


Abstract
Homing endonucleases (HEs) cleave long (∼20 bp) DNA target sites with high site specificity to catalyze the lateral transfer of parasitic DNA elements. In order to determine whether comprehensive computational design could be used as a general strategy to engineer new HE target site specificities, we used RosettaDesign (RD) to generate 3200 different variants of the mCreI LAGLIDADG HE towards 16 different base pair positions in the 22 bp mCreI target site. Experimental verification of a range of these designs demonstrated that over 2/3 (24 of 35 designs, 69%) had the intended new site specificity, and that 14 of the 15 attempted specificity shifts (93%) were achieved. These results demonstrate the feasibility of using structure-based computational design to engineer HE variants with novel target site specificities to facilitate genome engineering.
INTRODUCTION
Homing endonucleases (HEs) are native proteins found in all domains of life that use their highly site-specific endonucleolytic activity to initiate and target the lateral transfer of parasitic DNA elements. These parasitic DNAs are often self-splicing inteins or introns that encode their cognate HEs (1–3). HE proteins can be subdivided into five families based on shared protein sequence motifs. The LAGLIDADG homing endonucleases (LHEs) comprise the largest of these families with over 400 predicted members (4), and typically have DNA target sites of 22 bp together with a shared 3D-fold (Figure 1A) (3).
Figure 1.

mCreI and DNA target site structure. (A) mCreI is a monomerized version of the native I-CreI homodimeric LAGLIDADG homing endonuclease protein shown here (30). (B) Sequence of the native I-CreI/mCreI target site with positions of phosphodiester backbone cleavages in the top (downward filled triangle) and bottom (upward filled triangle) strands indicated on the sequence of the top strand of native target site DNA. Target site cleavage across the minor groove leaves 4-base, 3′-OH extended cohesive ends. Note the partially palindromic nature of the native target site sequence. (C) mCreI makes both direct and water-mediated contacts with target site DNA predominantly via amino acid residues located predominantly in β-sheets that lie in the major groove of target site DNA. The contact map is an updated and redrawn version of the original contact map shown in (34).
The combination of long target sites and high site specificity of cleavage make HEs potentially useful for genome engineering and therapeutic applications (5,6). However, realizing this potential requires the ability to generate HE variants that target specific genes or genomic targets in living cells. We and others have reported the use of experimental approaches to successfully redirect LAGLIDADG and His-Cys box HEs to new target site specificities (7–17). These efforts have relied chiefly on structure-driven experimental protocols in which large libraries of variant HE proteins are screened to identify members with the desired new cleavage specificity. Recent examples of this approach were the generation of I-CreI variant HEs with high specificity for target sites in genes responsible, when mutant, for human X-linked severe combined immune deficiency (X-SCID) or xeroderma pigmentosum group C (XPC) (14,15).
We recently reported an alternative, structure-based computational approach to alter HE target site specificity. This approach was used to design variants of the I-MsoI LHE that can cleave target sites containing 1, 3 or 4 interrelated base pair changes (18,19). Computational design provides an attractive alternative to the experimental approaches described above: New target site specificities can be generated rapidly, and without the need to create or screen large libraries of protein variants for each new target site/HE combination. Here we demonstrate the ability of computational design to generate variants of the LHE mCreI with altered specificities at all target site base pair positions that make direct or water-mediated contacts in the mCreI 22 bp target site. The high success rate of this approach indicates that computational design can provide a rapid and effective way to generate target site-specific variants of many HE proteins for genome engineering or clinical applications.
MATERIALS AND METHODS
Computational methods
mCreI designs were generated using RosettaDesign (RD), a macromolecular modeling and design suite (20). RD seeks to minimize the energy of a macromolecular system using a physically realistic all-atom energy function. In order to design novel mCreI target site specificities using RD, we simulated the desired change in target site DNA and then allowed RD to remodel and/or mutate nearby amino acids to accommodate the new DNA target (21–23). Only amino acids close enough to contact the simulated base pair change were allowed to mutate; other amino acids in the vicinity were allowed to alter their orientation if needed to accommodate local changes in the DNA interface, whereas more distant amino acids were held fixed. Amino acid side chain conformations and identities were sampled by randomly selecting a position, then substituting a randomly selected side chain rotamer conformation from the Dunbrack backbone-dependent rotamer library (24). RD also allowed water molecules to be inserted at canonical positions previously identified in high resolution structures of DNA–protein interfaces (22). Amino acid conformations and associated hydration patterns that improved the energy of the mCreI-target site DNA complex were preferentially accepted during design runs. With iteration, this process converged on amino acid substitution(s) that generate energetically favorable and specific designs for each target site position and base pair possibility. These designs were then rank-ordered based on their predicted specificity for the altered DNA target site and their structural plausibility, as described below.
This design approach was applied to generate mCreI models towards all single base pair variant possibilities in the DNA target site (Figure 1B and C). The central 4 bp positions in the mCreI target site (positions −2 to +2) were excluded, because mCreI makes no sequence-specific contacts to these positions that could serve as a basis for target specificity (Figure 1B and C). Designs for base pair positions ±11 were also attempted, but did not converge on useful design solutions. These sites are more distant from the protein backbone and hence there are fewer opportunities for introducing new side chain–base interactions beyond the single water-mediated contact, from S32 to guanine, at this base pair position (Figure 1C). Fifty independent iterations of the above protocol were performed against the remaining 16 bp target positions in order to ensure adequate sampling of design space to yield 3200 designs (16 target site positions × 4 bp possibilities/position × 50 iterations).
Overall design favorability was assessed by a combination of a RD-generated specificity score and by visual inspection of molecular models of designs to assess structural plausibility. The specificity of HE designs was estimated by computationally repacking each design with its cognate (design) target site and competitor target sites that contained the remaining 3 bp possibilities at each design position. The predicted binding energies of designs bound to cognate or to competitor target sites were then used to estimate the binding preference of designs for each target site (25). Visual inspection of the DNA interface of designs allowed us to identify energetically favorable designs that had the best (i.e. most structurally plausible and well-oriented) hydrogen bond and/or non-polar interactions with target DNA base pairs, and to discard designs that despite high predicted specificities made no contacts to the design base pair. We discarded ∼25% of designs with >80% predicted specificity on the basis of unfavorable structural properties. The added value of visual inspection for structural plausibility with the incorporation of human intuition has recently been highlighted by the success of the multiplayer online game for protein structural prediction ‘FoldIt,’ based on RD (http://fold.it/portal/) (26).
Recombinant protein purification
The symmetry of the mCreI DNA–protein interface allowed us to analyze one design for each base pair and position as representative of both ‘minus’ and ‘plus’ half site design solutions (Figure 1). Thus we used site-directed mutagenesis to generate the open reading frames for all ‘minus’ half site design variants. In brief, oligonucleotides encoding design amino acid substitutions were used to amplify the mCreI gene in PCR reactions that contained 200 µM dNTPs (New England Biolabs), 0.4 nM of each primer, ∼30–100 ng of the template mCreI gene in pET-15bHE, and 1 unit of Phusion thermophilic DNA polymerase in 1 × High Fidelity Phusion buffer (Finnzymes). Amplified fragments were purified by agarose gel electrophoresis and silica DNA binding (Qiagen), then digested with NcoI and NotI prior to ligation into the T7 expression vector pET-15bHE. Plasmids were then transformed into expression-competent C2566 Escherichia coli cells (New England Biolabs), followed by growth on plates containing 100 μg/ml carbenicillin and 0.2% (w/v) glucose. Protein expression was performed by inoculating individual colonies into 100 ml auto-induction medium [10 g/l tryptone, 5 g/l yeast extract, 0.5% glycerol, 0.05% glucose, 0.2% α-lactose, 0.5 M (NH4)2SO4, 1 M KH2PO4, 1 M Na2HPO4, 1 mM MgS04, 50 µM FeCl3, 20 µM CaCl2, 10 µM MnCl2, 10 µM ZnSO4, 2 µM CoCl2, 2.0 µM CuCl2, 2.0 µM NiCl2, 2.0 µM Na2MoO4, 2.0 µM Na2SeO3, 2.0 µM H3BO3], followed by growth for 8–12 h at 37°C and then a shift to 18°C for an additional 24 h when progressive glucose depletion from the growth medium lead to mCreI expression (27).
Recombinant mCreI proteins were purified using 1 ml HisTrap Ni protein purification columns and an AKTAExpress protein purification machine (GE Healthcare). In brief, bacterial pellets were resuspended in binding buffer (300 mM NaCl, 50 mM NaPO3 pH 8.0, 20 mM imidazole, 2 mM PMSF, 2.5 mM benzamidine), lysed and filtered, then applied to columns and washed with 20 ml wash buffer (300 mM NaCl, 50 mM NaPO3 pH 8.0, 20 mM imidazole). Protein was eluted using a three-step, 19 ml non-linear gradient of wash and elution buffers. Elution buffer consisted of 300 mM NaCl, 50 mM NaPO3 pH 8.0, 500 mM imidazole. The elution steps consisted of a 2 ml linear gradient of 0–30% elution buffer, a 15 ml linear gradient of 30–60% elution buffer and a final 2 ml linear gradient of 60–100% elution buffer. Fractions (1 ml) were monitored by UV absorption, and those containing mCreI were identified by gel electrophoresis of 10 µl aliquots of each fraction. Native mCreI and mCreI designs eluted at ∼312 mM imidazole (corresponding to 40% elution/60% wash buffer). mCreI-containing fractions were pooled, concentrated using a 10 kD protein concentrator (Millipore) to ∼100 µl, and then buffer exchanged by diluting with 7 ml of protein buffer (300 mM NaCl, 50 mM NaPO3 pH 8.0, 5% glycerol) followed by reconcentration to ∼100 µl. Protein concentrations were quantified by Bradford assay (Bio-Rad), then stored in 150 mM NaCl, 25 mM NaPO3 pH 8.0, 52% glycerol at −20°C.
In vitro cleavage assays
A competitive in vitro ‘bar code’ cleavage assay was developed to determine mCreI specificity at each target site position for all four base pair possibilities (Figure 2). The target site library consisted of 61 plasmids, each containing a different mCreI target site with one of four base pair possibilities at each target site position from −10 to +10 (Figure 1B) cloned into pDR–GFP-univ, a modified version of the pDR–GFP recombination reporter plasmid (28); (see http://depts.washington.edu/monnatws/plasmids/pDR-GFP%20univ.pdf for details). This provided a common target site library that could be used for both in vitro and in vivo cleavage assays. Sequence-verified target site plasmids were used as substrates to amplify different target sites for in vitro cleavage analyses. In brief, PCR primers were chosen to place each target site position at the center of an amplicon that could be readily distinguished on the basis of amplicon size from fragments containing the other three base pair possibilities at each target site base pair position (Figure 2). PCR reactions contained 200 µM dNTPs (New England Biolabs), 0.4 nM of each primer, 50 ng of the highly purified pDR–GFP-Cre template, 1.5 M betaine and 1 U of Taq thermophilic DNA polymerase in 1× Thermopol buffer (New England Biolabs). Betaine was required for successful amplification, and necessitated that amplifications be performed immediately after addition. PCR fragments were purified (Qiagen PCR Cleanup) and quantitated by UV spectrometry (Nanodrop), then combined to form substrate pools in which all four base pair possibilities at each target site base pair position were present in equimolar amount.
Figure 2.

In vitro cleavage assay. (A) In vitro ‘bar code’ cleavage assay developed to determine simultaneously cleavage specificity and activity for all four base pair possibilities at single target site positions in a single tube/single gel lane assay. Four primer pairs were used to amplify cleavage substrates from target site plasmids with the target site at the center of the resulting PCR fragment (left panel). Fragment lengths specify the base pair at the target site query position (ranging from 2200 base pairs for ‘A’ sites to 1320 base pairs for ‘T’ sites; right panel). (B) Pools of four substrates were cleaved in a single tube digest prior to separating substrates and cleavage products in a single lane of an agarose gel. Cleavage of substrate molecules at the centrally located mCreI target site generates two equal length cleavage products and a ‘bar code’ linking substrate and cleavage band intensities that reports cleavage activity and specificity simultaneously for all four base pair possibilities at single target site base pair positions. Examples of base-specific (central panel) and non-specific or degenerate cleavage patterns (right panel) are depicted.
Cleavage assays were performed in a final volume of 10 μl that contained the four pooled substrates for each target site base pair position (total cleavage substrate concentration was 65 nM) in 10 mM MgCl2, 20 mM TrisCl pH 8.0 and from 20 to 66 nM mCreI protein. Digestions were performed for 60 min at 37°C, then stopped by adding 100 mM EDTA, 0.1% SDS and 1.3% Ficoll 400 prior to electrophoresis through a 1% agarose/TAE gel to separate substrate and cleavage products. Gels were stained in 1 µg/ml ethidium bromide for 40 min followed by destaining in water for 10 min prior to digital imaging under 302 nm UV light. The intensities of substrate and cleavage product bands were quantified using TotalLab Quant image analysis software (www.totallab.com/products/totallabquant/) (Supplementary Table S1).
RESULTS
Cleavage specificity profiling of mCreI
We used the in vitro bar code assay described above to systematically determine the DNA cleavage specificity of mCreI. This provided systematic and quantitative data to guide and assess our design efforts. mCreI is a monomerized version of the well-characterized homodimeric I-CreI LHE that we generated in order to facilitate both protein and genome engineering applications (29,30). The target site library for these assays consisted of all single base pair variant mCreI DNA target sites cloned into a common target site plasmid (pDR–GFP-univ; Figure 2A). The same target site library was also used to determine cleavage specificity in human cells, where mCreI expression and target site cleavage leads to homologous recombination with the generation of GFP-positive cells that can be detected and quantified by flow cytometry (30) (H. Li and U. Ulge, unpublished data).
Target site specificity for mCreI ranged from highly specific at some base pair positions (e.g. ±3, 4, 9 and 10, where only a single base pair variant was cleavage sensitive), to near-complete degeneracy at other positions (e.g. −2, −5 and ±8, where all four base pair variant target sites displayed detectable cleavage) (Figure 2B; Supplementary Figure S1 top row and additional results not shown). These results systematically extended existing data on mCreI target site specificity by providing quantitative data over a range of protein concentrations, and thus substantially extended our prior analysis of highly complex libraries of potential target sites that were performed at a single protein concentration, and in which only the most cleavage-sensitive sites are likely to have been reliably identified (31).
Generation and experimental verification of mCreI designs
RD was used as a comprehensive computational framework for biomolecular modeling and design to determine the degree to which mCreI could be computationally engineered to cleave single base pair variant DNA target sites. We used RD to generate 3200 mCreI designs from which we selected a subset of 117 for further analysis. These represented the most energetically stable designs, and were the designs predicted to be most specific, of the 50 designs generated for each design target. In instances where these two designs were identical we excluded one from further analysis, thus reducing 128 initial high ranking designs to 117.
We based our choice of designs for further analysis on a combination of RD-predicted specificity, favorable energies and structural plausibility at all design base pair positions and across a range of predicted specificities (Figure 3). We experimentally characterized the cleavage specificity and activity of 35 mCreI design variants representing 13 different single base pair-variant target sites, as well as four different designs against the native mCreI target site (Table 1). Some of the included designs had small amino acid variations at the same residue because in silico calculations did not provide a single best solution despite iterative design attempts (see, e.g. Designs 5–8 against target −9C).
Figure 3.

Graphical representation of mCreI computational design output. A library of 3200 mCreI designs was generated using RD against all 4 base pair possibilities at each target site position from ±3 to ±11 (see text). The RD-predicted specificities and energies of 117 designs are plotted that represent the most energetically stable or the most specific of the 50 designs generated for each design target. Only a single design is plotted for instances in which the most stable and most specific design were the same. Experimentally validated design specificities are represented by squares labeled with the design base pair and position. Useful designs for target site positions ±11 did not emerge and are not represented, nor is a design that cleaved −8C that was an unanticipated—albeit a sequence-specific—outcome of an attempt to design for −8G (Table 1, Design 12).
Table 1.
Summary of experimentally validated mCreI computational designs
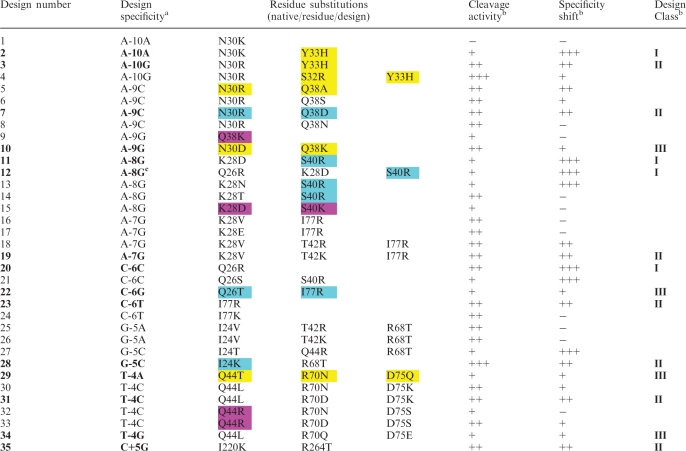 |
Cleavage activity and specificity are shown as ‘−’ for no activity/specificity; ‘+’ for reduced activity/specificity; ‘++’ for levels comparable to native mCreI; and ‘+++’ for greater than native levels. Activity and specificity assessments were made at 20 nM protein concentrations with the exception of designs against native base pairs (e.g. Designs 1, 2, 20 and 21) where specificity and activity were assessed using data across all protein concentrations tested (20–66 nM). The most successful designs for each novel specificity are shown in bold, with Class designations given to the right (see text). Cleavage and specificity data used to prepare Table 1 are summarized in Supplementary Figure S1. The ‘Design specificity’ column lists the native base pair specificity of mCreI followed by the target base pair position number and the design specificity to the right. Color shading of design substitutions indicates residue substitutions previously identified by Seligman and colleagues (yellow-boxed substitutions) (7,12), or by Pâques and colleagues (magenta-boxed substitutions) (9,10,15). Both of these groups used structure-guided random mutagenesis at selected positions in the native I-CreI DNA interface. The blue-boxed substitutions of Gao and colleagues (17) were generated after visual inspection of the I-CreI structure. Of note, our Design 4 32R and 33H substitutions were not combined by Seligman and colleagues and 30D and 28D in Designs 10 and 15 both appeared as glutamates in previous work in contrast to our computationally predicted aspartates. Color shading of design substitutions indicates residue substitutions previously identified by Seligman and colleagues (yellow-boxed substitutions) (7,12), or by Pâques and colleagues (magenta-boxed substitutions) (9,10,15). Both of these groups used structure-guided random mutagenesis at selected positions in the native I-CreI DNA interface. The blue-boxed substitutions of Gao and colleagues (17) were generated after visual inspection of the I-CreI co-crystal structure.
aDesign specificity is indicated by the native base at the numbered design position followed by the design base at that position.
bCleavage activity of native mCreI is defined as ‘++’, whereas cleavage specificity equivalent to native mCreI at a given position is defined as ‘++’. Class designations encompass both cleavage activity and specificity, with Class I designs being more specific, Class II as specific albeit altered, and Class III designs as less specific/more relaxed than native mCreI.
cDesign 12 was originally directed at −8C but was found to have a different specificity, for −8G, when characterized.
The in vitro specificity and activity of individual mCreI designs were assayed using the competitive ‘bar code’ cleavage assay shown in Figure 2 (Supplementary Figure S1). We used a simple and intuitive metric for specificity that assessed the number of single base pair variant target sites at a given base pair position that could be cleaved by a protein design at low protein concentrations (20 or 33 nM). ‘Highly specific’ mCreI design variants cleaved only one target site base pair at the design position, whereas ‘degenerate’ or ‘non-specific’ design variants detectably cleaved all four single base pair variants at the design position. In Table 1, we list the specificity of each design in comparison to WT, with ‘++’ designating designs that cleave as many target variants at the design position as the WT enzyme does.
Among the 13 novel single base pair specificity shifts that were attempted, only one (for −5A) was not achieved for an overall success rate of 93%. Eight novel specificities (for −10G, −9C, −9G, −7G, −6T, −5C, −4C and +5G) were achieved while maintaining cleavage activity comparable to native mCreI (Designs 3, 7, 10, 19, 23, 28, 31 and 35, respectively; Supplementary Figure S1). Four additional designs had the intended novel specificity, but displayed reduced cleavage activity. Two designs against the native mCreI target site showed increased specificity at native target site positions −10 and −6 that is most readily apparent at high enzyme concentrations (Designs 2 and 20; Supplementary Figure S1). Two-thirds of our designs (24 of 35, 69%) thus exhibited the intended specificity shift in conjunction with catalytic activity. Design 12, for −8G, represented an interesting 15th novel specificity: from −8A to C, as opposed to the original design target −8G. This was the sole design that was specific and catalytically active though did not exhibit the intended specificity shift.
The mCreI design successes summarized above represent three classes of outcome with different combinations of specificity and activity that each may be useful for specific engineering applications. Class I, containing four designs, had the highest average RD-predicted specificity of 87% (Figure 7; data not shown). These designs were more specific than mCreI, especially at high-protein concentrations, but were generally less active than native mCreI (Table 1, Supplementary Figure S1). Of note, our prior analyses of mCreI and mMsoI (30) emphasized that even modest levels of catalytic activity are sufficient to promote in vivo cleavage-dependent recombination in human cells. Molecular modeling of Class I designs indicated two different strategies that conferred high specificity: suppressing cleavage of a native base pair while favoring cleavage of an alternative base pair and designing toward the native base pair while suppressing cleavage of other tolerated base pairs. Design 11 is an example of the first of these strategies (Figure 4).
Figure 7.

Predicted and experimentally determined cleavage specificities of successful novel mCreI variants. Fourteen designs are shown color coded by Class of outcome and as a function of their RD-predicted and experimentally determined cleavage specificities (see text). Designs predicted to be more specific typically had higher observed specificities, although the relationship between predicted and observed specificities was modest (R2 value of ∼0.5). Observed specificities were calculated by quantifying the cleavage product intensity in gel images (Supplementary Figure S1), then dividing the intensity of the intended cleavage band by the sum of intensities of all cleavage products at the most specific enzyme concentration (usually 20 nM) (see Supplementary Table S1 for quantified cleavage band intensities). Again, as in Figure 3, a design with novel, but unintended, specificity for −8C has been excluded from the plot.
Figure 4.

Designs with enhanced cleavage specificity at a degenerate target site position. (A) Native mCreI cleaves all four base pair possibilities at the −8 target site position. (B) This lack of specificity reflects the presence of a single water-mediated bond from 28K to −8A. (C) Design 11, in contrast, cleaves only −8G even at high enzyme concentrations. This and comparable designs with enhanced specificities are referred to as Class I designs (see text). (D) The enhanced specificity of Design 11 appears to reflect the ability of residue substitutions to specify a G:C base pair at this position: 40T→R donates two hydrogen bonds to guanine, and 28K→D accepts a hydrogen bond from the complementary −8C. Neither of these interactions was possible with the native target site A:T base pair. Native amino acid residues and the native target site base pair are shown in yellow, design residue substitutions and variant target site base pairs in green, and water molecules as blue spheres (not to scale). The structure of the native enzyme bound to native target site DNA is from the co-crystal structure of I-CreI determined by Chevalier and colleagues (PDB ID 1G9Y). The corresponding structures for designs were computationally generated molecular models.
Class II designs included seven mCreI variants that cleaved design base pairs with specificities comparable to mCreI. The average RD-predicted specificity of Class II enzymes was 77%, and all seven Class II designs were approximately as active as native mCreI (Table 1; Supplementary Figure S1). Design 28, a representative example of Class II designs (Figure 5), preferentially cleaved −5C and to a lesser extent −5T. Native mCreI, in contrast, preferentially cleaved −5G and to a lesser extent −5A (Supplementary Figure S1 top row).
Figure 5.

Designs with altered cleavage specificity at a target site position. (A) Native mCreI preferentially cleaves target sites with −5G, and to a lesser extent −5A followed by −5C or T. (B) Recognition of the −5 position by native mCreI is mediated by 2 contacts made by residue 68R to −5G, and non-polar contact of 24I with the complementary C. (C) Design 28 cleaves −5C to near-completion even at 20 nM, with minor activity on −5T. (D) Recognition of −5C in Design 28 is mediated by 24I→K that contacts to the complementary G at −5, and by 68R→T that prevents potentially deleterious contacts with −5C. Designs with comparably altered specificities are referred to as Class II designs.
Class III designs included four mCreI variants with broader cleavage specificities relative to native mCreI, and an average RD-predicted specificity of 65%. All four Class III designs cleaved both their design base pair and the native base pair at the design position. Design 22, for −6G, is representative of Class III designs (Figure 6). Designs with lower overall specificity may be practically useful when a desired novel specificity can be achieved despite retaining the ability to cleave the native target site base pair.
Figure 6.

Designs that selectively broaden mCreI cleavage specificity. (A and D) Native mCreI cleaves −6C and to a lesser extent −6T at low protein concentrations, whereas Design 22 permits nearly equal cleavage of the −6G design target base pair as well as −6C or T. (B and C) The 26Q sidechain in native mCreI contacts the guanine complementary to −6C (B), but cannot make productive contacts with −6G either from 77I or 26Q (C). (E) In Design 22, 77R contacts both the −6G design target base as well as the adjacent −7A. (F) In the presence of the native −6C:G base pair, 77R pivots to make less favorable contacts to −7A and −8A that permit cleavage but with little base selectivity. Designs with comparably broadened specificities are referred to as Class III designs.
DISCUSSION
We used the Rosetta protein design methodology to generate variants of the monomeric LAGLIDADG homing endonuclease (LHE) mCreI that were specific for target sites containing a broad range of single base pair substitutions. Twenty-four of 35 mCreI design variants (69%) that were experimentally characterized had the intended new site specificities and 14 of 15 (93%) of attempted specificity shifts were achieved. This work represents the first systematic application of structure-based computational design to generate large numbers of catalytically active homing endonuclease proteins, with novel specificities at many different positions, in an LHE DNA–protein interface.
The 15 engineered specificities described above were identified among 3200 computational designs that were rank ordered on the basis of RD-predicted specificity and structural plausibility. This approach was largely successful in predicting the specificity of individual mCreI designs (Figure 7). The average predicted specificity of Class I designs was 10% higher than that of Class II designs, and 22% higher than Class III designs. Because the mCreI DNA–protein interface is highly symmetric, all of our ‘minus’ half site designs should be readily transferrable to comparable positions in the ‘+’ or right mCreI half site (Figure 1). We demonstrated this prediction explicitly with Design 35, in which the design to +5G was shown to have the same specificity/activity profile as its mirror image Design 28 to −5C.
We hypothesized at the outset that there might be a correlation between overall free energy, energetic stability and catalytic activity of designs on their design target sites. However, we found no correlation that would allow us to computationally predict design activity (Figure 3, Supplementary Figure S1). There are at least two explanations for this low concordance. First, different regions of the DNA–protein interface may have different tolerances for residue substitutions. When this source of variability was removed by comparing multiple designs for the same target site and base pair, RD was much better at predicting activity as well as specificity for a design target. Examples include Designs 2 through 4 for target site position −10A, where successive mCreI designs were progressively more active (Table 1 and Supplementary Figure S1); and Designs 16 and 19 for −7G which were more active than Designs 17 or 18. In both instances, RD successfully predicted the relative activity levels of the enzymes based on their calculated energies (Supplementary Figure S1; additional data not shown).
A second potential reason for the inconsistent relationship between predicted energy and activity is related to the design process: designs were selected on the basis of favorable energy, predicted specificity and structural plausibility. This approach makes engineering direct and transparent, but favoring designs with direct contacts to design base pairs may sacrifice favorable interactions with neighboring residues that influence catalysis. It should be possible to address this issue by developing multi-state design protocols to track both specificity and energy simultaneously, in order to explicitly optimize trade-offs during the design process.
Many of our most successful designs involved G:C as opposed to A:T base pairs. RD is particularly good at designing new guanine contacts because major groove electronegative atoms—the N7 and the carbonyl oxygen of C6—represent ideal targets for basic residue substitutions and contacts made by lysine or arginine side chains. The complementary base in a G:C base pair, cytosine, contains a single electropositive group that can be bound by any amino acid containing an electronegative R-group. A:T base pairs, in contrast, contain an electronegative atom and either an electropositive group (adenine) or a methyl group (thymine) that are more difficult to accommodate by residue substitutions in the DNA–protein interface. Adenine, for example, can be contacted by asparagine or glutamine, but only if both the carbonyl oxygen and the amine groups are correctly aligned. In turn, contacting a complementary base pair thymine requires the simultaneous positioning of a polar group over the carbonyl oxygen and a non-polar group over the methyl group. Despite these constraints, it should be possible to improve the rate of design success against A:T base pairs by improving RD’s energy function and by better sampling methods. One improvement would be to allow the protein backbone to move during the design process. This would increase the allowable atomic positions for side chains, and substantially increase the likelihood of identifying favorably aligned substitutions that make new, high specificity contact(s) with design base pairs.
One appeal of the computational design approach taken here is that it can be immediately extended as a general HE engineering approach to other HE proteins for which there are high resolution co-crystal structures. Other protein-specific variables that may facilitate design success include a DNA–protein interface that is ‘modular’ (i.e. a small number of contacts mediate recognition at many of the base pair positions); a large number of G:C base pair design targets; and design target base pair positions where residue substitutions will have little or no direct effect on scissile phosphates.
Our results compare favorably with—and substantially extend—previous efforts to modify I-CreI specificity: only 7 of our 35 computational design solutions (Designs 5, 7, 9, 10, 15, 22 and 29) had been previously identified in prior attempts to engineer I-CreI specificity (Table 1), and 11 of our 15 specificity shifts have not been reported before. Two interesting prior reports that overlap with our results include that of Redondo and colleagues who reported crystallographic structural data that are very similar to our Design 15 structural predictions (15) (additional data not shown), and 5 individual amino acid substitutions that appear in our designs with additional unique substitutions that enhanced cleavage specificity (see, e.g. our designs for −4C; Table 1). All of these previous reports used random mutagenesis and screening or selection to identify novel specificity variants of I-CreI (7,9,10,12,15), or they introduced amino acid mutations based on predictions from visual inspection of the DNA interface (17,32).
A major practical challenge at present is how to combine individual base pair designs to generate HE proteins with high specificity and activity against physiologic targets that contain multiple base pair differences from the native target site. There has already been limited success in combining experimentally identified I-CreI residue substitutions to allow the recognition of target sites with multiple base pair differences [see, e.g. (15,16)]. Iterative or directed application of the general design protocol described here should enable the design of HE proteins with high specificity and activity against even these more challenging target sites. The feasibility of a computational approach has already been demonstrated by using RD to generate variants of the I-MsoI LHE to recognize target sites with 1, 3 or 4 contiguous target site base pair changes (19).
The computational design strategies we employed have two important practical advantages for the engineering of HEs or other proteins: they are not protein specific, and they are fast. RD provides a comprehensive computational framework for modeling and design that can be applied immediately to other HE proteins for which there is a high-resolution structure [examples include I-MsoI (18,19) as mentioned above and I-AniI (33)]. RD design protocols are also fast: our comprehensive library of 3200 mCreI designs was generated in automated fashion in under 12 h using modest computational resources. In contrast, experimental approaches to HE engineering are time and materials intensive, and require a substantial new investment for each new HE protein/target site pair. Moreover, experimental approaches often fail to identify why problems arise when they do, in contrast to computational design strategies that make use of prior successes and failures in a rational and directed manner to facilitate protein engineering.
Merging computational and experimental approaches to HE engineering should provide a particularly powerful, fast and reliable way to generate HE variants with desired target site specificities. This can be most readily achieved at present by using computational design to generate small pools of high-quality designs that can then be rapidly subjected to selection or screening to identify or optimize a desired specificity. The resulting HE protein designs should be useful as highly site-specific catalysts to enable a wide range of biological, medical and industrial applications that require precise genome engineering.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. National Institutes of Health Interdisciplinary Training Grant in Cancer Research (T32 CA80416), F30 (DK083223) and Poncin Fund awards (to U.Y.U.); U.S. National Institutes of Health Interdisciplinary Research Roadmap and Bill and Melinda Gates Foundation/Foundation for the National Institutes of Health Grand Challenges in Global Health awards (to D.A.B); U.S. National Institutes of Health U54 Interdisciplinary Research Roadmap award (1RL1 CA133831) and Bill and Melinda Gates Foundation/Foundation for the National Institutes of Health Grand Challenges in Global Health awards (to R.J.M.). Funding for open access charge: National Institutes of Health grant (1RL1 CA133831).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Justin Ashworth, Jordan Jarjour, Barry Stoddard and Sheila Teves for help with conceptual and experimental aspects of this project. Stefan Pellenz generated the pDR-GFP-univ plasmid used for target site library constructions, whereas Alden F.M. Hackmann assembled figures and proof-read the final manuscript.
REFERENCES
- 1.Belfort M, Roberts R. Homing endonucleases - keeping the house in order. Nucleic Acids Res. 1997;25:3379–3388. doi: 10.1093/nar/25.17.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimble FS. Invasion of a multitude of genetic niches by mobile endonuclease genes. FEMS Microbiol. Lett. 2000;185:99–107. doi: 10.1111/j.1574-6968.2000.tb09046.x. [DOI] [PubMed] [Google Scholar]
- 3.Stoddard BL. Homing endonuclease structure and function. Q Rev. Biophy. 2005;38:1–47. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 4.Grishin A, Fonfara I, Alexeevski A, Spirin S, Zanegina O, Karyagina A, Alexeyevsky D, Wende W. Identification of conserved features of LAGLIDADG homing endonucleases. J. Bioinformatics Comput. Biol. 2010;8:453–469. doi: 10.1142/s0219720010004665. [DOI] [PubMed] [Google Scholar]
- 5.Pâques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives on gene therapy. Curr. Gene Ther. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- 6.Stoddard BL, Scharenberg AM, Monnat RJ., Jr . Advances in engineering homing endonucleases for gene targeting: Ten years after structures. In: Bertolotti R, editor. Progress in Gene Therapy. Singapore: World Scientific Publishing; 2007. [Google Scholar]
- 7.Seligman LM, Chisholm KM, Chevalier BS, Chadsey MS, Edwards ST, Savage JH, Veillet AL. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002;30:3870–3879. doi: 10.1093/nar/gkf495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Zhao H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005;33:e154. doi: 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, Stricher F, Petit AS, Patin A, Guillier S, et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J. Mol. Biol. 2006;355:443–458. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 10.Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J. Am. Chem. Soc. 2006;128:2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- 12.Rosen LE, Morrison HA, Masri S, Brown MJ, Springstubb B, Sussman D, Stoddard BL, Seligman LM. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. 2006;34:4791–4800. doi: 10.1093/nar/gkl645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eklund JL, Ulge UY, Eastberg J, Monnat RJ., Jr Altered target site specificity variants of the I-PpoI His-Cys box homing endonuclease. Nucleic Acids Res. 2007;35:5839–5850. doi: 10.1093/nar/gkm624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnould S, Perez C, Cabaniols JP, Smith J, Gouble A, Grizot S, Epinat JC, Duclert A, Duchateau P, Pâques F. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. J. Mol. Biol. 2007;371:49–65. doi: 10.1016/j.jmb.2007.04.079. [DOI] [PubMed] [Google Scholar]
- 15.Redondo P, Prieto J, Munoz IG, Alibes A, Stricher F, Serrano L, Cabaniols JP, Daboussi F, Arnould S, Perez C, et al. Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases. Nature. 2008;456:107–111. doi: 10.1038/nature07343. [DOI] [PubMed] [Google Scholar]
- 16.Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N, Villate M, Thomas S, Lemaire L, Montoya G, et al. Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Res. 2009;37:5405–5419. doi: 10.1093/nar/gkp548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao H, Smith J, Yang M, Jones S, Djukanovic V, Nicholson MG, West A, Bidney D, Falco SC, Jantz D, et al. Heritable targeted mutagenesis in maize using a designed endonuclease. Plant J. 2010;61:176–187. doi: 10.1111/j.1365-313X.2009.04041.x. [DOI] [PubMed] [Google Scholar]
- 18.Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ, Stoddard BL, Baker D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, Baker D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010;38:5601–5608. doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R, Baker D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 21.Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J. Mol. Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 22.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das R, André I, Shen Y, Wu Y, Lemak A, Bansal S, Arrowsmith CH, Szyperski T, Baker D. Simultaneous prediction of protein folding and docking at high resolution. Proc. Natl Acad. Sci. USA. 2009;106:18978–18983. doi: 10.1073/pnas.0904407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashworth J, Baker D. Assessment of the optimization of affinity and specificity at protein-DNA interfaces. Nucleic Acids Res. 2009;37:e73. doi: 10.1093/nar/gkp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, Leaver-Fay A, Baker D, Popovic Z, and players Predicting protein structures with a multiplayer online game. Nature. 2010;466:756–760. doi: 10.1038/nature09304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Studier FW. Protein purification by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Develop. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dürrenberger F, Rochaix J-D. Characterization of the cleavage site and the recognition sequence of the I-CreI DNA endonuclease encoded by the chloroplast ribosomal intron of Chlamydomonas reinhardtii. Mol. Gen. Genet. 1993;236:409–414. doi: 10.1007/BF00277141. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Pellenz S, Ulge U, Stoddard BL, Monnat RJ., Jr Generation of single-chain LAGLIDADG homing endonucleases from native homodimeric precursor proteins. Nucleic Acids Res. 2009;37:1650–1662. doi: 10.1093/nar/gkp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Argast GM, Stephens KM, Emond MJ, Monnat RJ., Jr I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J. Mol. Biol. 1998;280:345–353. doi: 10.1006/jmbi.1998.1886. [DOI] [PubMed] [Google Scholar]
- 32.Epinat JC, Arnould S, Chames P, Rochaix P, Desfontaines D, Puzin C, Patin A, Zanghellini A, Paques F, Lacroix E. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003;31:2952–2962. doi: 10.1093/nar/gkg375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, Stoddard BL, Baker D. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–1304. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurica MS, Monnat RJ, Jr, Stoddard BL. DNA recognition and cleavage by the LADLIDADG homing endonuclease I-CreI. Mol. Cell. 1998;2:469–476. doi: 10.1016/s1097-2765(00)80146-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.