


Abstract
In eukaryotes, it is generally assumed that translation initiation occurs at the AUG codon closest to the messenger RNA 5′ cap. However, in certain cases, initiation can occur at codons differing from AUG by a single nucleotide, especially the codons CUG, UUG, GUG, ACG, AUA and AUU. While non-AUG initiation has been experimentally verified for a handful of human genes, the full extent to which this phenomenon is utilized—both for increased coding capacity and potentially also for novel regulatory mechanisms—remains unclear. To address this issue, and hence to improve the quality of existing coding sequence annotations, we developed a methodology based on phylogenetic analysis of predicted 5′ untranslated regions from orthologous genes. We use evolutionary signatures of protein-coding sequences as an indicator of translation initiation upstream of annotated coding sequences. Our search identified novel conserved potential non-AUG-initiated N-terminal extensions in 42 human genes including VANGL2, FGFR1, KCNN4, TRPV6, HDGF, CITED2, EIF4G3 and NTF3, and also affirmed the conservation of known non-AUG-initiated extensions in 17 other genes. In several instances, we have been able to obtain independent experimental evidence of the expression of non-AUG-initiated products from the previously published literature and ribosome profiling data.
INTRODUCTION
Translation initiation is the only step during protein biosynthesis where an incoming aminoacyl tRNA is bound directly in the ribosomal P-site (1,2). There is a greater flexibility for mismatches in the codon:anticodon duplex in the P-site (3,4) in comparison with the A-site where, in contrast, proper geometry of messenger RNA (mRNA):tRNA interactions is strictly monitored by the decoding center for the first two positions of a codon (5). As a result, initiator Met-tRNAi can be incorporated into the ribosome at a wider range of codons than in the case of elongator Met-tRNA whose incorporation is strictly limited to AUG codons. Such a flexible usage of initiation codons by Met-tRNAi requires additional control mechanisms for discrimination between potential start sites.
These control mechanisms differ between eukaryotes and bacteria. In fact, the initiation step represents the biggest mechanistic difference in the process of protein biosynthesis between organisms from different domains of life. In bacteria, with a single notable exception of psyllid endosymbiont Carsonella (6), the small subunit of the ribosome binds the mRNA a short distance upstream of the initiation codon through interaction between the 3′-end of the 16S rRNA and a complementary sequence, termed Shine–Dalgarno (7). In contrast, in eukaryotes, the scanning model for the initiation of translation postulates that the small ribosomal subunit, in complex with initiation factors and Met-tRNAi, binds first to the 5′ cap. Then it scans 5′–3′ until it finds a suitable initiator codon (8). Exceptions include internal ribosome entry sites, shunting and reinitiation mechanisms, see references (9) and (10) for invigorating discussion of contrasting viewpoints. During scanning, the efficiency of initiation is dependent on the nucleotide context surrounding the initiator codon with the optimal context being known as the Kozak motif (11).
The initiation context—comprising the 6 nt before and the 1 nt immediately following a potential initiation codon—has significant influence on the recognition of that codon as an initiation site, through as yet poorly understood mechanisms requiring the activity of eIF1 (12,13). The optimal context in mammals is GCCRCCAUGG (14) with the identity of the underlined nucleotides in the −3 and +4 positions (relative to the ‘A’ of the AUG) being the most important. An ‘A’ in the −3 position is preferred over a ‘G’ and a purine in that position is more important than a ‘G’ in the +4 position (15). The identity of the nucleotides in the other positions plays an important role only when there is no purine in position −3 and no ‘G’ in position +4 (15).
Although relatively rare, initiation of translation of eukaryotic mRNAs can occur at non-AUG codons that differ from AUG at a single nucleotide position. Initiation on the nine possible such codons occurs with a varying degree of efficiency that is partially taxon dependent. For example, in mammals and plants CUG appears to be the most efficient non-AUG initiation codon, while AAG and AGG are the least efficient (16). For initiation at non-AUG codons, the presence of a good Kozak context is crucial (17–19). The rules for the most favorable context are believed to be identical to those for an AUG codon. In addition, a strong RNA secondary structure starting ∼15 nt downstream of the initiation site significantly increases the efficiency of initiation at non-AUG codons (20).
In mammals, at least 38 mRNAs from 23 genes have been reported to have or else have annotated non-AUG initiation codons in GenBank (21). In most cases, the non-AUG initiation provides an alternative longer isoform in addition to an isoform resulting from initiation at a standard AUG codon downstream via a process termed ‘leaky scanning’ (22). Where alternative isoforms are produced as a result of leaky scanning, the longer isoform frequently contains a signal for subcellular localization that is absent in the shorter form (23–26). In some cases of non-AUG-initiated genes, the principles of leaky scanning ensure that the non-AUG initiation codon is the sole initiation site of the main CDS even when downstream in-frame AUG codons exist. This happens when the region between the non-AUG initiation codon and the first available downstream in-frame AUG codon is populated by one or even many out-of-frame AUG codons, especially if some of them occur in a good initiation context as, for example, in the case of EIF4G2 (27). Conversely, another peculiar feature of eukaryotic initiation can be exploited to suppress initiation on intervening out-of-frame AUG codon(s). Translation of open reading frames (ORFs) shorter than approximately 35 codons results in only partial dissociation of terminating ribosomes from the mRNA, with the 40S subunit able to resume scanning downstream (11,28). Scanning following translation of such an ORF, however, precludes efficient reinitiation until initiation factors have been reloaded onto the 40S subunit. As a result AUG codons, even in perfect (Kozak) context, are inefficiently recognized if they are present ∼100–200 nt downstream of an efficiently translated short upstream ORF (29). Translation of an ORF longer than approximately 35 codons usually precludes resumption of scanning downstream. An additional layer of complexity comes from the fact that the efficiency of ‘reloading’ the scanning 40S subunit following translation of a short upstream ORF (uORF) is under regulatory control through the phosphorylation status of Ser-51 of eIF2α (29). Leaky scanning, combined with the peculiar features of reinitiation following translation of short uORFs, can result in diverse and complex scenarios for predicting which codons, and under what conditions, are recognized as initiation sites for translation on eukaryotic mRNAs (22).
Earlier in vitro work with rabbit reticulocyte lysates showed that the concentration of Mg2+ negatively regulates the fidelity of translation initiation (18). A recent study found that non-AUG-initiated uORFs regulate expression of ornithine decarboxylase homologs in many eukaryotic organisms (30). In one of the mammalian homologs, and perhaps in all other homologs that have this particular regulatory feature, polyamines induce initiation on an AUU codon. It is noteworthy that polyamines are essentially organic polycations and, as such, their biophysical role overlaps with that of Mg2+ ions which, at least in vitro, show an identical effect on the fidelity of initiation. A recently developed technique for profiling the ribosomal density on mRNAs revealed the presence of translating ribosomes on >200 non-AUG-initiated uORFs in yeast. Moreover, initiation on these uORFs was shown to be upregulated under amino acid starvation conditions (31). Another recent study showed that variations in the intracellular levels of eIF1 lead to significant differences in the stringency of start codon selection (32). These results suggest that the fidelity of initiation is variable under physiological conditions and strongly supports the idea that non-AUG initiation might perform regulatory roles in eukaryotic cells. Therefore, the utilization of non-AUG initiation in mammalian mRNAs merits further exploration, as the regulated alternative usage of start codons provides an opportunity for global alterations of a proteome in a coordinated manner. An earlier finding that sequences in the 5′-untranslated regions (UTRs) are highly conserved and that the level of conservation globally increases toward the UTR/CDS boundaries (33) is suggestive that this conservation could be due, in part, to the 3′-ends of a portion of 5′-UTRs encoding N-terminal extensions to the annotated AUG-initiated proteins. The protein coding potential of 5′-UTRs and modes of its utilization have been discussed in detail elsewhere (34).
Here we performed a systematic analysis of the 5′-UTRs of human GenBank RefSeq mRNAs to investigate the extent of non-AUG initiation in humans. Our methodology is based on the analysis of codon substitution rates in pairwise alignments of human and mice orthologous sequences that allows us to detect 5′-UTR fragments evolving under the constraints of protein coding evolution. If a segment of a 5′-UTR evolves under such constraints, it is likely that the corresponding protein sequence is produced and is positively functional. Candidates selected by this criterion were subjected to further computational analysis using multiple alignments of sequences from other vertebrate species followed by rigorous manual evaluation.
MATERIALS AND METHODS
A pipeline for identification of candidates for non-AUG translation initiation
A schematic outline of the pipeline for potential 5 extension of CDS (P5EC) detection is illustrated in Figure 2. A total of 46 500 human mRNA sequences were downloaded from the NCBI RefSeq database (35) on 12 February 2009 (release 33) using the Refseq ftp site: ftp://ftp.ncbi.nih.gov/refseq/release/. We chose RefSeq annotations to minimize the effect from erroneous and inconsistent misannotations as well as to avoid redundancy in our dataset. All sequences that have in-frame stop codons within 50 codons upstream of annotated AUG initiator codons were discarded. For the remaining mRNA sequences, the region surrounding the annotated AUG codon was translated from the nearest upstream in-frame stop codon to the 100th codon downstream of the AUG. The inclusion of the 5′-terminal 100 codons of the annotated CDS was a necessary element to ensure detection of correct mouse orthologs in the following steps. This procedure resulted in a set of 3437 peptide sequences. This set of peptide sequences was then queried against the mouse Refseq mRNA database using tblastn as implemented in NCBI BLAST client netblast-2.2.19. The best hits for each of the peptides were considered as candidate orthologs. Those sequences that produced alignments with a span of at least 50 codons upstream of the annotated CDS were extracted for further processing; 2194 human–mouse orthologous pairs were obtained. Sequences were conceptually translated, re-aligned using muscle (36) and back-translated using t_coffee utilite (37) to yield nucleotide alignments. A custom perl script was designed to trim P5EC alignments prior to Ka/Ks calculation in the following way. Columns containing gaps in either the human or mouse sequence were removed. Because the alignment of sequences was carried out at the amino acid level, all gaps at the nucleotide level occur in multiples of 3 and therefore their removal does not affect reading frame. For alignments containing in-frame stop codons in the mouse sequence, columns corresponding to the stop codons and all columns upstream were removed. This was done based on the assumption that, in general, functionally important P5ECs and alternative translation initiation starts (ATISs) should be conserved between mouse and human sequences and that the presence of a stop codon in a mouse sequence indicates that the sequence is not translated in mouse. If, after trimming, the resulting alignment was shorter than 25 codons, then the corresponding mouse P5EC could not be longer than 25 codons. Also it would not be practical to include such short alignments into further analysis. Therefore, only alignments with a length of at least 25 codons upstream of the annotated AUG (1193 alignments) were subjected to Ka/Ks analysis as implemented in the codonml program of the PAML package (38). Then the alignments were scored based on Ka/Ks values, length of the pairwise alignments and identity at the protein level.
Figure 2.

Pipeline of RefSeq mRNA analysis for the identification of conserved 5′ CDS extensions (P5ECs). White boxes indicate annotated CDSs. Black boxes correspond to 5′ in-frame codon extensions up to the closest in-frame stop codon. Xs correspond to the deleted regions of human–mouse alignments prior to Ka/Ks analysis.
Identification of candidates with known alternative transcript variants
An additional step, as discussed in the ‘Results’ section, was the identification of those P5EC-containing mRNAs whose genes are known to have alternative transcripts containing 5′-extensions to the annotated CDS. For this purpose, annotations of mRNA sequences from the pool above (1193) were searched for the following text strings: ‘transcript variant’ or ‘shorter’ preceding ‘N-terminal’ within the same annotation field of GenBank format RefSeq entries. Due to the lack of uniformity in annotation descriptions, this procedure does not guarantee identification of all mRNAs with alternatively spliced transcripts where the CDS is 5′-terminally extended. However, this step allowed us to achieve two goals. First, we eliminated a large portion (204 instances) of genes where P5ECs exist because they correspond to translated regions in alternative transcripts and hence most likely do not utilize non-AUG initiation. Second, we created a set of sequences that could be utilized as a positive control, because these genes represent cases where sequences upstream of annotated start codons are known to be translated (see ‘Results’ section).
As described in the Results section, a relatively high Ka/Ks ratio for the entire P5EC does not necessarily exclude the possibility that at least the 3′ part of the P5EC might be evolving under purifying selection. Therefore, we used a relatively relaxed threshold for choosing the candidates for further detailed semi-manual analysis, i.e. Ka/Ks <1, and the existence of a significant human–mouse pairwise alignment that spans at least 25 codons in length. Identity at the nucleotide or protein level was not taken into account at this point. This resulted in 742 candidates for further analysis.
Analysis of the approximately 700 candidates using deep phylogenetic analysis and manual evaluation
As it has been mentioned above, our filter for RefSeq mRNAs containing P5ECs due to alternative splicing was not exhaustive because of idiosyncrasies in RefSeq annotation comments. Therefore, all 742 candidates were subjected to further examination for the presence of alternative splice forms using the Entrez Gene database (39). All candidates containing alternative splice or transcription initiation forms resulting in noticeably variable 5′-ends were excluded from further analysis. The sequences of those candidates that passed this step were queried against human ESTs from dbEST (40) using blastn. Retrieved ESTs were used to search for the possible existence of non-annotated splice variants in the vicinity of the 5′-UTR-CDS junctions in the candidates under examination. Sequences that showed multiple 5′ variants, either resulting from alternative transcription initiation or alternative splicing, not included in RefSeq, but deemed likely to be real were, with a few exceptions, removed from further consideration. The exceptions were made for alternative transcripts that nevertheless lacked any upstream in-frame AUG codons. Among the set of genes that passed the filters above, we found numerous cases where the 5′-end extensions contained in-frame AUG codons not present in the annotated RefSeq sequence. Such examples were also excluded from further analysis. For the candidates that passed this step, the human extension up to the upstream in-frame stop codon was conceptually translated and queried against the dbEST, wgs and RefSeq_rna databases to retrieve sequences from non-human species that share significant similarity with the query. If the coding sequence was interrupted either by an in-frame stop codon or a frameshift in sequences from multiple organisms (i.e. >20% of the total available), especially if the conservation at the amino acid level was weak, such candidates were deemed questionable and were excluded from further analysis. The final criterion used to select for positives in the screen was the presence of a well-conserved ‘near cognate’ initiation codon for the putative extension. In total, 36 genes passed all these tests.
Multiple sequence alignments were constructed for the final list of candidates, plus the previously identified cases of upstream non-AUG-initiated extensions that passed the same selection criteria, and analyzed using PAML codonml (38,41) and MLOGD (42). Codonml was used to recalculate Ka/Ks, now using the multiple species alignments instead of just human–mouse and restricting to the region between the annotated AUG and the predicted upstream non-AUG initiation codon. MLOGD calculates coding potential by using the pattern of substitutions observed within an input sequence alignment to compare a coding model with a non-coding model via a likelihood ratio test. Although Ka/Ks could not be calculated with MLOGD (it is fixed via the BLOSUM62 matrix used within the coding model), MLOGD could be used to produce graphs of coding potential versus sequence position and could therefore be used to help identify the most probable non-AUG initiation sites.
These alignments were also used to calculate the probability, P (no stop codons), that the ORF of the extension would be preserved by chance if the sequence were non-coding. For each alignment, nucleotide columns were randomized 10 000 times and the proportion of randomizations in which the ORF was preserved (i.e. no stop codons in the zero-frame in any sequence) was taken as an estimate for P (no stop codons). The procedure controls for the phylogenetic non-independence of aligned nucleotides and any gene-specific nucleotide biases in the extension region (e.g. GC-rich regions tend to have fewer stop codons and therefore tend, by chance, to have longer ORFs). It also controls for gene-specific conservation biases (e.g. in a 5′-UTR that happens to be highly conserved due to some non-coding functional element, a chance ORF in one sequence is more likely to be preserved throughout the alignment). One caveat with interpretation of these P-values is that, for a small number of alignments, stop-codon-containing sequences from a small number of species had been previously discarded from the alignment either as presumed sequencing errors or on the assumption that the extension has been lost in some species. The P (no stop codons) value was not used as part of the selection criteria. In fact, most of the non-AUG-initiated extensions identified here are not sufficiently long to have a statistically significant P (no stop codons) value. However, it is useful for a small number of cases where we identified a very long extension that, nonetheless, was not subject to strong purifying selection and hence had weak Ka/Ks and MLOGD scores.
A similar randomization procedure was used to determine P-values for the MLOGD and Ka/Ks scores. Because MLOGD and Ka/Ks are codon-based statistics, alignment columns containing gaps were removed prior to randomization (otherwise, after randomization of nucleotide columns, alignment gaps would no longer occur in groups of three so reading frames would not be conserved between species with and species without alignment gaps). Due to CPU constraints, only 1000 randomizations were used for each alignment, which restricts the resolution of raw P-values to ∼0.001. In many cases, however, the observed MLOGD and log(Ka/Ks) values (recalculated for the same degapped alignments) were many standard deviations away from the corresponding mean values for the randomizations. To obtain higher resolution P-values (shown in Table 1), a normal approximation was assumed for the distribution of the randomization statistics for any particular alignment (after log transform for Ka/Ks; MLOGD is already on a log scale) and the probabilities of obtaining the observed log(Ka/Ks) and MLOGD statistics in randomized alignments were calculated with respect to this distribution.
Table 1.
Ranking of newly identified and known non-AUG-initiated N-terminal extensions
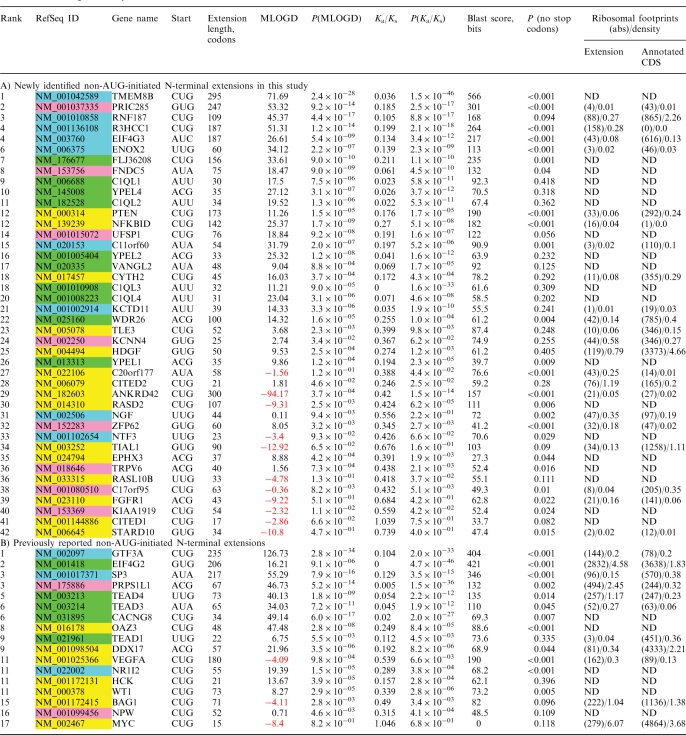 |
Column 1, combined ranking; column 2, GenBank accession number; column 3, gene name; column 4, predicted non-AUG initiation codon in human (see Supplementary Dataset 1 for full details); column 5, length in codons of predicted N-terminal extension in human; column 6, MLOGD score (negative values are shown in red and indicate candidates subject to weak or no purifying selection); column 7, P-value for MLOGD score based on randomizations; column 8, Ka/Ks ratio (with gap-containing columns removed from the alignment); column 9, P-value for Ka/Ks based on randomizations; column 10, BLAST bits score measured on the alignment of the human and mouse extensions; column 11, probability of the ORF of the extension being preserved by chance if non-coding. Note that the MLOGD score scales with alignment length and divergence and the BLAST bits score scales with length. The final ranking is based on the average of rankings by the individual scores (MLOGD, Ka/Ks and BLAST bits). Columns 12 and 13 give information on the number of mRNA fragments protected by ribosomes for extension (column 10) and annotated CDSs (column 11). The absolute number of footprints and density of footprints are separated by a backslash. Density is calculated as the absolute number divided by the length of the mRNA fragment (extension or CDS). ‘ND’, not detected. A) Ranking of the 42 newly identified non-AUG extensions. GenBank accession numbers highlighted in green represent extensions that are conserved beyond mammals and for which all available sequences from vertebrates appear to utilize non-AUG initiation codon(s); accession numbers highlighted in light blue represent extensions that are conserved beyond mammals and for which some or all non-mammalian sequences appear to utilize AUG instead of non-AUG initiation; accession numbers highlighted in magenta represent extensions that are initiated by AUG codons in at least some mammals; accession numbers highlighted in yellow represent extensions that are conserved only in mammals (in some cases only eutherian mammals) and which are never initiated by AUG codons. B) Ranking of previously reported non-AUG extensions.
Analysis of ribosomal profiling data
Short reads generated during ribosomal profiling experiments in HeLa cells described in Guo et al. (43) were obtained from the Gene Expression Omnibus (accession GSE22004). Short reads from all available experiments (SRR057511, SRR057512, SRR057516, SRR057517, SRR057521, SRR057522, SRR057526, SRR057529, SRR057532) were aggregated and aligned to the 59 mRNA sequences using Bowtie short read aligner (44), allowing zero mismatches in the first 25 nt ‘seed’ region. According to Guo et al. (43), on average, mRNA nucleotide positions corresponding to the 5′-end of the short reads in the alignments are located 15-nt upstream of the P-site tRNA in the ribosome. Therefore, coordinates of ribosome positions were calculated accordingly, i.e. shifted by 15-nt downstream from the beginning of the alignment between a short read and mRNA sequence. During this analysis, we did not check whether particular reads could be potentially aligned to other positions within the human genome and therefore the absolute numbers of footprints may be skewed. The density of ribosomes in CDSs and non-AUG extensions was calculated as the absolute number of footprints corresponding to a particular region divided by its length.
RESULTS
Analysis of P5ECs
Most protein-coding sequences evolve under purifying selection and this feature can be used for detection of protein coding regions in nucleotide sequences (45). If, in a particular gene, translation initiates upstream of the annotated start codon, then the sequence located between the annotated start codon and the actual (upstream) start codon should evolve under constraints of purifying selection. For brevity, we will refer to such regions as P5ECs. We used the existence of P5ECs as an initial indicator of utilization of alternative initiator codons. In principle, the presence of a P5EC does not guarantee that an alternative in-frame start codon is used for initiation. Figure 1 illustrates five possibilities (examples for which are known) where the sequence upstream of, and in-frame with, an annotated start codon would evolve under purifying selection: (i) Initiation at an upstream in-frame non-AUG codon (ATIS), the subject of this study. (ii) Programmed Ribosomal Frameshifting (PRF), where initiation occurs at the start codon of a uORF and then ribosomes enter the main protein-coding ORF (pORF) by shifting reading frames at a specific location. The most prominent human examples are the three antizyme paralogs (46). (iii) Stop Codon Readthrough (SCR). Similar to the above but where the uORF and the pORF occur in the same translational phase and are separated by a single Stop Codon. No chromosomal genes are known to utilize this phenomenon in humans but, in flies, three examples have been experimentally identified (47) and >100 additional candidates have been identified by comparative sequence analysis (48). (iv) RNA editing. A start codon is generated post-transcriptionally by the insertion of a U between an A and a G, as has been suggested for the linker histon H1F0 and HMGN1 protein genes (49). (v) Alternative Splicing. An exon containing a start codon in one transcript variant could be skipped in another transcript variant. In this case the latter transcript would use an initiator codon located downstream of the start codon of the former transcript. However the region between the 3′-end of the Alternatively Spliced Exon (ASE) and the start codon used in the second transcript would still evolve under the constraints of protein coding sequence, because this region is translated in the first transcript variant. This fifth class of P5EC-containing mRNAs is the largest class. Therefore, during our analysis we paid particular attention to discriminate P5ECs occurring as a result of alternative splicing from those resulting from non-annotated translation events. The pipeline for the initial computational analysis of human Refseq mRNAs is outlined in Figure 2 and is described in detail in the ‘Materials and Methods’ section.
Figure 1.

Five known molecular mechanisms responsible for the initiation of translation upstream of the first 5′ in-frame AUG codon. mRNAs are shown as horizontal lines. Dark grey boxes represent annotated CDS regions. Light grey boxes represent extensions of CDSs upstream of annotated AUG codons up to the closest in-frame stop codon. Black boxes denoted as P5EC represent upstream regions where codons in-frame with annotated CDSs evolve under purifying selection. Diagonal stripes are used to denote alternatively spliced exons.
Comparison of P5ECs due to alternative splicing with the other P5ECs
After obtaining Ka/Ks values for P5EC regions, we generated a set of sequences where the presence of a P5EC is due to the existence of alternative splice variants (see ‘Materials and Methods’ section). We compared the distribution of Ka/Ks values for these P5ECs with the Ka/Ks values for the remaining P5ECs. The distribution is shown in Figure 3 (see Supplementary Data for the actual values for each sequence). The distribution of Ka/Ks values for mRNAs with known alternative transcript variants (containing CDS 5′ extensions) is significantly sharper than the distribution of Ka/Ks values for the rest of the P5ECs, with the great majority of them falling under 0.2. Therefore, Figure 3 clearly illustrates that Ka/Ks can be used as a predictor of bona fide CDS 5′ extensions. Figure 4 shows scatter plot distributions of Ka/Ks ratios for sequences from both data sets in relation to the length of P5EC (upper panels) and the level of identity between mouse and human orthologs at the protein level. While P5ECs from both datasets have highly variable length, it is clear that those resulting from alternative transcript variants have, on average, higher identity at the protein level as well as lower Ka/Ks values. While low Ka/Ks ratio and high protein identity are good indicators of translated P5ECs, high Ka/Ks ratio and low protein identity does not necessarily means that a P5EC is not translated. This is because, at this stage of the analysis, the statistics were calculated for the entire region between the annotated AUG codon and the nearest in-frame stop codon in the 5′-UTR. However, if alternative initiation occurs closer to the 3′-end of a P5EC, then the region of the P5EC upstream of the ATIS would not evolve under the constraints of protein coding evolution and the cumulative values of Ka/Ks and of protein identity for the entire region would be intermediate between values typical of coding and non-coding sequences. Therefore, we used a relatively relaxed Ka/Ks ratio threshold for selecting the candidates for further detailed analysis (see ‘Materials and Methods’ section).
Figure 3.

Histogram of Ka/Ks values for mRNA sequences with known 5′ extensions. White bars represent mRNAs for which alternative transcripts with extended CDSs are known and therefore corresponding extensions are known to be translated in alternative transcripts. Sequences of these extensions are expected to evolve as protein coding sequences and were used as an internal control in this study. Black bars represent the remaining mRNAs for which it is not known whether alternative mRNA isoforms exist. Curves indicate the number of genes (y-axis) with Ka/Ks below a particular value (x-axis).
Figure 4.

Scatter plots of Ka/Ks ratios for the alignments of the sequences corresponding to P5ECs from different mRNAs (y-axis) in relation to the level of protein identity (bottom panels), and the lengths of P5ECs (top panels). The right-hand panels correspond to mRNAs for which transcript variants with 5′-extended CDSs are known. The left panels correspond to the remaining mRNAs.
Identification and analysis of candidates
After a series of automated, semi-automated and manual steps that are described in detail in ‘Materials and Methods’, 36 candidates satisfied the criteria of our search. One of them, Wilms tumor 1 homolog, has a reported non-AUG initiation in mouse which is homologous to the extension in human and, therefore, the gene was grouped with the known examples of non-AUG initiation discussed elsewhere in this study. In almost all cases, the putative 5′ extensions in the candidates are bracketed upstream by in-frame stop codons in at least one and often many species, thus precluding the existence of an upstream AUG codon for initiation of translation for the extension. In some of the 35 examples, it appears that the occurrence of non-AUG initiation is conserved throughout vertebrates. This is also the case for the known non-AUG extensions in TEAD1, TEAD3, TEAD4 (50) and EIF4G2 (51). Vertebrate genes have numerous paralogs that originated as a result of large-scale DNA duplications in an early chordate (52). For example, TEAD1 has three human paralogs in two of which a non-AUG-initiated N-terminal extension is present and conserved throughout vertebrates. When utilization of non-AUG initiation is ancient for a particular gene, as evident by conservation up to the root of the vertebrate tree, it can be expected that its paralogs may also employ this mechanism. We investigated whether non-AUG extensions are present in paralogs of the 35 provisional positives in our search, especially deeply conserved ones. This led to the identification of seven additional candidates that had escaped initial detection due to limitations in our rigorous selection criteria at various stages of the pipeline.
In the next step, the nature and depth of conservation of the extension in the 42 candidates were probed in non-human/mouse species. In 10 cases (C1QL1, C1QL2, C1QL3, C1QL4, YPEL1, YPEL2, YPEL4, WDR26, FLJ36208 and VANGL2), 7 of them belonging to two paralogous clusters, both the extension and its non-AUG initiation appear to be conserved in all vertebrates for which sequence is available (examples highlighted in green in Table 1). In nine other cases, the extension also appears to be conserved in most or all vertebrates but the non-AUG initiation is conserved only in mammals (highlighted in blue in Table 1). In a further eight cases, the extension appears to be conserved only in mammals and in some of them (usually a minority) it is initiated by AUG either at the position of the putative human non-AUG initiation codon or in its vicinity (highlighted in magenta in Table 1). In two of them the non-AUG initiation evolved recently as all non-primate species have AUG initiation in place of the human non-AUG initiation codon. In one of these, FNDC5, the putative non-AUG initiation is specific to humans while in chimp and gorilla an AUG codon appears to be utilized instead. In the final 15, the extension is conserved only in mammals and is always initiated by a non-AUG codon (highlighted in yellow in Table 1).
In half (i.e. 21) of the newly identified non-AUG-initiated extensions, one to six out-of-frame AUG codons exist between the putative non-AUG start site and the next available in-frame AUG codon downstream. In the example with six out-of-frame AUG codons, R3HCC1 and perhaps in some others, the downstream in-frame AUG would not be available for initiation, and translation of the main CDS would occur solely via the upstream non-AUG initiation site. Conversely, in the 21 extensions lacking any out-of-frame AUG codons, a significant proportion of ribosomes would be predicted to reach the in-frame AUG codon via leaky scanning, resulting in the translation of multiple C-terminally coincident isoforms.
In addition to the 23 human non-AUG utilizing genes compiled previously (21), we found six others either in the published literature or with annotated non-AUG initiation in RefSeq. To help with the evaluation of the candidates reported here for the first time, these 29 known human cases of non-AUG initiation were subjected to the same qualitative analysis as described above for the new candidates. Seventeen known extensions exhibit a conservation pattern similar to the 42 new candidates, while the remaining 12 do not pass the rigorous qualitative tests for conservation used to identify the 42 new candidates. This analysis also showed that in TEAD1, a UUG codon further upstream, in addition to the previously identified AUU codon (53), is perfectly conserved and is likely to also serve as an initiation codon. In DDX17, the previously proposed CUG initiator (54) is not well conserved; however, tandem ACG-GUG codons downstream are perfectly conserved and are the more likely initiation site(s).
The total size of all 42 newly identified extensions is 3374 codons; the average size is 80.3 codons and the median is 51 codons. In the 17 known cases that passed the qualitative test, the average extension is 87.7 codons and the median is 65 codons. Among the newly identified non-AUG-initiated extensions, the shortest is 17 codons and the longest is 300 codons. Among the 17 known and conserved non-AUG-initiated extensions, the shortest is 15 codons and the longest is 235 codons, see Figure 5 for distribution of extension lengths. The annotated sequences of mRNAs described in Table 1 are available in the Supplementary Dataset 1.
Figure 5.

Boxplots of non-AUG CDS extension length distributions for previously known cases and those identified in this study.
Of the 9 possible codons that differ from AUG in a single position, 5 are used to initiate the extensions of the 17 known cases that passed our qualitative analysis for conservation of the extension. By far the most commonly used is CUG, 10 times (59%), followed by ACG, AUA and UUG with two occurrences of each, and GUG with one occurrence. The distribution of the putative initiation codons of the 42 new candidates in humans is not radically different (notwithstanding the potential for observer bias in locating the precise initiation codon in a small number of cases). Seven of the nine possible codons appear to be utilized, with only the very inefficiently recognized AGG and AAG not used. The distribution of the 7 used is: 15 CUGs (36%), 7 ACGs, 6 GUGs, 5 AUUs, 4 each of UUG and AUA and, finally, 1 AUC. This order correlates with the efficiency of initiation for each non-AUG codon, with CUG the most efficient of them all (16). In addition to the identity of the initiation codon, the initiation context of the 17 previously known examples that passed the screening process and the 42 newly identified candidates were examined (Figure 6). Once again the pattern is similar in both cases though the previously identified cases are closer to the optimal context for mammalian genes.
Figure 6.

Weblogo representation of the region surrounding the known and putative conserved non-AUG initiation sites in humans. Numbering is relative to the first nucleotide of the start codon. (A) Representation for the 42 sequences with newly identified extensions. (B) Representation for the 17 sequences with previously identified and conserved extensions. (C) Representation of all AUG start sites of humans [the frequencies for nucleotide occurrence at each position for the human mRNAs were obtained from the Transterm database (73)].
The 42 candidates and the 17 known examples were subjected to a more precise quantitative analysis, including calculation of Ka/Ks (38,41) and MLOGD (42) scores for multiple sequence alignments of up to 32 vertebrate species, and blast bits scores for human–mouse alignments, as described in ‘Materials and Methods’. Candidates were ranked separately by the Ka/Ks ratio, MLOGD score and the BLAST bits score and then the rankings were averaged. The results of these analyses are shown in Table 1. As a general rule, extensions that rank near the top and that show conservation beyond mammals are far more likely to be physiologically significant. Conversely, extensions that rank near the bottom are conserved only in mammals, are never initiated by AUG codons in any species and are less likely to be physiologically significant even if they are in fact translated.
Independent experimental evidence for the non-AUG extensions from Western blot data
For 6 of the 42 candidates identified in our analysis, published western blot data provide independent experimental support for translation of the non-AUG-initiated extension in vivo (see Supplementary Dataset 2). These include:
Ankyrin repeat domain 42 (ANKRD42), also known as several ankyrin repeat protein (SARP), is known to interact with protein phosphatase 1. Browne et al. (55) described the original cloning of the human gene and reported the presence of two protein isoforms in vivo, one with an apparent molecular weight of 92–95 kDa and another with a molecular weight of 65 kDa. Without supporting evidence, they assumed that the two products result from translation on two alternatively spliced mRNA variants represented by two cloned complementary DNAs (cDNAs): DQ508934 (for the longer isoform) and DQ508815 (for the shorter isoform). This, however, is contradicted by a northern blot presented in the same report [Figure 3F in (55)] which shows a single mRNA species of approximate size 3.6 kb. The authors also noted that their longest clone, DQ508934, does not have an in-frame AUG that could initiate a 92- to 95-kDa protein. They then assumed that the AUG must be present in the genomic region just upstream of the 5′-end of the longer cDNA clone. Such an AUG is indeed present in the genomic DNA of humans, 16 nt upstream of the 5′-end of DQ508934, but there is no evidence that this region is transcribed. In fact, although approximately 50 human ESTs exist for the 5′-end of ANKRD42, not a single one extends beyond the 5′-end of DQ508934. In the orthologs in mouse, dog, orangutan, cattle and other mammals, delimiting upstream in-frame stop codons exist that unambiguously occur 3′ of any in-frame AUG codons that might come from the upstream genomic region. The simplest explanation for the protein species observed in vivo is that they correspond to alternative translation initiation from a conserved CUG codon in a good initiation context (producing the extension ranked 29th in Table 1), and the next in-frame AUG codon located ∼900 nt downstream. These products have expected molecular weights of 89.6 and 57.3 kDa, respectively, close to the observed 92–95 and 65 kDa. It is significant that, in the 900 nt space between the conserved CUG and first in-frame AUG, there is a single out-of-frame AUG in humans that is immediately followed by a stop codon and is therefore unlikely to significantly affect scanning ribosomes that failed to initiate on the CUG. In mice and rats, however, six out-of-frame AUG codons are present between the CUG and the first in-frame AUG. Thus, in these species, one would predict synthesis of only a single protein isoform, i.e. the longer CUG-initiated form (unless alternative transcripts are produced).
Surprisingly, considering the direct evidence for its in vivo expression, the data shown in Table 1 suggest that in this case the 5′ extension does not actually evolve under strong purifying selection (Ka/Ks ∼ 0.4). Yet the existence of such a long extension (lacking any in-frame stop codons) in most mammalian orthologs strongly argues that the extension is indeed translated.
Hepatoma-derived growth factor (HDGF), also known as high-mobility group protein 1-like, is a developmentally regulated pulmonary endothelial cell-expressed angiogenic factor. HDGF northern blots identified a single 2.3-kb mRNA in many rat tissues (56). Western blot analysis on human microvascular endothelial cells revealed the presence of at least two protein species—one major with apparent size of 43 kDa and one minor with apparent size of 48 kDa (57). Examination of available ESTs aligning with human HDGF reveals evidence for alternative mRNA variants resulting from different transcription initiation sites and a different exon 1, but none of the ESTs contains an additional in-frame AUG upstream of the previously identified initiation codon. Our analysis predicts that the human HDGF mRNA encodes a 50-amino acid extension initiated by a GUG codon in good context, with both the GUG codon and the context conserved in mammals (ranked 25th in Table 1). In several species, e.g. pig and dog, an upstream delimiting stop codon exists. Initiation on the GUG and on the downstream AUG codon is expected to produce protein products with estimated molecular weights of 31.6 and 26.8 kDa, respectively. Neither of these coincides with the observed products on SDS western blots (48 and 43 kDa), but the difference between the two observed products (∼5 kDa) is nearly identical to the difference between the two predicted products. No out-of-frame AUG codons exist between the GUG and AUG codons. Thus, it appears that standard leaky scanning is sufficient to account for the observed protein products.
Mammalian eukaryotic translation initiation factor 4 γ 3 (EIF4G3, more commonly known as eIF4GII) is a part of the cap-binding protein complex of eukaryotic translation initiation factor 4F. EIF4GII has two known paralogs, eIF4G1 (also known as eIF4GI) and eIF4G2. The latter is known to be initiated at a non-AUG codon (27,51). The study reporting the cloning and initial characterization of eIF4GII identified a single mRNA species by northern blot in humans with a size of 6.0 kb (58). The measured size of the endogenous protein was ∼220 kDa. Another band is present on western blots of the native eIF4GII which is ∼20 kDa above the main band. This band is unremarked in the main text, but it is clearly visible in Figure 3B lane 2 of Gradi et al. (58). The size difference between the two native bands is ∼20 kDa. This is close to the difference between the predicted sizes of the AUC- and AUG-initiated products (extension ranked fourth in Table 1). The proposed AUC initiation codon and its context are conserved in all mammals for which sequence information is available. Curiously, the 4 nt surrounding the putative AUC codon, AAAAUCC (−3 and +4 positions underlined), which include the initiation context, are identical to the equivalent positions of AUU-initiated uORFs in mammalian antizyme inhibitor and the 5′-CUG-initiated N-terminal extension of mammalian antizyme 3 (30).
The non-AUG initiation in EIF4G2 and EIF4G3 is very intriguing because it implies possible autoregulation and hence a role for these two proteins in modulating the stringency of start codon selection. If such autoregulation exists, it would be analogous to the recently discovered autoregulation of eIF1 (see ‘Discussion’ section). So far, however, neither protein has a known role in the stringency of start codon selection.
RASD family, member 2 (RASD2, alternatively known as Rhes) is a Ras homolog and is also related to other Ras GTP-binding proteins. It is predominantly expressed in the striatum of mammalian brains. Mouse experiments with RASD2 reveal behavioral abnormalities when the gene is inactivated (59). Our analysis identified a CUG-initiated extension encoded by the RASD2 mRNA that is conserved in mammals and ranked 30th in Table 1. The knockout analysis is accompanied by western blots on striatum extracts from wild-type and Rasd2−/− animals (59). In mice the CUG-initiated peptide has an expected size of 41.2 kDa while downstream in-frame AUG initiation would result in a protein with a predicted molecular weight of 30.2 kDa. The western blots show three protein products present in the wild-type animals but not in the knockout. One, with an apparent molecular weight of 31 kDa, is nearly identical to a band that appears in HeLa cells transfected with a Rasd2 construct lacking the extension, thus suggesting that this is the product of initiation from the AUG codon. The two other Rasd2-specific products on the western blot have apparent sizes of 39 and 48 kDa. It seems likely that one (or both) of these corresponds to the CUG-initiated protein product. Available human and mouse ESTs do not provide support for the existence of alternative transcripts that could be used to explain the presence of the larger product.
Neurotrophin 3 (NTF3) is a target-derived neurotrophic factor. NTF3 and its paralogs, nerve growth factor, brain-derived neurotrophic factor, neurotrophin 4/5 and neurotrophin 6, bind to both low- and high-affinity receptors on target cells to elicit a cascade of intracellular responses to produce their biological effects. Disruption of NTF3 leads to severe sensory and sympathetic deficits that are incompatible with postnatal life in mice (60). Our analysis indicates the presence of a UUG-initiated extension in human NTF3 (ranked 33rd in Table 1). An antibody for NTF3 is commercially available from Abcam. The product description page shows a western blot with NTF3 antibody detecting two bands—one with an apparent size of 30 kDa and one with a size of 32 kDa, closely matching the predicted sizes of NTF3 initiated at the UUG (33.3 kDa) and at the downstream in-frame AUG (30.8 kDa).
WD repeat domain 26 (WDR26) is a WD40 repeat-containing protein that might be involved in signal transduction and might affect transcriptional regulation (61). The protein is highly conserved from humans to yeast. Our analysis identified a well-conserved ACG/CUG-initiated extension in vertebrates that is present from human to fish, and ranked 22nd in Table 1. In humans the AUG- and ACG-initiated proteins have expected molecular weights of 72.1 and 81.3 kDa, respectively. An antibody for WDR26 is commercially available from Abcam. The product description page shows a western blot that displays two different bands corresponding to proteins with apparent sizes of 80 kDa, for the main product, and 90 kDa for the minor product. The ∼10 kDa difference between the two corresponds closely to the difference expected for initiation on the conserved ACG and on the first in-frame AUG codon.
Curiously, the published annotations of this gene (and the previous version of the Refseq mRNA NM_025160.5) not only omit the non-AUG initiated extension but also the well-conserved first in-frame AUG, and instead assume that initiation occurs on the second in-frame AUG codon (61,62). (Note, however, that the latest version of the Refseq entry NM_025160.6 [updated during preparation of this article on 21 July 2010] has the CDS starting from the first in-frame AUG.)
Orthologs of WDR26 in non-vertebrate metazoans were identified and investigated for the presence of the non-AUG-initiated extension. The gene in Drosophila has an AUU-initiated extension that is conserved in other flies though not in mosquitoes. It appears that the extension arose independently in vertebrates and in flies.
Evidence of non-AUG initiation from ribosomal profiling data
The second set of experimental data supporting our predictions was obtained from recently published ribosomal profiling experiments carried out in human cells (43). In this work, mRNA fragments protected by ribosomes were converted into a library of oligonucleotides and subjected to massively parallel sequencing. The alignment of corresponding sequences onto sequences of mRNAs allows detection of ribosomal locations and the density of the ribosomes on mRNA. We have aligned raw sequences obtained by Guo et al. (43) using the bowtie program (44) and quantified locations of the ribosomes on the mRNA sequences where non-AUG initiation was predicted (see ‘Materials and Methods’ section). We have been able to extract reads for 32 of 59 genes. Examples of ribosomal profiles for NM_004494 and NM_001010858 are shown in Figure 7. For the other sequences, the number of footprints corresponding to CDS and non-AUG extensions, as well as ribosomal density in these regions, are given in Table 1. The presence of ribosomal profiles in the extensions upstream of annotated CDSs indicates that initiation takes place upstream of annotated AUG initiation codons and supports our predictions regarding non-AUG initiation.
Figure 7.

Plots showing density of mRNA fragments protected by ribosomes for NM_004494 and NM_001010858. The position of the annotated AUG codon was taken as zero; relative coordinates of stop codons and predicted non-AUG initiators are indicated. Regions corresponding to annotated CDSs are highlighted in dark grey; regions corresponding to non-AUG-initiated extensions are highlighted in light grey. The presence of ribosomal footprints in the region of an extension indicates that the initiation of translation takes place upstream of the annotated CDS.
Other noteworthy candidates for non-AUG initiation
Ecto-NOX disulfide-thiol exchanger 2 (ENOX2, also known as tNOX) is a hydroquinone and NADH oxidase with protein disulfide-thiol interchange activity (63). It is associated with the outer leaflet of the plasma membrane at the surface of cancer cells and in sera of cancer patients but is absent from the surface of non-cancer cells and from sera of healthy individuals (64). Full-length tNOX mRNA is present in both normal and cancer cells, but its translation is apparently inhibited in normal cells. Our analysis identified an N-terminal extension of 60 amino acids, likely initiated at an upstream UUG codon. The non-AUG initiation is conserved only in mammals, but the extension itself appears conserved in most non-mammalian vertebrates where it is initiated by an AUG codon. The gene ranks sixth in the analysis shown in Table 1 and is strongly supported by evolutionary evidence derived from codon substitutions in multiple alignments.
The mRNA of tNOX has a feature that perhaps explains the low level of expression of this protein in normal cells. The region between the UUG and the downstream in-frame AUG contains an out-of-frame AUG codon in very strong initiation context which starts a 22-codon ORF. The length of this ORF is close to that considered to prevent reinitiation following its translation. Therefore, the role of this short ORF is likely to inhibit AUG initiation of the main CDS thus ensuring that translation of the main CDS is almost solely dependent on initiation at the upstream inefficient UUG.
Western blots for tNOX are available but are not informative about the utilization of the extension in vivo. The detected product is 34 kDa which is only a fraction of the predicted size, 66.6 kDa, of even the AUG-initiated polypeptide, thus indicating extensive post-translational processing.
Not much is known about the function of the gene R3H domain and coiled coil containing 1 (R3HCC1). The protein is well conserved in mammals. Two features of R3HCC1 make it particularly striking with regard to its proposed non-AUG initiation. The first is the existence of six out-of-frame AUG codons between the putative CUG initiation codon in humans, present in perfect Kozak context, and the next available in-frame AUG codon, as mentioned above. This configuration is also present in the other six available mammalian sequences which, on average, have 5.2 out-of-frame AUG codons in this region. The second is the fact that the position of this first available in-frame AUG codon is variable and, in cattle (Bos taurus), it is just eight codons away from the stop codon. In cattle, as indeed in all other mammals, an upstream in-frame delimiting stop codon exists, suggesting that translation of the main CDS would occur solely via the non-AUG initiation site. Considering the large number of out-of-frame AUGs following the conserved CUG (in near perfect Kozak context) and the next in-frame AUG, and the variable position of the AUG, it seems very likely that the CUG codon is the sole position at which translation of this CDS is initiated in most or all mammals.
Ufm1-specific peptidase 1 (UFSP1) is responsible for release of the ubiquitin-like protein Ufm1 from Ufm1-conjugated cellular proteins as well as for activating the Ufm1 precursor (65). UFSP1 is a cysteine protease in which Cys53 (numbering relative to the mouse sequence) is part of the catalytic center and is believed to carry out the nucleophilic attack on the carbonyl carbon of the substrate (66). UFSP1 is a single-exon gene in mammals. Unlike the gene in Mus musculus and three other species, the other 16 available mammalian sequences do not appear to have in-frame AUG codons capable of initiating translation of the full-length protein. The first available in-frame AUG is located at a position equivalent to Met85 of the mouse sequence—i.e. more than 30 residues downstream of the critical Cys53. This means that, even if UFSP1 initiated with Met85 has some biological activity, it would be predicted to be non-functional as a cysteine protease. In fact because of this the human ortholog is currently designated as ‘non-functional’ although our analysis suggests that initiation at a CUG codon (in a good Kozak context and in a position nearly identical to Met1 of mouse Ufsp1) would produce a fully functional enzyme.
DISCUSSION
In this study, we identified 42 novel candidates for conserved non-AUG-initiated N-terminal extensions in RefSeq human mRNAs. The purpose of this study was to find the most reliable candidates for non-AUG initiation and the approach used was highly conservative. Therefore, a large number of filters were used to increase selectivity at the expense of decreased sensitivity. First, the initial pool of data were limited only to those human mRNAs that derive from genes with detectable homologs in mouse. This limitation was needed to ensure sufficient depth of phylogenetic analysis required for MLOGD and Ka/Ks calculations. Therefore all instances of non-AUG initiation utilization in human genes with no orthologs in mouse would be missed by this study. The second limitation was the length of P5EC. All sequences with in-frame stop codons closer than 50 codons to the annotated CDS start site were removed by our pipeline. In addition, all sequences that do not produce an ungapped alignment of mouse-human P5ECs of at least 25 codons in length were removed. The minimal alignment length was set to ensure a sufficient number of substitutions for Ka/Ks estimation. Therefore it is likely that we missed some cases where a non-AUG initiator is located close to the annotated CDS start. In other cases, alternative splicing or alternative transcription initiation sites create mRNA species that are missing the first annotated AUG codon. It is conceivable that some of these transcripts could be translated from a non-AUG codon prior to the next available in-frame AUG. Our search deliberately excluded these potential cases. The currently available RefSeq sequences are often missing significant portions of the 5′-terminal regions of the corresponding mRNAs. This became evident during our manual analysis of the 742 automatically selected candidates. In fact, in one case of a candidate that made it among the 42 finalists (NM_025160, version from 26 June 2007), the originally aligned sequence did have an upstream in-frame AUG codon. However, after the analysis of available ESTs for the locus we built a gene model with an extended 5′-end beyond this AUG that itself turned out to have a conserved non-AUG-initiated extension. The extension appeared to have a candidate non-AUG start, and the protein sequence generated by conceptual translation starting from this non-AUG codon is phylogenetically conserved. In this context, it should be noted that it is conceivable that the extensions of some of the candidates identified here are after all initiated by AUG codons resulting from rare transcript variants for which there is currently no evidence in existing EST databases. Another recently identified case of non-AUG initiation [CUG in thioredoxin reductase 3, TXNRD3 (67)] escaped our analysis for a similar reason—the corresponding Refseq entry XM_001130163 lacks the 5′ region where the CUG codon is situated. Cases of N-terminal extensions under positive selection would also have been missed. Finally, only extensions that can be traced near the root of the mammalian tree were scored as candidates in our analysis but it is likely that smaller phylogenetic branches also have conserved and physiologically active non-AUG-initiated extensions. For example our analysis indicates that the known non-AUG extension of CDKN2B is conserved in primates but not in more distantly related mammals. For these and probably other reasons the cases of physiologically important non-AUG initiation are likely to be more numerous than those identified in the current and previous studies. Furthermore, there could be non-AUG-initiated coding regions in the 5′-UTRs of mRNAs that are in a different reading frame or separated by a stop codon from the main CDS. Examples include a uORF that regulates expression of AZIN1 in vertebrates (30), and others found by ‘wet’ proteomics analysis (68). Such cases of non-AUG initiation would have been completely undetected by our search. Despite all these limitations, our analysis suggests the existence of at least 59 human genes, ∼0.3% of the total, with conserved non-AUG-initiated N-terminal extensions.
Previous examples of non-AUG initiation were almost always found serendipitously after experimental data showed mismatches between expected and observed protein masses or sequences. As our analysis has shown, often such additional protein products were treated as artifacts or simply ignored because identification of their nature was a problem remote from the primary goals of these studies. This study has addressed the issue systematically by employing the power of comparative genomics to predict completely overlooked conserved potential N-terminal extensions.
A very exciting possible reason for utilization of non-AUG initiation is alternative regulation of initiation at AUG and non-AUG start codons. Eukaryotes have developed elaborate mechanisms for recognition of the correct initiation codon. Mutations are known that reduce the fidelity of initiation in eukaryotes—specifically the ‘Sui-’ mutants in Saccharomyces cerevisiae (69–70). Many of these mutations occur in SUI1, also known as eukaryotic initiation factor 1 (eIF1). Within the ribosomal machinery ensuring fidelity of initiation in eukaryotes, eIF1 seems to play a central role (13,71). eIF1 has been implicated in all three proofreading processes during initiation: discrimination between AUG and non-AUG codons; discrimination between AUG codons in good and poor (Kozak) context and discrimination between AUG codons close (<20 nts) to the 5′ cap and AUG codons that are further away from the 5′ cap (the latter being preferred for initiation in eukaryotes). eIF1 appears to confer conformational changes to the scanning 40S ribosome (72). When bound to it, eIF1 induces an open conformation conducive to scanning (13). Upon eIF1 release, the 40S subunit is believed to take a closed conformation that precludes further scanning and promotes initiation of translation. If non-AUG initiation is widely used for regulation, it seems likely that eIF1 plays a major role in the mechanism. Remarkably, eIF1 in most eukaryotes appears to be under translation autoregulation utilizing its ability to inhibit initiation at poor start sites (32). In this case, an AUG codon in a poor context is used instead of non-AUG initiation, but experiments in the same study demonstrated that the amplitude of repression associated with high levels of eIF1 is even more pronounced at non-AUG start codons. Although the existence of a translational regulation mechanism that exploits variability in the stringency of start codon selection is now proven, little is known about the extent of its utilization in humans. Tripling the number of known conserved human non-AUG initiation sites would certainly aid in addressing this issue.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Science Foundation Ireland (grant number 06/IN.1/B81 to P.V.B., 08/IN.1/B1889 to J.F.A.); Wellcome Trust (grant number 088789 to A.E.F.); National Institute of Health (grant number GM079523 to J.F.A.). Funding for open access charge: Science Foundation Ireland.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs Nick Ingolia and Jonathan Weismann for introducing us to the details of the ribosomal profiling technique.
REFERENCES
- 1.Ramakrishnan V. Ribosome structure and the mechanism of translation. Cell. 2002;108:557–572. doi: 10.1016/s0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- 2.Simonetti A, Marzi S, Myasnikov AG, Fabbretti A, Yusupov M, Gualerzi CO, Klaholz BP. Structure of the 30S translation initiation complex. Nature. 2008;455:416–420. doi: 10.1038/nature07192. [DOI] [PubMed] [Google Scholar]
- 3.Potapov AP, Triana-Alonso FJ, Nierhaus KH. Ribosomal decoding processes at codons in the A or P sites depend differently on 2′-OH groups. J. Biol. Chem. 1995;270:17680–17684. doi: 10.1074/jbc.270.30.17680. [DOI] [PubMed] [Google Scholar]
- 4.Baranov PV, Gesteland RF, Atkins JF. P-site tRNA is a crucial initiator of ribosomal frameshifting. RNA. 2004;10:221–230. doi: 10.1261/rna.5122604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 6.Baumann L, Thao ML, Hess JM, Johnson MW, Baumann P. The genetic properties of the primary endosymbionts of mealybugs differ from those of other endosymbionts of plant sap-sucking insects. Appl. Environ. Microbiol. 2002;68:3198–3205. doi: 10.1128/AEM.68.7.3198-3205.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- 8.Kozak M. Evaluation of the “scanning model” for initiation of protein synthesis in eucaryotes. Cell. 1980;22:7–8. doi: 10.1016/0092-8674(80)90148-8. [DOI] [PubMed] [Google Scholar]
- 9.Kozak M. New ways of initiating translation in eukaryotes? Mol. Cell. Biol. 2001;21:1899–1907. doi: 10.1128/MCB.21.6.1899-1907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider R, Agol VI, Andino R, Bayard F, Cavener DR, Chappell SA, Chen JJ, Darlix JL, Dasgupta A, Donze O, et al. New ways of initiating translation in eukaryotes. Mol. Cell. Biol. 2001;21:8238–8246. doi: 10.1128/MCB.21.23.8238-8246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell SF, Lorsch JR. Should I stay or should I go? Eukaryotic translation initiation factors 1 and 1A control start codon recognition. J. Biol. Chem. 2008;283:27345–27349. doi: 10.1074/jbc.R800031200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 16.Peabody DS. Translation initiation at non-AUG triplets in mammalian cells. J. Biol. Chem. 1989;264:5031–5035. [PubMed] [Google Scholar]
- 17.Chen SJ, Lin G, Chang KJ, Yeh LS, Wang CC. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J. Biol. Chem. 2008;283:3173–3180. doi: 10.1074/jbc.M706968200. [DOI] [PubMed] [Google Scholar]
- 18.Kozak M. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol. Cell. Biol. 1989;9:5073–5080. doi: 10.1128/mcb.9.11.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Portis JL, Spangrude GJ, McAtee FJ. Identification of a sequence in the unique 5′ open reading frame of the gene encoding glycosylated Gag which influences the incubation period of neurodegenerative disease induced by a murine retrovirus. J. Virol. 1994;68:3879–3887. doi: 10.1128/jvi.68.6.3879-3887.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozak M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl Acad. Sci. USA. 1990;87:8301–8305. doi: 10.1073/pnas.87.21.8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tikole S, Sankararamakrishnan R. A survey of mRNA sequences with a non-AUG start codon in RefSeq database. J. Biomol. Struct. Dyn. 2006;24:33–42. doi: 10.1080/07391102.2006.10507096. [DOI] [PubMed] [Google Scholar]
- 22.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natsoulis G, Hilger F, Fink GR. The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell. 1986;46:235–243. doi: 10.1016/0092-8674(86)90740-3. [DOI] [PubMed] [Google Scholar]
- 24.Chatton B, Walter P, Ebel JP, Lacroute F, Fasiolo F. The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J. Biol. Chem. 1988;263:52–57. [PubMed] [Google Scholar]
- 25.Souciet G, Menand B, Ovesna J, Cosset A, Dietrich A, Wintz H. Characterization of two bifunctional Arabidopsis thaliana genes coding for mitochondrial and cytosolic forms of valyl-tRNA synthetase and threonyl-tRNA synthetase by alternative use of two in-frame AUGs. Eur. J. Biochem. 1999;266:848–854. doi: 10.1046/j.1432-1327.1999.00922.x. [DOI] [PubMed] [Google Scholar]
- 26.Chang KJ, Wang CC. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J. Biol. Chem. 2004;279:13778–13785. doi: 10.1074/jbc.M311269200. [DOI] [PubMed] [Google Scholar]
- 27.Imataka H, Olsen HS, Sonenberg N. A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J. 1997;16:817–825. doi: 10.1093/emboj/16.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajkowitsch L, Vilela C, Berthelot K, Ramirez CV, McCarthy JE. Reinitiation and recycling are distinct processes occurring downstream of translation termination in yeast. J. Mol. Biol. 2004;335:71–85. doi: 10.1016/j.jmb.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 29.Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 30.Ivanov IP, Loughran G, Atkins JF. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc. Natl Acad. Sci. USA. 2008;105:10079–10084. doi: 10.1073/pnas.0801590105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanov IP, Loughran G, Sachs MS, Atkins JF. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1) Proc. Natl Acad. Sci. USA. 2010;107:18056–18060. doi: 10.1073/pnas.1009269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shabalina SA, Ogurtsov AY, Rogozin IB, Koonin EV, Lipman DJ. Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res. 2004;32:1774–1782. doi: 10.1093/nar/gkh313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kochetov AV. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays. 2008;30:683–691. doi: 10.1002/bies.20771. [DOI] [PubMed] [Google Scholar]
- 35.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 39.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;33:D54–D58. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boguski MS, Lowe TM, Tolstoshev CM. dbEST—database for “expressed sequence tags”. Nat. Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 42.Firth AE, Brown CM. Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics. 2006;7:75. doi: 10.1186/1471-2105-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nekrutenko A, Makova KD, Li WH. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res. 2002;12:198–202. doi: 10.1101/gr.200901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanov IP, Atkins JF. Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. 2007;35:1842–1858. doi: 10.1093/nar/gkm035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bekaert M, Firth AE, Zhang Y, Gladyshev VN, Atkins JF, Baranov PV. Recode-2: new design, new search tools, and many more genes. Nucleic Acids Res. 2010;38:D69–D74. doi: 10.1093/nar/gkp788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin MF, Carlson JW, Crosby MA, Matthews BB, Yu C, Park S, Wan KH, Schroeder AJ, Gramates LS, St Pierre SE, et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Res. 2007;17:1823–1836. doi: 10.1101/gr.6679507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zougman A, Ziolkowski P, Mann M, Wisniewski JR. Evidence for insertional RNA editing in humans. Curr. Biol. 2008;18:1760–1765. doi: 10.1016/j.cub.2008.09.059. [DOI] [PubMed] [Google Scholar]
- 50.Stewart AF, Richard CW, 3rd, Suzow J, Stephan D, Weremowicz S, Morton CC, Adra CN. Cloning of human RTEF-1, a transcriptional enhancer factor-1-related gene preferentially expressed in skeletal muscle: evidence for an ancient multigene family. Genomics. 1996;37:68–76. doi: 10.1006/geno.1996.0522. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi K, Maruyama M, Tokuzawa Y, Murakami M, Oda Y, Yoshikane N, Makabe KW, Ichisaka T, Yamanaka S. Evolutionarily conserved non-AUG translation initiation in NAT1/p97/DAP5 (EIF4G2) Genomics. 2005;85:360–371. doi: 10.1016/j.ygeno.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 52.McLysaght A, Hokamp K, Wolfe KH. Extensive genomic duplication during early chordate evolution. Nat. Genet. 2002;31:200–204. doi: 10.1038/ng884. [DOI] [PubMed] [Google Scholar]
- 53.Xiao JH, Davidson I, Matthes H, Garnier JM, Chambon P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell. 1991;65:551–568. doi: 10.1016/0092-8674(91)90088-g. [DOI] [PubMed] [Google Scholar]
- 54.Uhlmann-Schiffler H, Rossler OG, Stahl H. The mRNA of DEAD box protein p72 is alternatively translated into an 82-kDa RNA helicase. J. Biol. Chem. 2002;277:1066–1075. doi: 10.1074/jbc.M107535200. [DOI] [PubMed] [Google Scholar]
- 55.Browne GJ, Fardilha M, Oxenham SK, Wu W, Helps NR, da Cruz ESOA, Cohen PT, da Cruz ESEF. SARP, a new alternatively spliced protein phosphatase 1 and DNA interacting protein. Biochem. J. 2007;402:187–196. doi: 10.1042/BJ20060600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Everett AD. Identification, cloning, and developmental expression of hepatoma-derived growth factor in the developing rat heart. Dev. Dyn. 2001;222:450–458. doi: 10.1002/dvdy.1204. [DOI] [PubMed] [Google Scholar]
- 57.Everett AD, Narron JV, Stoops T, Nakamura H, Tucker A. Hepatoma-derived growth factor is a pulmonary endothelial cell-expressed angiogenic factor. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L1194–L1201. doi: 10.1152/ajplung.00427.2003. [DOI] [PubMed] [Google Scholar]
- 58.Gradi A, Imataka H, Svitkin YV, Rom E, Raught B, Morino S, Sonenberg N. A novel functional human eukaryotic translation initiation factor 4G. Mol. Cell. Biol. 1998;18:334–342. doi: 10.1128/mcb.18.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spano D, Branchi I, Rosica A, Pirro MT, Riccio A, Mithbaokar P, Affuso A, Arra C, Campolongo P, Terracciano D, et al. Rhes is involved in striatal function. Mol. Cell. Biol. 2004;24:5788–5796. doi: 10.1128/MCB.24.13.5788-5796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou XF, Rush R. Sympathetic neurons in neonatal rats require endogenous neurotrophin-3 for survival. J. Neurosci. 1995;15:6521–6530. doi: 10.1523/JNEUROSCI.15-10-06521.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Wang Y, Xia C, Li D, Li Y, Zeng W, Yuan W, Liu H, Zhu C, Wu X, et al. WDR26: a novel Gbeta-like protein, suppresses MAPK signaling pathway. J. Cell. Biochem. 2004;93:579–587. doi: 10.1002/jcb.20175. [DOI] [PubMed] [Google Scholar]
- 62.Wei X, Song L, Jiang L, Wang G, Luo X, Zhang B, Xiao Overexpression of MIP2, a novel WD-repeat protein, promotes proliferation of H9c2 cells. Biochem. Biophys. Res. Commun. 2010;393:860–863. doi: 10.1016/j.bbrc.2010.02.099. [DOI] [PubMed] [Google Scholar]
- 63.Chueh PJ, Kim C, Cho N, Morre DM, Morre DJ. Molecular cloning and characterization of a tumor-associated, growth-related, and time-keeping hydroquinone (NADH) oxidase (tNOX) of the HeLa cell surface. Biochemistry. 2002;41:3732–3741. doi: 10.1021/bi012041t. [DOI] [PubMed] [Google Scholar]
- 64.Cho N, Chueh PJ, Kim C, Caldwell S, Morre DM, Morre DJ. Monoclonal antibody to a cancer-specific and drug-responsive hydroquinone (NADH) oxidase from the sera of cancer patients. Cancer Immunol. Immunother. 2002;51:121–129. doi: 10.1007/s00262-001-0262-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K, et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem. 2007;282:5256–5262. doi: 10.1074/jbc.M610590200. [DOI] [PubMed] [Google Scholar]
- 66.Ha BH, Ahn HC, Kang SH, Tanaka K, Chung CH, Kim EE. Structural basis for Ufm1 processing by UfSP1. J. Biol. Chem. 2008;283:14893–14900. doi: 10.1074/jbc.M708756200. [DOI] [PubMed] [Google Scholar]
- 67.Gerashchenko MV, Su D, Gladyshev VN. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J. Biol. Chem. 2010;285:4595–4602. doi: 10.1074/jbc.M109.070532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oyama M, Kozuka-Hata H, Suzuki Y, Semba K, Yamamoto T, Sugano S. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol. Cell Proteomics. 2007;6:1000–1006. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- 69.Donahue TF, Cigan AM, Pabich EK, Valavicius BC. Mutations at a Zn(II) finger motif in the yeast eIF-2 beta gene alter ribosomal start-site selection during the scanning process. Cell. 1988;54:621–632. doi: 10.1016/s0092-8674(88)80006-0. [DOI] [PubMed] [Google Scholar]
- 70.Donahue TF. Genetic approaches to translation initiation in Saccharomyces cerevisiae. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational Control of Gene Expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. pp. 595–614. [Google Scholar]
- 71.Nanda JS, Cheung YN, Takacs JE, Martin-Marcos P, Saini AK, Hinnebusch AG, Lorsch JR. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 2009;394:268–285. doi: 10.1016/j.jmb.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Passmore LA, Schmeing TM, Maag D, Applefield DJ, Acker MG, Algire MA, Lorsch JR, Ramakrishnan V. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell. 2007;26:41–50. doi: 10.1016/j.molcel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 73.Jacobs GH, Chen A, Stevens SG, Stockwell PA, Black MA, Tate WP, Brown CM. Transterm: a database to aid the analysis of regulatory sequences in mRNAs. Nucleic Acids Res. 2009;37:D72–D76. doi: 10.1093/nar/gkn763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.