM, Houtz RL, Hurley JH. Mechanism of multiple lysine methylation by the SET domain enzyme Rubisco LSMT. Nat. Struct. Biol. 2003;10:545–552. doi: 10.1038/nsb946. [DOI] [PubMed] [Google Scholar]
- 45.Guo HB, Guo H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc. Natl Acad. Sci. USA. 2007;104:8797–8802. doi: 10.1073/pnas.0702981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross CT, McGinnis W. DEAF-1, a novel protein that binds an essential region in a Deformed response element. EMBO J. 1996;15:1961–1970. [PMC free article] [PubMed] [Google Scholar]
- 47.Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 48.Ansieau S, Leutz A. The conserved Mynd domain of BS69 binds cellular and oncoviral proteins through a common PXLXP motif. J. Biol. Chem. 2002;277:4906–4910. doi: 10.1074/jbc.M110078200. [DOI] [PubMed] [Google Scholar]
- 49.Scarr RB, Sharp PA. PDCD2 is a negative regulator of HCF-1 (C1) Oncogene. 2002;21:5245–5254. doi: 10.1038/sj.onc.1205647. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Chen W, Gaudet J, Cheney MD, Roudaia L, Cierpicki T, Klet RC, Hartman K, Laue TM, Speck NA, et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO's activity. Cancer Cell. 2007;11:483–497. doi: 10.1016/j.ccr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawada K, Yang Z, Horton JR, Collins RE, Zhang X, Cheng X. Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J. Biol. Chem. 2004;279:43296–43306. doi: 10.1074/jbc.M405902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.