

Abstract
Brain electrical activity is largely composed of oscillations at characteristic frequencies. These rhythms are hierarchically organized and are thought to perform important pathological and physiological functions. The slow wave is a fundamental cortical rhythm that emerges in deep non-rapid eye movement sleep. In animals, the slow wave modulates delta, theta, spindle, alpha, beta, gamma and ripple oscillations, thus orchestrating brain electrical rhythms in sleep. While slow wave activity can enhance epileptic manifestations, it is also thought to underlie essential restorative processes and facilitate the consolidation of declarative memories. Animal studies show that slow wave activity is composed of rhythmically recurring phases of widespread, increased cortical cellular and synaptic activity, referred to as active- or up-state, followed by cellular and synaptic inactivation, referred to as silent- or down-state. However, its neural mechanisms in humans are poorly understood, since the traditional intracellular techniques used in animals are inappropriate for investigating the cellular and synaptic/transmembrane events in humans. To elucidate the intracortical neuronal mechanisms of slow wave activity in humans, novel, laminar multichannel microelectrodes were chronically implanted into the cortex of patients with drug-resistant focal epilepsy undergoing cortical mapping for seizure focus localization. Intracortical laminar local field potential gradient, multiple-unit and single-unit activities were recorded during slow wave sleep, related to simultaneous electrocorticography, and analysed with current source density and spectral methods. We found that slow wave activity in humans reflects a rhythmic oscillation between widespread cortical activation and silence. Cortical activation was demonstrated as increased wideband (0.3–200 Hz) spectral power including virtually all bands of cortical oscillations, increased multiple- and single-unit activity and powerful inward transmembrane currents, mainly localized to the supragranular layers. Neuronal firing in the up-state was sparse and the average discharge rate of single cells was less than expected from animal studies. Action potentials at up-state onset were synchronized within ±10 ms across all cortical layers, suggesting that any layer could initiate firing at up-state onset. These findings provide strong direct experimental evidence that slow wave activity in humans is characterized by hyperpolarizing currents associated with suppressed cell firing, alternating with high levels of oscillatory synaptic/transmembrane activity associated with increased cell firing. Our results emphasize the major involvement of supragranular layers in the genesis of slow wave activity.
Keywords: current source density, unit activity, laminar recording, slow wave activity, sleep
Introduction
Brain rhythms, a prominent characteristic of EEG discovered in its initial recordings by Berger (1929), are thought to organize cortical activity (Buzsaki and Draguhn, 2004). Especially prominent in sleep (Loomis et al., 1937), microphysiological studies of their neural basis have until now relied on animal models (Steriade, 2006). Presurgical diagnostic procedures in epilepsy may allow the experimenter to open an invasive window on the brain and record local field and action potentials in order to investigate the fine scale generators of electrical brain oscillations (Worrell et al., 2004, 2008; Jirsch et al., 2006; Clemens et al., 2007; Urrestarazu et al., 2007; Axmacher et al., 2008; Fabo et al., 2008; Cash et al., 2009; Jacobs et al., 2009; Schevon et al., 2009; Crepon et al., 2010). Traditionally, cortical oscillations have been divided into distinct bands, with more or less distinct roles in, for example, vigilance states, various cognitive functions and pathology. Most importantly, the slow (delta) and especially the very fast rhythms (ripple and fast ripple) have been found with fine scale intracranial observations to influence pathological excitability effectively, and may serve as a basic substrate underlying paroxysmal activity (Bragin et al., 2002; Worrell et al., 2004, 2008; Jirsch et al., 2006; Urrestarazu et al., 2007; Fabo et al., 2008; Jacobs et al., 2009; Schevon et al., 2009; Crepon et al., 2010). More generally, during normal cortical function, oscillations are hierarchically organized and this oscillatory hierarchy can effectively control neuronal excitability and stimulus processing (Lakatos et al., 2005; Steriade, 2006). Low-frequency oscillations seem to play an important role in cognitive functions even in the awake state (Lakatos et al., 2008; Schroeder and Lakatos, 2009), despite the fact that under other circumstances, slow rhythms are usually good signatures of compromised cerebral functions (Ebersole and Pedley, 2003) and sleep (Achermann and Borbely, 1997). In this work, we attempt to link the slow- and higher-frequency cortical oscillations to gain a better insight into the intricate mechanisms of human cortical electrical activity, and show the organizing principles that may govern the structure of human cortical electrical rhythms in sleep.
A fundamental mode of cortical activity in mammals is the predominance of slow (<1 Hz) oscillations during the deepest stage of non-rapid eye movement (REM) sleep (Achermann and Borbely, 1997; Steriade et al., 2001; Timofeev et al., 2001; Luczak et al., 2007). In humans, this stage (the third and deepest stage of non-REM sleep; N3, also called slow wave sleep) is reached when 20% or more of an epoch consists of slow wave activity (0.5–2 Hz) in the frontal EEG, having peak-to-peak amplitudes larger than 75 μV, and accompanied by the behavioural signs of sleep (Iber et al., 2007). Intracellular recordings in cats during natural slow wave sleep have revealed that slow oscillations are composed of rhythmically recurring phases of increased cellular and synaptic activity (up-states) followed by hyperpolarization and cellular silence (down-states) (Steriade and Timofeev, 2003). In human slow wave sleep, the surface positive slow wave activity half-wave (up-state) contains increased alpha and beta power compared with the surface negative slow wave activity half-wave (down-state), suggesting that their basic neurophysiology may be similar to animal findings (Molle et al., 2002; Massimini et al., 2004). While the slow oscillation in animals is limited to below 1 Hz (Steriade et al., 1993b), the recent American Academy of Sleep Medicine guidelines suggest the 0.5–2 Hz range for slow wave activity in humans (Iber et al., 2007).
Studies into the neural mechanisms of slow waves have been motivated by reports that they underlie restorative sleep functions and serve memory consolidation (Huber et al., 2004; Marshall et al., 2006; Vyazovskiy et al., 2008) via ensemble reactivation (Born et al., 2006; Euston et al., 2007) and synaptic strength normalization (Vyazovskiy et al., 2008). Slow oscillation can be induced artificially by various anaesthetics in vivo (Steriade et al., 1993b; Volgushev et al., 2006; Luczak et al., 2007) and ionic environments in vitro (Sanchez-Vives and McCormick, 2000; Shu et al., 2003; Haider et al., 2006). Slow oscillations are generated in cortico-cortical networks, since they survive thalamectomy (Steriade et al., 1993a), but not the disruption of cortio-cortical connections (Amzica and Steriade, 1995). However, recent data suggest a complex thalamocortical interplay in slow oscillation generation (Crunelli and Hughes, 2010). Fine scale laminar analysis of neuronal firing activity revealed that artificial slow oscillations in slice preparations are the earliest and most prominent in the infragranular layers, where they are initiated, and spread toward the superficial layers with a long ∼100 ms inter-laminar delay (Sanchez-Vives and McCormick, 2000). Subthreshold membrane potential fluctuations giving rise to local field potentials (LFPs) clearly precede neuronal firing at up-state onset; thus, firing may be the consequence rather than the cause of up-state initiation (Chauvette et al., 2010). Current source density (CSD) analysis of the low-frequency (<1 Hz) components of the artificial, anaesthesia-induced slow oscillations (Steriade and Amzica, 1996) localized the most prominent up-state-related sinks to the middle and deepest cortical layers (most probably layers III and VI). In contrast, the fast (30–40 Hz) components were more distributed, composed of ‘alternating microsinks and microsources’ along the whole cortical depth (Steriade and Amzica, 1996). In another publication the same authors reported a massive up-state-related sink in layers II–III, besides weaker ones in the deeper layers during spontaneous and evoked K-complexes (Amzica and Steriade, 1998). In still another cat study, the maximal up-state-related sink in natural sleep was located in the middle and deep layers (Chauvette et al., 2010). The laminar distribution of the major up-state-related sink in the rat primary auditory cortex was variable (Sakata and Harris, 2009). On average across animals, the maximal up-state-related sink was located in middle and deep layers (most probably layers III–V) in natural sleep, whereas it was located in superficial layers (most probably layers II and III) under urethane anaesthesia (Sakata and Harris, 2009). In intact animals the up-state onset-related initial firing, intracellular membrane potential and LFP changes could be detected in any layer in a probabilistic manner, with a short inter-laminar delay (∼10 ms); however, on average, the earliest activity was found in the infragranular layers (Sakata and Harris, 2009; Chauvette et al., 2010). Although the cellular and synaptic/transmembrane mechanisms of slow waves during natural sleep are thus under intense investigation in animals, these mechanisms have not previously been studied in humans.
Here we show the basic correspondence between the surface positive phase of the slow wave in humans and the up-state described in animals. Besides spindle and beta band oscillations (Molle et al., 2002), we found that the up-state in humans strongly groups action potentials, alpha, gamma (30–150 Hz) and ripple oscillations (100–200 Hz), which have been implicated in attention, memory and epilepsy (Grenier et al., 2003; Molle et al., 2004; Jensen et al., 2007). Despite these basic similarities, we found that the neural mechanisms of natural slow wave activity in humans show several differences compared with previous studies in animals. Specifically, the cortical synaptic/transmembrane generators of the slow wave activity slow (<2 Hz) components, as well as the associated high-frequency (10–200 Hz) oscillations, were all strongly and consistently localized in the supragranular layers, in partial contrast to previous proposals based on studies in animals (Steriade and Amzica, 1996; Sakata and Harris, 2009; Chauvette et al., 2010). In addition, we found that, as measured by cellular discharges, the onset of the up-state was rather synchronous across cortical layers, as in intact animals (Sakata and Harris, 2009; Chauvette et al., 2010) but unlike in slices from ferrets, where long inter-laminar firing delays at up-state onset were found (Sanchez-Vives and McCormick, 2000). Furthermore, the average firing rate of human cortical neurons in the up-state was a fraction of what has generally been observed in animal studies (Steriade et al., 2001; Isomura et al., 2006; Luczak et al., 2007). We consider the experimental, cytoarchitectonic, pathological and phylogenetic aspects that may have contributed to these important differences between the slow waves in humans versus lower mammals.
Materials and methods
Patients and electrodes
Five patients with intractable epilepsy underwent chronic clinical subdural grid and strip electrode implantation (Fig. 1) as a standard procedure for localization of the seizure focus and eloquent areas. Fully informed consent was obtained from each subject under the auspices of the Hungarian Medical Scientific Council and local ethical committee, National Institute of Neuroscience, Budapest, Hungary according to the World Medical Association Declaration of Helsinki. Conventional clinical subdural electrocorticography (ECoG) electrode strips and grids were implanted to cover the frontal, temporal and parietal gyri. In addition to the surface electrodes, a 350 µm diameter, 24-contact experimental laminar multichannel microelectrode array was implanted perpendicular to the cortical surface, underneath the clinical grids (Ulbert et al., 2001a, b, 2004a; Cash et al., 2009; Keller et al., 2009). The 40 µm diameter Platinum/Iridium contacts were spaced evenly at 150 µm providing LFP recordings from a vertical, 3.5 mm long cortical track, spanning from layer I to layer VI. A silicone sheet attached to the top of the microelectrode array shank prevented the first contact from sliding more than 100 µm below the pial surface (Ulbert et al., 2001a). In each case, the explanted microelectrode array was visually inspected under a microscope for structural damage, and we did not find any alteration, indicating intact structure throughout the recordings. The location and duration of the clinical electrode implantation were determined entirely by clinical considerations, and the microelectrode array was placed in cortex that was likely to be removed at the definitive surgery.
Figure 1.
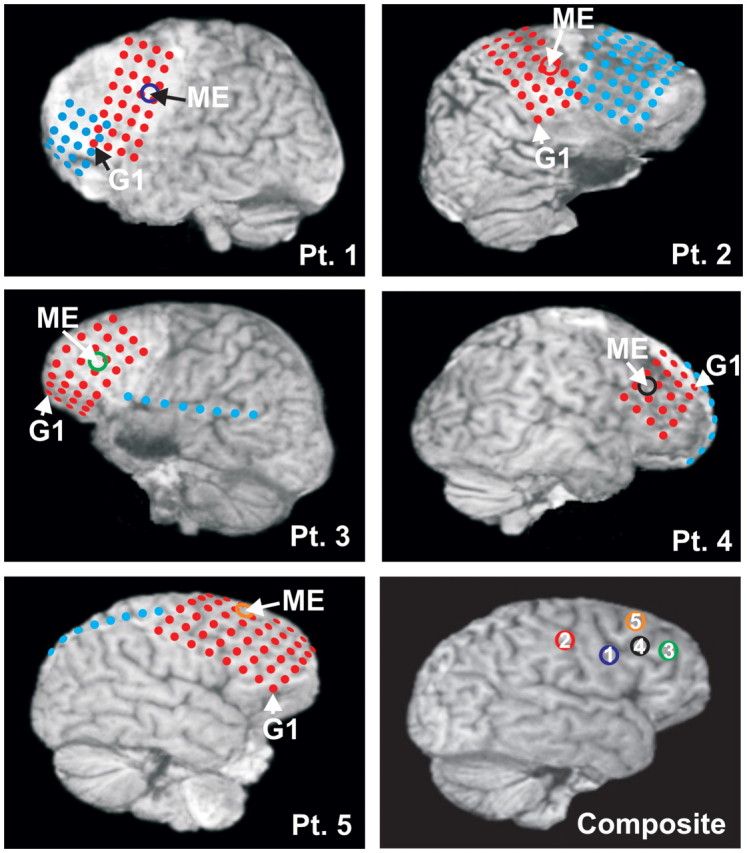
Grid, strip and multichannel microelectrode array (ME) locations in all patients. Locations are superimposed on 3D reconstructions of MRIs taken with the electrodes in place, aided by intraoperative navigation and photographs. Grid and strip electrode contacts are depicted in red and blue colours; the first grid contact is marked with G1. Microelectrode array locations of Patient (Pt) 1 (blue), left Brodmann area (BA) 9; Patient 2 (red), right BA 2; Patient 3 (green), left BA 46; Patient 4 (black), right BA 9; Patient 5 (orange), right BA 8 are marked with circles.
Histology
The positions of the electrodes were confirmed by intraoperative navigation, co-localization of intraoperative photographs, pre- and postoperative magnetic resonance scans and 3D magnetic resonance reconstructions (Fig. 1). Photographs were also taken during the resective surgery to confirm that the surface electrodes did not shift during monitoring. The brain tissue containing the electrode track in Patients 4 and 5 was removed en bloc for further anatomical analysis (Ulbert et al., 2004b; Fabo et al., 2008). It was cut into 2–5 mm blocks and immersed into a fixative containing 4% paraformaldehyde, 0.1% glutaraldehyde and 0.2% picric acid in 0.1 M phosphate buffer (pH 7.4). The fixative was changed every hour to a fresh solution during constant agitation for 6 h, and then the blocks were post-fixed in the same fixative overnight. Vibratome sections (60 µm thick) were cut from the blocks and photographs were taken from the electrode tracks. Following washing in phosphate buffer, sections were immersed in 30% sucrose for 1–2 days and then frozen three times over liquid nitrogen. Endogenous peroxidase was blocked by 1% H2O2 in phosphate buffer for 10 min. Sections containing the electrode track were processed for immunostaining against the neuron marker NeuN (Fig. 2A), calretinin (Fig. 2B, reconstructed from camera lucida), SMI-32 (Fig. 2C) and glial fibrillar acidic protein (Fig. 2D) to stain every neuron, a subset of interneurons, pyramidal cells and glia, respectively. Phosphate buffer was used for all the washes (3 × 3–10 min between each step) and dilution of the antisera. Non-specific immunostaining was blocked by 5% milk powder and 2% bovine serum albumin. Monoclonal mouse antibodies against NeuN (1:3000, Chemicon, Temecula, CA, USA), SMI-32 (1:3000, Covance, Princeton, NJ, USA), glial fibrillar acidic protein (1:2000, Novocastra, Newcastle, upon Tyne, UK) and calretinin (1:5000, SWANT, Bellinzona, Switzerland) were used for 2 days at 4°C. Specificity of the antibodies has been thoroughly tested by the manufacturers. For visualization of immunopositive elements, biotinylated anti-mouse immunoglobulin G (1:300, Vector) was applied as secondary antiserum followed by avidin-biotinylated horseradish peroxidase complex (ABC; 1:300, Vector). The immunoperoxidase reaction was developed by 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma), as a chromogen. Sections were then osmicated (0.25% OsO4 in phosphate buffer, 30 min) and dehydrated in ethanol (1% uranyl acetate was added at the 70% ethanol stage for 30 min) and mounted in Durcupan (ACM, Fluka). Layers of the neocortex were outlined using all of the above stains and a shrinkage correction factor published earlier (Turner et al., 1995; Wittner et al., 2006).
Figure 2.

Electrode track histology, representative examples from Patient 4. (A) The microelectrode array electrode track (black contour line) and inferred contact locations (black dots) are shown relative to cortical layers (Roman numbers) revealed by the NeuN stain. Laminarization appears to be intact. (B) Camera lucida reconstruction of calretinin immunopositive (CR+) cell bodies and processes next to the electrode track. (C) Well preserved pyramidal (SMI-32 stain) and (D) glial cells, stained with glial fibrillar acidic protein (GFAP) next to the electrode track.
Cell counting was performed in Patients 4 and 5 using camera lucida drawing (Fig. 2B) of calretinin immunopositive cells (two sections per patient). The normalized (between 0 and 1) calretinin immunopositive cell density laminar depth profile (number of cells over unit area of cortex) was calculated in each consecutive 150 µm wide and variable length (1–3 mm) horizontal cortical stripes to match the depth structure of the electrophysiology measurements.
Recordings
After electrode placement, the patients were transferred to the intensive monitoring unit for 5–7 days, where continuous 24 h video-EEG observation took place in order to localize the seizure focus. ECoG from clinical strip and grid electrodes (32–92 channels, mastoid reference) was recorded concurrently with patient video using the standard hospital system (band-pass: 0.1–200 Hz, acquisition rate: 400–5000 Hz/16 bit). Video-EEG data for the duration of monitoring were stored on hard disks for later analysis. The spatial LFP gradient, the voltage difference between consecutive laminar electrode contacts, was provided by a special preamplifier placed inside the head bandage of the patient (Ulbert et al., 2001a). For simplicity, throughout the text the spatial potential gradient is expressed in microvolts rather than the formally correct microvolt per inter-contact distance (150 µm). This reference-independent measurement method was proven to be effective in minimizing the motion-related and electro-magnetic artefacts (Ulbert et al., 2001a). The LFP gradient was split into the EEG range (0.1–300 Hz) and single- and multiple-unit activity frequency range (300–5000 Hz) by analogue band-pass filtering at the level of a custom-made main amplifier (Ulbert et al., 2001a). An EEG range signal was sampled at 2 kHz/16 bit; the multiple-unit activity range was sampled at 20 kHz/12 bit and stored on a hard drive.
Slow wave activity detection
We have analysed the LFP gradient, multiple-unit activity, single-unit activity and ECoG data acquired from each patient during one to three nocturnal recording sessions. Since the sleep of the patients was fragmented due to medical care and distress from the hospitalization and head wound, we cannot provide standard hypnograms that are usually obtained from healthy subjects without a recent craniotomy. Craniotomies may also distort the scalp distribution of the EEG due to the lack of bone and excessive fluid accumulation below the scalp; furthermore, if scalp electrodes are placed close to the frontal craniotomy wounds, they may induce infection, and therefore we avoided placing more than two frontal scalp EEG electrodes. Partial sleep staging was performed based on readings of the available scalp EEG and ECoG electrodes by expert neurologists. In this study, we have analysed electrophysiological data obtained only from the deepest stage of non-REM sleep (N3, or slow wave sleep) (Iber et al., 2007). Behavioural sleep was confirmed by the video recording, while slow wave sleep was electrographically identified in accordance with the recent American Academy of Sleep Medicine guidelines (Iber et al., 2007). Slow wave sleep periods were identified when 20% or more of an epoch consisted of slow wave activity (waves in the 0.5–2 Hz frequency range with peak-to-peak amplitude larger than 75 μV, measured over the frontal regions) (Iber et al., 2007). Data containing interictal spikes (within 1 min) and seizures (within 60 min) were excluded from the study to avoid epileptic contamination.
In addition to spectral (Fig. 3A) and autocorrelation analyses (Fig. 3B), slow wave activity cycle detection was based on phase and amplitude information, extracted from the narrow-band filtered (0.3–3 Hz, 24 dB/octave, zero phase shift) layer II LFP gradient (Fig. 4A) and ECoG (Fig. 4B) data. Instantaneous phase of the filtered signal was calculated by the Hilbert transformation. In our implementation, a single slow wave activity cycle was defined between –180° and +180° phase. The –180° phase value corresponded to the trough of the negative half-wave (down-state) preceding the 0° phase, which corresponded to the peak of the positive half-wave (up-state) and finally the +180° phase corresponded to the following negative half-wave trough (down-state). At each +180° crossing, the phase was wrapped –360° for better visualization. To avoid the detection of higher-frequency (e.g. theta) oscillations, waves with shorter than 500 ms cycle lengths (corresponding to higher than 2 Hz frequency) were excluded from the analysis. Waves with non-monotonic phase runs were also excluded, since phase inversions may indicate higher frequency contamination. In addition to phase constraints, valid slow wave activity cycles had to fulfil the following amplitude criteria: the up-state peak amplitude had to be more positive than +50 μV, and the preceding or following down-state trough amplitude had to be more negative than –50 μV. The slow wave activity detection algorithm parameters were tuned and carefully validated by expert electroencephalographers. Similar algorithmic parameters were used for all of the patients. To facilitate comparison of our results with previous animal studies, the threshold level was set to +50 μV on the filtered (0.3–3 Hz, 24 dB/octave, zero phase shift) upper layer III LFP gradient, and the wave triggered (up-state locked) averages were calculated on the unfiltered LFP gradient and multiple-unit activity (Supplementary Fig. 10).
Figure 3.
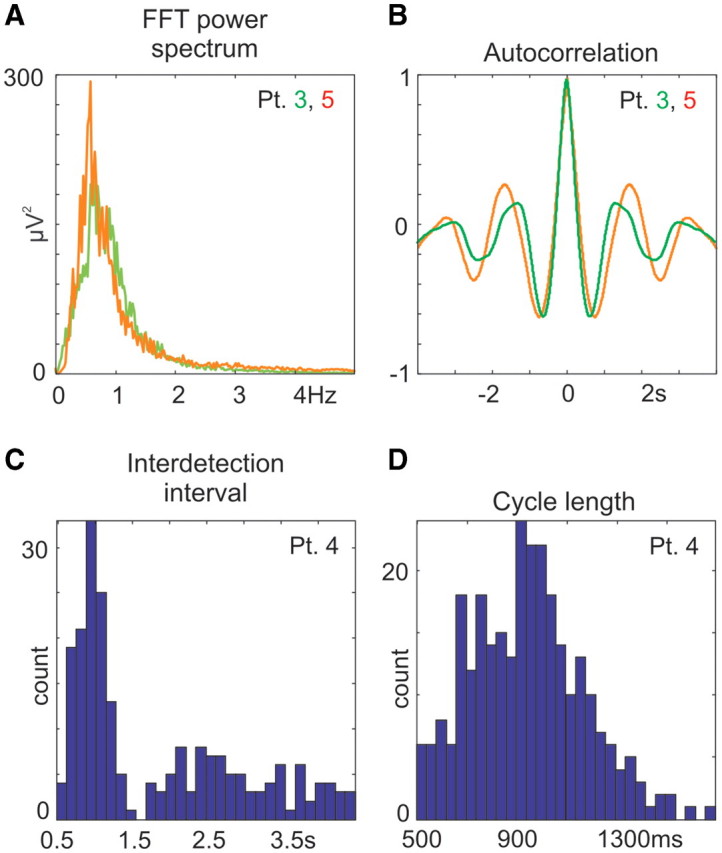
Spectral and temporal properties of slow wave activity cycles. (A) Representative examples of the fast Fourier transformation power spectrum and (B) autocorrelation of supragranular LFP gradient in Patients 3 and 5. For additional fast Fourier transformation and autocorrelations see Supplementary Fig. 5G and H. (C) Representative examples of interdetection interval histograms (y-axis: counts, x-axis: time between up-state detections, 166 ms bin) and (D) cycle length histogram (y-axis: counts, x-axis: valid cycle lengths, 33 ms bin) in Patient 4. For additional data see Supplementary Figs 3 and 4.
Figure 4.

Similarity between LFP gradient recorded with a microelectrode array within the cortex and ECoG recorded from macrocontacts subdurally. (A) Upper: ‘raw’ traces of single sweeps containing slow wave activity; broadband (0.1–300 Hz) LFP gradient from layer II. Middle: ‘filtered’ traces after band-pass (0.3–3 Hz) filtering. Lower: ‘phase’ traces showing the instantaneous phase of the ‘filtered’ trace above derived by the Hilbert transform. Grey rectangles indicate automatically detected up-states (surface positive half-waves). Patient 4. (B) Same as (A), but recorded from neighbouring ECoG contacts. (C) Colour map of LFP gradient and (D) ECoG spectral power during the slow wave activity shown in (A) and (B). x-axis: time, y-axis: frequency, z-axis: colour coded relative spectral power in dB, compared with the mean of the entire interval in each frequency band (relative spectrogram). For more examples of single sweep traces, see Supplementary Figs 1 and 2.
To quantify and compare slow wave activity parameters with other studies, the frequency of slow wave activity occurrence (detected valid cycles per minute), the interdetection interval histogram (Fig. 3C) and the cycle length histogram (Fig. 3D) were calculated (Massimini et al., 2004). The single sweep (Fig. 4C and D) and averaged (Fig. 5A) time-frequency content of the slow wave activity signal was also computed using wavelet transforms (Delorme and Makeig, 2004). In addition, we attempted to describe the laminar distribution of the slow wave activity in more detail using the LFP gradient fast Fourier transformation power spectrum depth profile (Fig. 5B), the pairwise linear coherence between each LFP gradient trace in the slow wave activity (0.3–3 Hz) frequency range (Fig. 5C) and the depth profile of the LFP gradient autocorrelation (Fig. 5D).
Figure 5.
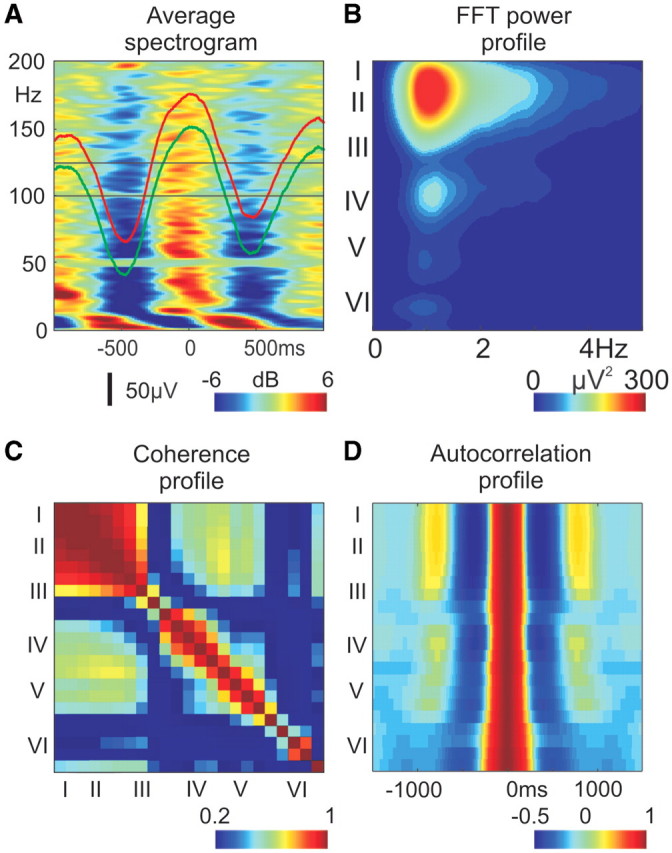
Spectro-temporal and spatial properties of slow wave activity, representative data from Patient 3. (A) Increased broadband spectral activity during up-states: up-state locked, averaged, relative spectrogram of layer II LFP gradient (x-axis: time, y-axis: frequency, z-axis: colour-coded averaged relative spectral power in dB). For the neighbouring ECoG, the averaged relative spectrogram is depicted in Supplementary Fig. 5A. Red (LFP gradient) and green (ECoG) traces show the average potentials. (B) Depth distribution profile of the LFP gradient fast Fourier transformation power spectrum (EEG range: 0.1–300 Hz data, no additional digital filtering was used, x-axis: frequency, y-axis: cortical depth, with corresponding layers, z-axis: colour-coded fast Fourier transformation power). For more power spectrum examples, see Supplementary Fig. 5B. (C) Depth distribution profile of pairwise coherence of LFP gradient channels in different cortical layers. x-axis: cortical depth, with corresponding layers, y-axis: cortical depth, with corresponding layers, z-axis: colour-coded pairwise coherence of the band-pass (0.3–3 Hz) LFP gradient. For more pairwise coherence examples, see Supplementary Fig. 5C. (D) Depth distribution profile of LFP gradient autocorrelation. x-axis: time, y-axis: cortical depth, with corresponding layers, z-axis: colour-coded autocorrelation of the LFP gradient. For more laminar autocorrelation examples, see Supplementary Fig. 5D.
Current source density analysis
CSD analysis identifies synaptic/transmembrane generators of LFP in laminated neural structures (Freeman and Nicholson, 1975; Nicholson and Freeman, 1975). The negative of the second spatial derivative of the LFP closely approximates the macroscopic current density over a unity cell membrane area. Since the LFP gradient (Fig. 6A) is the first spatial derivative of LFP, one additional spatial derivation yielded the CSD (Fig. 6C) for the EEG range (0.1–300 Hz) data. Inhomogeneous conductivity and electrode spacing were not taken into account (both were substituted by the dimensionless number 1 in the calculations); high spatial frequency noise and boundary effects were reduced by Hamming-window smoothing and interpolation (Ulbert et al., 2001a), and thus CSD was expressed in microvolts. It was shown previously (Ulbert et al., 2001a, b, 2004a, b; Wang et al., 2005; Halgren et al., 2006; Knake et al., 2007; Fabo et al., 2008; Cash et al., 2009; Steinvorth et al., 2009; Wittner et al., 2009) that our recording and analysis techniques can reliably detect CSD activity in each layer of the human cortex and hippocampus.
Figure 6.

Role of supragranular layers in slow wave activity generation. Representative depth profile map examples from Patient 4 (A–D) and grand averages of all patients (E–H). (A) LFP gradient (LFPg), (B) multiple-unit activity and (C) CSD depth profile maps. x-axis: time, y-axis: cortical depth, with corresponding laminarization, z-axis: colour-coded amplitude of LFP gradient, multiple-unit activity and CSD units. Positive values are red, negative are blue, except for CSD, where sink is depicted in red and source in blue. (D) LFP gradient spectrograms (SPC) from nine representative channels in layers I–VI. Axes are similar to Fig. 5A. Box-whisker plots of (E) LFP gradient, (F) multiple-unit activity, (G) CSD, (H) LFP gradient (red) and CSD (blue) gamma power (30–150 Hz); normalized grand average of all patients at the peak of the up-state in each layer. Mean: small box, standard error (SE): large box, standard deviation (SD): whisker. For detailed statistical analysis see Supplementary Fig. 9.
Statistical analysis of electrophysiology and histology
ANOVA with Tukey’s honestly significant difference test were applied to the normalized values (LFP gradient and CSD: between –1 and +1; multiple-unit activity, gamma band LFP gradient and CSD: between 0 and 1). Normalized values were grouped by layers (I–VI), and the grand average (across all patients) of LFP gradient, multiple-unit activity, CSD, gamma band (30–150 Hz) LFP gradient and gamma band CSD power depth profile at the up-state peak were tested to determine statistically significant differences (P < 0.01) between electrophysiological activations in different layers of the cortex (Fig. 6E–H). Results are depicted on box-whisker plots [small box = mean; big box = standard error (SE); whisker = standard deviation (SD)]. For detailed statistical data, see Supplementary Fig. 9.
In Patients 4 and 5 (with available histology), the averaged, normalized calretinin immunopositive cell density (Fig. 8A) and averaged, normalized electrophysiology depth profiles (consecutive values at each cortical depth) were constructed. Average depth profiles of the LFP gradient (Fig. 8B) and CSD (Fig. 8C) at the peak of the up-state were normalized between –1 and +1, while average depth profiles of multiple-unit activity (Fig. 8D) and spectral measures (Fig. 8E and F) (gamma band LFP gradient and CSD) at the peak of the up-state were normalized between 0 and 1. The calretinin immunopositive cell density depth profile was normalized between 0 and 1. SE is marked by whisker in this case. The normalized cell density and electrophysiology measures were compared using the Pearson r correlation method with P < 0.01 significance level criterion.
Figure 8.

Depth profiles of calretinin immunopositive (CR+) cell density and slow wave activity. (A) Averaged normalized calretinin immunopositive cell density profile of Patients 4 and 5, whiskers represent standard errors. The number in the upper right corner indicates the Pearson r correlation between the two patients. (B) LFP gradient (LFPg), (C) CSD, (D) multiple-unit activity, (E) LFP gradient gamma power and (F) CSD gamma power of the averaged normalized depth profile of up-state in Patients 4 and 5 with standard error (whisker). Number in the upper right corner indicates the Pearson r correlation between calretinin immunopositive density and (B–F).
Single- and multiple-unit activity analysis
A continuous estimate of population neuronal firing rate was calculated from the multiple-unit activity range (300–5000 Hz) data. The signal was further filtered (500–5000 Hz, zero phase shift, 48 dB/octave), rectified and decimated at 2 kHz, applying a 0.5 ms sliding average rectangular window, followed by a final, smoothing low-pass filter (20 Hz, 12 dB/octave) (Fig. 6B). Putative single units (Fig. 10B, Supplementary Fig. 7) were analysed by conventional threshold detection and clustering methods using Dataview and Klustawin (Heitler, 2006) and custom-made MATLAB software. Putative single units from Patients 1, 4 and 5 were included in the analyses, which were recorded stably for at least 600–1000 s in slow wave sleep. In Patient 2, multiple-unit activity was not recorded for technical reasons, while Patient 3 showed no discriminable single units. After threshold recognition (mean ± 3–5 SD) (Csicsvari et al., 1998) at a given channel, three representative amplitude values were assigned to each unclustered spike waveform. These triplets were projected into 3D space and a competitive expectation-maximization based algorithm (Harris et al., 2000) was used for cluster cutting (Heitler, 2006). If the autocorrelogram of the resulting clusters contained spikes within the 2 ms refractory interval, it was reclustered. If reclustering did not yield a clean refractory period, the cell was regarded as multiple units and omitted from the single cell analysis. Given the gradient recording, spikes at neighbouring traces appeared as mirror images, thus from adjacent channels (150 µm apart) only one channel (the one that yielded the better signal to noise ratio) was included in the analysis. To reveal double detection, the cross-correlogram was constructed (Staba et al., 2002b) for next to adjacent (300 µm apart) pairs of putative single cells. No coincident interactions [99% confidence limit at 0 ms (Staba et al., 2002b)] were found. A spike train was determined as a burst, if at least three consecutive spikes occurred within a maximum 20 ms long interval, which was preceded and followed by at least 20 ms long intervals with neuronal silence (Staba et al., 2002a).
Figure 10.

Single-unit firing in slow wave activity. (A) Superimposed (40 consecutive sweeps) and (B) individual single sweeps of the simultaneously recorded supragranular LFP gradient (LFPg) and multiple-unit activity/single-unit activity (SUA) (Patient 5). Solid and dashed red lines represent LFP gradient mean and standard deviation. (C) Representative normalized (from 0 to 1) firing rate versus phase histograms (from –180° to +180°, in 30° bins) of clustered cells from different layers and patients. Red line: positive half-wave (up-state), green line: negative half-wave (down-state). (D) Columns represent individual slow wave activity cycles (1–252), rows represent clustered neurons (cell 1–12 of Patient 4) and colour represents the firing of a given cell. Blue: no firing in the given up-state for the given cell, green: one, yellow: two, red: three or more action potentials. (E) Histogram of the overall number of spikes for all the 12 clustered cells during up-states. (F) Histogram of the number of active cells, firing at least one action potential during up-states. These data illustrate sparse firing in up-states, only a small fraction of the clustered cells fire and these cells together generate only a few action potentials. For additional data, see Supplementary Figs 7 and 8.
Phase dependence of single cell firing rate (Fig. 10C) was computed for 30° phase bins; the total number of firing in a given bin was divided by the total time that the cortex spent in that phase bin (thus producing a phase histogram). The Rayleigh test (P < 0.01) was used to judge if the resulting circular distribution was significantly different from the uniform distribution.
We have shown previously (Ulbert et al., 2001a, b, 2004a, b; Wang et al., 2005; Halgren et al., 2006; Fabo et al., 2008; Cash et al., 2009; Wittner et al., 2009) that our single-unit activity, multiple-unit activity recording and analysis techniques can reliably detect task or epilepsy-related modulation of neuronal firing from each layer of the human cortex and hippocampus.
Results
Previous studies of slow wave activity in humans (Achermann and Borbely, 1997; Massimini et al., 2004, 2005, 2007; Molle et al., 2004; Marshall et al., 2006) have been limited to macroelectrode recordings that superimpose activity from several squared centimetres of cortex. These recordings are ambiguous as to the circuits involved, are not sensitive to neuronal firing and do not distinguish between excitatory and inhibitory mechanisms. We used laminar multichannel microelectrode array recordings (Ulbert et al., 2001a, b, 2004a, b; Wang et al., 2005; Halgren et al., 2006; Knake et al., 2007; Fabo et al., 2008; Cash et al., 2009; Steinvorth et al., 2009; Wittner et al., 2009) to estimate neuronal firing and synaptic/transmembrane currents in different cortical layers. Since cortical neuronal populations and synaptic inputs are organized into distinct layers, these recordings allowed us to resolve the cortical generators underlying slow wave activity in humans.
General features of human slow wave activity
Clinical subdural strip and grid electrodes and multichannel microelectrode arrays were implanted into the frontal and parietal cortices of patients (n = 5) with intractable epilepsy (Fig. 1) in order to identify the seizure focus and eloquent cortex prior to surgical therapy. The focus was eventually localized to the frontal lobe in four patients and to the temporal lobe in one. Histology of the microelectrode array penetration track was recovered in two patients; it showed intact laminarization (Fig. 2A) and well-preserved interneurons, pyramidal cells and glia (Fig. 2B–D), indicating no discernable epilepsy or implantation-related damage of the examined cortex. None of the patients had pre-operative pathological MRI findings in the 1–2 cm vicinity of the microelectrode array implantation site.
Automatic slow wave activity cycle detection was based on amplitude and phase information using an LFP gradient (Fig. 4A) and ECoG (Fig. 4B) recorded during slow wave sleep (also see Supplementary Figs 1 and 2). On average, 20 slow wave activity cycles (mean = 20 1/min, range = 12–26 1/min, SD = 7 1/min) were detected per minute (Supplementary Figs 3 and 4). Cycle length peaked on average at 0.8 s (mean = 0.8 s, range = 0.6–1.4 s, SD = 0.3 s) (Fig. 3D). Interdetection interval (Fig. 3B) peaked on average at 1.1 s (mean = 1.1 s, range = 0.8–1.2 s, SD = 0.4 s), all comparable to healthy subjects (Massimini et al., 2004). LFP gradient (Fig. 3A and C) and ECoG (not shown) fast Fourier transformation power spectrum and autocorrelation (Fig. 3A and B, Supplementary Fig. 5G and H) also corresponded well to previous human (Achermann and Borbely, 1997) and animal (Isomura et al., 2006) findings, indicating correct slow wave activity cycle identification and relatively normal slow wave activity production.
The LFP gradient recorded in layer II (Fig. 4A) closely resembled the locally recorded ECoG (Fig. 4B), with Pearson r > 0.9 (P < 0.01) in all patients (Supplementary Fig. 1). Time-locked averages to the peak of the surface positive half-wave (up-state) showed similar LFP gradient and ECoG waveforms regardless if time locking was based on the LFP gradient or ECoG (Fig. 5A, red and green traces). Both LFP gradient and ECoG (Figs 4C and D and 5A, Supplementary Fig. 5A) showed broadband (10–200 Hz) spectral increases during up-states and decreases during down-states.
Laminar distribution of slow wave activity
To estimate the laminar contribution of various activities, microelectrode array channels were assigned into six putative layers (I–VI) based on the histological findings (Fig. 2A) when available and cortical depth when not. This analysis revealed a substantial concentration of the 0.3–3 Hz band LFP gradient fast Fourier transformation power within layers I–III (Fig. 5B, Supplementary Fig. 5B) in each patient, indicating strong supragranular synaptic/transmembrane activity. The slow wave activity shape similarities between electrode contacts were significantly greater in supragranular versus infragranular layers in each patient (0.634 versus 0.423, grand average pairwise coherence, Kruskal–Wallis ANOVA, P < 0.01) (Fig. 5C, Supplementary Fig. 5C), while autocorrelation profiles revealed a more precisely paced rhythm supragranularly (Fig. 5D, Supplementary Fig. 5D) in each patient.
Several measurements, both in individual patients (Fig. 6A–D) and in grand averages (Fig. 6E–H), reflecting different aspects of population synaptic/transmembrane and firing activity, were maximal in supragranular layers (for detailed statistical analysis, see Supplementary Fig. 9) at the up-state peak. Normalized, grand average depth profiles of LFP gradient (Figs 6E and 8B) were marked by maximally positive deflections in layers I–III, inverting in layers V and VI into a small negativity. Multiple-unit activity was also maximal in layer III (Figs 6F and 8D). The CSD depth profile at the peak of the slow wave activity up-state showed a maximal source (outward current) in layer I and maximal sink (inward current) in layers II and III, and only very small CSD deflections were observed infragranularly (Figs 6G and 8C). In contrast, the CSD depth profile of a population of interictal spikes detected manually and locked to the surface positive LFP gradient (similarly as in the case of the up-state) were markedly different, exhibiting a large sink–source pair in the infragranular layers (Supplementary Fig. 11). Significant increases [bootstrap analysis (Delorme and Makeig, 2004), P < 0.01] in LFP gradient spectral power were detected in all layers at 10–100 Hz frequencies during up-states (Fig. 6D). Gamma power of LFP gradient and CSD was maximal in layer III (Figs 6H, 8E and F). Separate averages of different slow wave activity cycle lengths corresponding to appropriate (0.6–0.8 Hz, 0.8–1 Hz, 1–1.3 Hz and 1.3–2 Hz) oscillation frequencies also yielded qualitatively similar LFP gradient, spectral LFP gradient, multiple-unit activity and CSD distribution (Fig. 7, Supplementary Fig. 6). We have found no statistically significant differences in any layers (ANOVA, Tukey’s honestly significant difference post hoc test, P > 0.3) in the CSD or multiple-unit activity at the peak of the up-state between any of the four frequency bands indicating similar cortical generator mechanisms above (up to 2 Hz) and below 1 Hz (down to 0.6 Hz).
Figure 7.
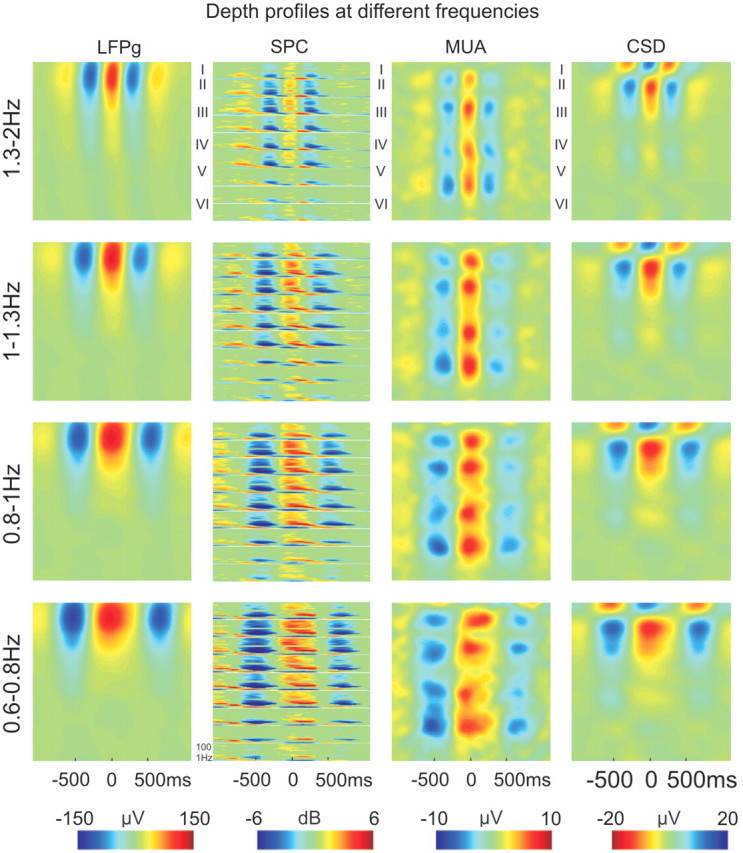
Depth profiles at different slow wave activity frequencies. Up-state-locked averages of LFP gradient (LFPg), LFP gradient spectrogram (SPC), multiple-unit activity and CSD in Patient 3 at four different slow wave activity frequencies. Frequencies 1.3–2 Hz, correspond to a cycle length of: 500–750 ms; 1–1.3 Hz to 750–1000 ms; 0.8–1 Hz to 1000–1250 ms and 0.6–0.8 Hz to 1250–1500 ms. Roman numerals mark putative cortical layers. Colour calibrations are on the bottom. CSD sink is depicted in red, source in blue. Each spectrogram window shows the spectral content (z-axis, colour coded) versus time (x-axis) of a representative LFP gradient channel from a given layer from 1 to 100 Hz (y-axis), measures are expressed in dB relative to a distant baseline (–2500 to –1500 ms).
Calretinin immunopositive cell density depth profiles (Fig. 8A) were calculated in two patients and correlated with the depth profile at the up-state peak of the LFP gradient, CSD, multiple-unit activity, LFP gradient and CSD gamma power (Fig. 8B–F). Calretinin immunopositive cell density between Patient 4 and 5 showed high similarity (r = 0.95, P < 0.01). The highest positive correlation was found between calretinin immunopositive cell density and CSD gamma power (r = 0.85, P < 0.01).
Multiple-unit activity timing at up-state onset
The time courses of multiple-unit activity were examined to determine if one layer may lead others. It was shown in ferret slices (Sanchez-Vives and McCormick, 2000) that layer V’s multiple-unit activity consistently led layers II and III by an average of over 100 ms. In our study, the up-state-associated multiple-unit activity peak-locked averages indicated no evident timing difference in any of the patients, between layers III and V, regardless of whether peak alignment was based on layer III or layer V activity (Fig. 9A and B, Supplementary Fig. 5E and F).
Figure 9.
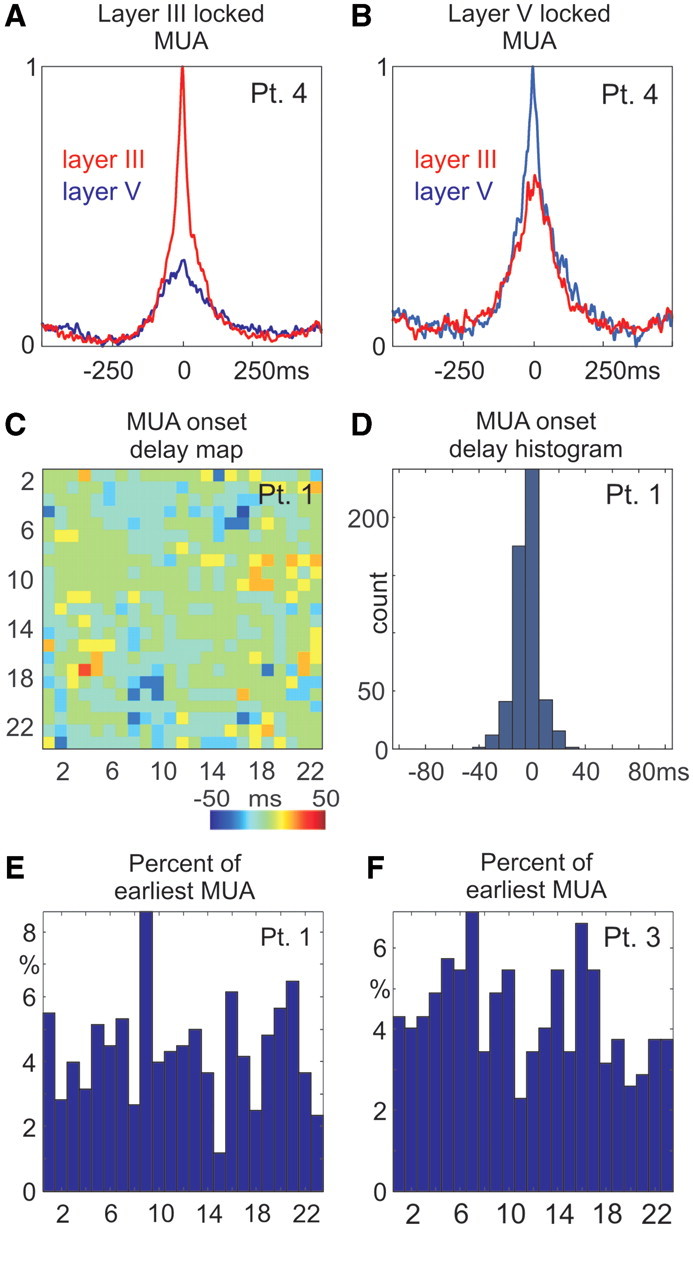
Timing of up-state-related multiple-unit activity in different layers. (A and B) Simultaneity of multiple-unit activity response in supra- and infragranular layers of Patient 4. Multiple-unit activity from layers III (red trace) and V (blue) are shown when aligned and averaged on the up-state-associated multiple-unit activity peak detected in (A) layer III and (B) in layer V. There is no visible multiple-unit activity delay between layers III and V regardless of which layer is used for time locking. (C) Multiple-unit activity cross-correlation peak latencies (x-axis versus y-axis) between each pair of channels in Patient 1. Positive latencies (red) indicate that x channel leads over y channel, while negative latencies (blue) represent lagging. (D) Histogram of leading and lagging values from (C). (E and F) Percentage of a given multiple-unit activity channel showing the earliest firing at up-state onset, representative data from Patients 1 and 3.
To characterize the multiple-unit activity timing between different layers further, it was cross-correlated (3 SD threshold, 10 ms bin size) between each pair of channels, within 200 ms of every up-state onset. In agreement with animal studies (Sakata and Harris, 2009; Chauvette et al., 2010), delay maps and histograms (Fig. 9C and D) indicated a short inter-laminar multiple-unit activity timing difference at up-state onset; most of the delays were within the ± 10 ms bin. We also calculated how often (in percentage of all sweeps) any given multiple-unit activity channel shows the earliest firing at up-state onset. In all patients (where multiple-unit activity was available), the initial firing was quite uniformly distributed across cortical depths. Unlike in a ferret in vitro study (Sanchez-Vives and McCormick, 2000), we found no evidence for long (∼100 ms) lead or lag times between different layers (Fig. 9E and F).
Single-unit correlates of slow oscillation
Recordings from three patients yielded good quality (Supplementary Fig. 7) single-unit activity (Fig. 10). Epochs (∼1000 s) showing the largest slow wave activity detection frequencies were selected for analysis from the first sleep cycle. Overall 33 single units were clustered (9, 12 and 12 from Patients 1, 4 and 5) with mean firing rate of 0.66 Hz (range = 0.12–2.0 Hz, SD = 0.48) and mean burst frequency of 3.1 1/min (range = 0–14 1/min, SD = 3.6). Both the average firing rate and the spontaneous burst rate were well below the reported epileptic threshold found in cortical and hippocampal structures (Staba et al., 2002a).
Nearly all cells (31 of 33) showed significantly non-uniform spiking (Fig. 10A and C, Supplementary Fig. 7D and E) over the slow wave activity cycle (Rayleigh test, P < 0.01), with a peak up-state firing rate mean of 1.63 Hz (range = 0.45–4.6 Hz, SD = 0.96). We found no significant differences between patients in mean firing rates (Kruskal–Wallis ANOVA, P > 0.2), indicating homogeneous distribution. Although mean firing rates grouped by supra- versus infragranular layers showed no significant differences (P > 0.1), supragranular peak up-state firing rates were significantly higher (2.2 Hz versus 1.2 Hz, Kruskal-Wallis ANOVA, P < 0.01) than the same measure for infragranular layers. We found the proportion of firing cells and the rate at which they fire in any given up-state (Fig. 10D–F, Supplementary Fig. 8) remarkably low. On average, only 27% of the clustered cells were active (firing at least one spike) during any given up-state (20%, 25% and 36% in each patient). Thus, an average neuron fired in every third to fifth up-state. Moreover, on average, each neuron fired only 0.32 spikes per up-state (0.44, 0.2 and 0.32 in each patient). As an example, out of the 12 clustered neurons in Patient 4, the most probable number of active cells in a given up-state was 2 (Fig. 10F, see also Supplementary Fig. 8C and F), and the most probable number of overall spikes the 12 cells fired within a given up-state was also 2 (Fig. 10E, see also Supplementary Fig. 8B and E).
Discussion
Our results establish a close similarity between human slow wave activity and the animal slow oscillation at the level of field potential, cellular firing activity and spectral measurements (Steriade, 2006), but they also reveal a number of novel, unexpected findings. Consistent with prior studies in animals, we have shown in humans that the up-state was associated with increased firing and elevated spindle, alpha, beta, gamma and ripple power during the surface-positive LFP half-wave, while the down-state was characterized by the widespread surface negative LFP half-wave with decreased firing and oscillatory activity (Cash et al., 2009). Differences from prior studies were found in the laminar distribution of the up-state, average firing rates during the up-state and the consistency of generators for oscillations above versus below 1 Hz. These contrasts could reflect cortical cytoarchitectonic differences or they could be due to the circumstances of the recordings, including natural sleep versus different types of anaesthesia, or in vivo versus in vitro preparations. They could also be due to epileptic pathology or to phylogenetic differences.
Epilepsy and slow wave activity
Epilepsy is a multi-causal disease with diverse aetiology. Focal epilepsies have circumscribed seizure initiating regions without severe pathological alterations in other areas. Surgical candidates are selected exclusively from this patient group during the careful pre-operative evaluation, based on several diagnostic findings (CT, MRI, functional MRI, PET, single-photon emission computed tomography, video-EEG, magnetoencephalography, functional neurophysiological tests, Wada-test and seizure semiology). In the present study we included only patients with evidence for focal disease.
The laminar LFP gradient, CSD, multiple-unit activity and spectral profile of interictal activity in vivo and in vitro have already been established by our group (Ulbert et al., 2004a, b; Fabo et al., 2008; Wittner et al., 2009). We have shown that the initial phase of the interictal discharges are large amplitude brief events characterized by substantial action potential, LFP gradient, CSD, multiple-unit activity and spectral surges, often emerging from the granular and infragranular (Supplementary Fig. 11) layers of the cortex (Ulbert et al., 2004a). These events are clearly distinct from the background activity and exquisitely visible in single sweeps. Based on our prior knowledge, we carefully excluded any suspicious pathological events from the analysis presented in this article, and we also carefully avoided analysing data derived from electrodes in the proximity of the seizure focus.
Several other considerations suggest that the current findings on the neuronal mechanisms underlying slow wave activity, although recorded in epileptic patients, might also apply to healthy subjects. Our slow wave activity morphology corresponded well to those oscillations collected from standard scalp sleep EEG recordings from healthy subjects. Similarities included not only the slow wave activity frequency and rhythmicity (Achermann and Borbely, 1997), but the asymmetric shape, the briefer and sharper deflection in the down-state (Massimini et al., 2004) and higher beta power content in the up-state (Molle et al., 2002). Our results of detection frequency, cycle length and interdetection interval histograms are all comparable to previous findings from healthy subjects (Massimini et al., 2004), despite both the recording and analysis methodologies being different. Minor deviations in the exact numbers are therefore natural and may reflect methodological differences rather than disease-related alterations. In addition, neither the firing rate nor the burst rate exceeded the pathological criteria found for single neurons in slow wave sleep (Staba et al., 2002a, b). Finally, the lack of any MRI abnormalities and intact laminarization of the excised tissue strongly suggests that we recorded from structurally intact regions, free of gross functional alterations.
Nevertheless, there are some observations in our study that may be related to the patients’ pathology. Out of the 33 clustered units in three patients, two single cells in one patient (Patient 1) showed uniform firing during the slow wave activity cycle (layer III unit #3, average firing rate = 1.17 Hz, layer V unit #6, average firing rate = 0.54 Hz). While some firing during the down-states could be expected due to biological variability of the slow wave activity or due to inaccuracy of the state detection algorithm, a lack of significant modulation by slow wave activity in these cells may reflect a pathological resistance by a small subgroup of cortical neurons to the network-wide deactivation occurring in the down-state.
Localization of cortical oscillations in slow wave activity
A consistent CSD pattern of our study was the prominent sink–source pair in the supragranular layers compared with the weak infragranular activation. This localization was true for both the low- (0.5–2 Hz) and the high-frequency (10–200 Hz) oscillations in all patients, in frontal and parietal areas.
In our interpretation, the prominent layer II and III sink during the up-state reflects the large active inward currents flowing across the distal dendritic membrane compartments of layers V and VI pyramidal cells and distal, proximal and basal dendritic membrane compartments and perhaps on the somatic membrane of layers II and III pyramidal cells. The corresponding passive, return, source currents are flowing in layer I, across the most superficial apical dendritic membrane compartments of the pyramidal cells. The spatial CSD pattern in the down-state is inverted, exhibiting a large active current source in layers II and III due to hyperpolarizing currents (likely outward potassium flows from the pyramidal cells) and a passive return sink in layer I (Cash et al., 2009).
Our CSD findings from the frontal and parietal areas in natural sleep are in contrast with a study in the cat suprasylvian area, albeit under ketamine/xylazine anaesthesia (Steriade and Amzica, 1996). At low frequencies (∼1 Hz), the maximal up-state-related sink in the cat was located in the middle rather than in the superficial layers, surrounded by not only a superficial but also a large deep source. In addition, a substantial up-state-related sink in the deepest layer was present in the cat, which was practically invisible in our human recordings. The same authors also showed a massive supragranular (layers II and III) up-state-related sink besides one or two deeper and weaker sinks, during the spontaneous and evoked K-complex (Amzica and Steriade, 1998). Moreover, at higher frequencies (∼35 Hz) and during the K-complex, a series of ‘alternating microsinks and microsources’ was found throughout the depth of the cat suprasylvian area (Steriade and Amzica, 1996; Amzica and Steriade, 1998). Such alternating patterns are best explained by insufficient spatial resolution in the LFP sampling (8 contact, 250 µm spacing electrode array) and corresponding spatial aliasing error, and not by neuronal sources (Tenke et al., 1993).
A recent study in the cat suprasylvian area in natural sleep with adequate spatial sampling (100 µm) revealed alternation free middle and deep layer sinks and a superficial source during the up-state (Chauvette et al., 2010). Thus, besides cytoarchitectonic differences between different types of cortices (e.g. frontal and parietal areas in humans versus suprasylvian area in the cat), and methodological errors, it is also plausible to assume that neuronal mechanisms of natural sleep and ketamine/xylazine anaesthesia may be different, further accounting for the divergent findings. Another recent study, on the rat primary auditory cortex using high spatial resolution (50 µm) CSD mapping, showed the maximal up-state-related sink to be in the presumed supragranular layers under urethane anaesthesia, while in natural sleep it is rather the presumed granular and probably infragranular layers that exhibited the largest up-state-related sink (see average data, Supplementary Figs 10 and 16 in Sakata and Harris, 2009). Our CSD results in natural sleep are quite close to the results of Sakata and Harris (2009) obtained under urethane anaesthesia, except for the large source deep in the infragranular layers.
Discrepancies between cat, rat and human data thus most probably stem from multiple sources, including but not limited to the recording methodology (spatial density of sampling, electrode types implanted), use of anaesthetics (ketamine/xylazine versus urethane versus natural sleep), cytoarchitectonics of the cortex (suprasylvian cortex in cat versus auditory cortex in rat versus frontal and parietal areas in humans), as well as species differences.
According to our observations, the slow wave activity depth profile, represented by the up-state peaks, was similar between the four investigated frequency ranges including the slow (<1 Hz) and delta band (up to 2 Hz). In our opinion, these frequency bands are thus substantially overlapping, hence a less strict distinction should be applied between activities above versus below 1 Hz. We agree that the slow activity and thalamic delta have obviously different neuronal mechanisms, but it seems that these waves cannot be distinguished using exclusively a frequency band criterion.
We have found similar signs of elementary hierarchical organization of low- and high-frequency oscillations in humans as it was shown in animal models (Lakatos et al., 2005; Steriade, 2006). The organizing substrate was the up-state of the slow wave activity, which gave rise to a wide variety of higher-frequency activity including spindle, alpha, beta, gamma and ripple oscillations. Each of these high-frequency oscillatory bursts was quite different from sweep to sweep, showing for example occasional spindle sequences or marked gamma or ripple band enhancements at various peak frequencies. These observations suggest that each slow wave activity cycle with unique oscillatory signature reflects individual information content coded differently in the oscillatory process. Given the variability of the high-frequency oscillatory activity during up-states, it is plausible to assume that different underlying neuronal populations might be responsible for the generation of each specific oscillatory pattern. This strategy might be beneficial in the configuration of functional connectivity between neurons to form stable ensembles that may promote the consolidation of memory in sleep.
Paroxysmal activity is known to emerge more often from non-REM sleep compared with REM (Steriade, 2003). Animal studies revealed that cortical hyperexcitability associated with ripple oscillations often results in pathological synchronization leading to epileptic seizures (Grenier et al., 2003). We have shown that up-states are characterized by a large increase in cortical excitability reflected in the increased power of gamma and ripple oscillations. Thus, we hypothesize that the active state of the slow oscillation may play an important role in the generation of seizures and other paroxysmal signs in the cortical epileptic network.
Laminar calretinin immunopositive interneuron density and slow wave activity
The relatively high correlation in laminar location between LFP gradient, CSD, multiple-unit activity and gamma power during up-states and calretinin immunopositive cell density may provide additional insights into the mechanism for the predominance of oscillatory activity in supragranular layers. Calretinin immunopositive cells are relatively numerous for inhibitory cells, comprising ∼8% of the total number of prefrontal neurons and ∼14.2–17.6% of layer II and III neurons, in human prefrontal cortex (Gabbott et al., 1997). Calretinin immunopositive cell density in the human, monkey, cat and rat cortex is highest in the supragranular layers (Fonseca and Soriano, 1995; Gabbott et al., 1997; Schwark and Li, 2000). The layer II and III population of calretinin immunopositive cells (78% of all calretinin immunopositive cells) is significantly more numerous (+31%) in humans than in the rat (Gabbott et al., 1997), and the supragranular calretinin immunopositive predominance in humans is not affected by epilepsy (Barinka et al., 2010). The relatively high density and remarkable vertically oriented dendritic alignment (Gabbott et al., 1997) of layers II and III calretinin immunopositive cells (Fig. 2B) suggest that this population on its own may contribute significantly to the CSD. Unlike basket cells that target principal cells (Somogyi et al., 1983) establishing local negative-feedback circuits, layers II and III calretinin immunopositive interneurons preferentially target other inhibitory cells locally in layers II and III and target pyramidal cells in layer V (Meskenaite, 1997), forming local positive-feedback circuits (Dantzker and Callaway, 2000) in the supragranular layers and negative-feedback circuits between supra- and infragranular layers. The negative feedback imposed by the population of calretinin immunopositive cells may relatively attenuate the infragranular activation, while the positive-feedback disinhibition (Tamas et al., 1998) may amplify the supragranular synaptic/transmembrane oscillations in the gamma band and cellular activity (Whittington et al., 1995).
In addition, GABAB receptors are more concentrated in the upper layers of the cortex, at least in rodents (Lopez-Bendito et al., 2002; Tamas et al., 2003), which might also contribute to the potassium current that may play an important role in the down-state generation (Timofeev et al., 2001).
Action potential activity in slow wave activity
In vitro slice studies in animals found that firing in infragranular layers consistently lead supragranular layers by over 100 ms at the onset of the up-state (Sanchez-Vives and McCormick, 2000). In contrast, we found that the onset of activity during up-states differs less than ±10 ms between layers. Although the slice preparation is a powerful tool, it severs connections that are present in the intact animal, removing background synaptic input and placing the cell in an artificial medium.
In healthy humans, it is believed that each individual slow wave cycle has a distinct origin and propagates uniquely across a number of brain areas (Murphy et al., 2009). Similar patterns were also found in animal models (Ferezou et al., 2007; Mohajerani et al., 2010). Thus, variable projections may be involved in its propagation, terminating in variable cortical layers making the laminar distribution of the initial unit firing also variable, as it was shown in our study.
The low average firing rate (0.6 Hz) found in here is consistent with a previous human report (Ravagnati et al., 1979). Animal studies using extracellular silicon probes and intracellular sharp electrodes report average firing rates in the 2–20 Hz range from the entire depth of the cortex (Steriade et al., 2001; Isomura et al., 2006; Luczak et al., 2007), and cell-attached and whole-cell patch-clamp studies from layers II and III neurons report average firing rates in the 0.01–0.3 Hz range (Margrie et al., 2002; Waters and Helmchen, 2006; Hromadka et al., 2008). Some of these differences may be related to the laminar location of the neurons and to ‘collateral damage’ inherent to the different techniques. Extracellular recordings might be biased toward higher average firing rates, because of the use of a minimum spontaneous firing rate (1–2 Hz) constraint (Luczak et al., 2007). We did not use such correction in our unit analysis, thus slower firing cells were also included. Sharp electrode intracellular recordings (Steriade et al., 2001; Isomura et al., 2006) disrupt the cell membrane and introduce leakage current, which may also alter the firing rate. In cell-attached recordings (Margrie et al., 2002; Hromadka et al., 2008), the membrane is partially covered by the recording pipette causing substantial mechanical stress, receptor, ion channel masking and membrane capacitance changes, while the whole-cell configuration (Waters and Helmchen, 2006) disrupts the membrane and causes cell dialysis. Indeed, when establishing the whole-cell configuration, the spontaneous firing rate may double compared with the cell-attached state (Margrie et al., 2002). Given these apparently strong effects of recording methodology on cell firing, it is hard to definitively relate the present findings to animal experiments. However, it seems reasonable to expect that any technique which physically contacts the cell would alter the firing rate to a greater extent than techniques which do not. To elucidate these differences further, unbiased extracellular action potential techniques need to be implemented in different animal models.
Concluding remarks
In summary, the differences between our recordings of slow wave activity and those previously described may be due to the observed cortical areas, the experimental preparation, the type of sleep induction and the neurological condition. We suggest that the strong supragranular activity may characterize slow wave activity, in contrast with certain types of epileptic discharges and some specific components of sensory and cognitive-evoked responses, which are mostly localized to the granular and infragranular layers (Ulbert et al., 2001b, 2004a; Wang et al., 2005; Halgren et al., 2006). The strong supragranular oscillatory activity in sleep may be beneficial for the local, higher-order processing of sensory experience and perhaps memory consolidation, since these layers are interconnected by dense cortico-cortical projections forming fine-scale functional networks to perform integrative functions (Yoshimura et al., 2005). The weaker infragranular activity may reflect the relatively suppressed cortical executive, output functions, which may prevent effective connectivity between distant cortical areas from developing in slow wave sleep (Massimini et al., 2005).
Some differences between humans and cats or rats might also be expected given that our last common ancestor was about 75 million years ago, and our prefrontal cortex is more than a hundred times larger, with a striking increase in pyramidal cell dendritic complexity (Elston, 2003; Elston et al., 2006). Our finding that the slow wave activity and corresponding high-frequency rhythms including spindle, alpha, beta, gamma and ripple oscillations may involve supragranular layers is consistent with this massive cortical expansion of neuronal number (Herculano-Houzel et al., 2007), inasmuch as increased dendritic complexity and cortico-cortical association fibres predominantly target these layers (Gonzalez-Burgos et al., 2000).
Funding
NeuroProbes IST-027017, Epicure LSH-CT-2006–037315, OTKA PD-77864, OTKA K-81357, NKTH-ANR Neurogen, EB9282, NS18741.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- CSD
current source density
- ECoG
electrocorticogram
- LFP
local field potential
- REM
rapid eye movement
References
- Achermann P, Borbely AA. Low-frequency (<1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience. 1997;81:213–22. doi: 10.1016/s0306-4522(97)00186-3. [DOI] [PubMed] [Google Scholar]
- Amzica F, Steriade M. Disconnection of intracortical synaptic linkages disrupts synchronization of a slow oscillation. J Neurosci. 1995;15:4658–77. doi: 10.1523/JNEUROSCI.15-06-04658.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amzica F, Steriade M. Cellular substrates and laminar profile of sleep K-complex. Neuroscience. 1998;82:671–86. doi: 10.1016/s0306-4522(97)00319-9. [DOI] [PubMed] [Google Scholar]
- Axmacher N, Elger CE, Fell J. Ripples in the medial temporal lobe are relevant for human memory consolidation. Brain. 2008;131:1806–17. doi: 10.1093/brain/awn103. [DOI] [PubMed] [Google Scholar]
- Barinka F, Druga R, Marusic P, Krsek P, Zamecnik J. Calretinin immunoreactivity in focal cortical dysplasias and in non-malformed epileptic cortex. Epilepsy Res. 2010;88:76–86. doi: 10.1016/j.eplepsyres.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Berger H. Über das Elektroenkephalogramm des Menschen. Arch Psychiatr Nervenkr. 1929;87:527–70. [Google Scholar]
- Born J, Rasch B, Gais S. Sleep to remember. Neuroscientist. 2006;12:410–24. doi: 10.1177/1073858406292647. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Staba RJ, Reddick M, Fried I, Engel J., Jr Interictal high-frequency oscillations (80–500 Hz) in the human epileptic brain: entorhinal cortex. Ann Neurol. 2002;52:407–15. doi: 10.1002/ana.10291. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–9. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Cash SS, Halgren E, Dehghani N, Rossetti AO, Thesen T, Wang C, et al. The human K-complex represents an isolated cortical down-state. Science. 2009;324:1084–7. doi: 10.1126/science.1169626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvette S, Volgushev M, Timofeev I. Origin of active states in local neocortical networks during slow sleep oscillation. Cereb Cortex. 2010 doi: 10.1093/cercor/bhq009. Advance Access published on March 3, 2010, doi:10.1093/cercor/bhq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens Z, Molle M, Eross L, Barsi P, Halasz P, Born J. Temporal coupling of parahippocampal ripples, sleep spindles and slow oscillations in humans. Brain. 2007;130:2868–78. doi: 10.1093/brain/awm146. [DOI] [PubMed] [Google Scholar]
- Crepon B, Navarro V, Hasboun D, Clemenceau S, Martinerie J, Baulac M, et al. Mapping interictal oscillations greater than 200 Hz recorded with intracranial macroelectrodes in human epilepsy. Brain. 2010;133:33–45. doi: 10.1093/brain/awp277. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci. 2010;13:9–17. doi: 10.1038/nn.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Buzsaki G. Reliability and state dependence of pyramidal cell-interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron. 1998;21:179–89. doi: 10.1016/s0896-6273(00)80525-5. [DOI] [PubMed] [Google Scholar]
- Dantzker JL, Callaway EM. Laminar sources of synaptic input to cortical inhibitory interneurons and pyramidal neurons. Nat Neurosci. 2000;3:701–7. doi: 10.1038/76656. [DOI] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Ebersole JS, Pedley TA. Current practice of clinical electroencephalography. Philadelphia, PA: Lippincott Williams & Wilkins; 2003. [Google Scholar]
- Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;13:1124–38. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Elston GN, Benavides-Piccione R, Elston A, Zietsch B, Defelipe J, Manger P, et al. Specializations of the granular prefrontal cortex of primates: implications for cognitive processing. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:26–35. doi: 10.1002/ar.a.20278. [DOI] [PubMed] [Google Scholar]
- Euston DR, Tatsuno M, McNaughton BL. Fast-forward playback of recent memory sequences in prefrontal cortex during sleep. Science. 2007;318:1147–50. doi: 10.1126/science.1148979. [DOI] [PubMed] [Google Scholar]
- Fabo D, Magloczky Z, Wittner L, Pek A, Eross L, Czirjak S, et al. Properties of in vivo interictal spike generation in the human subiculum. Brain. 2008;131:485–99. doi: 10.1093/brain/awm297. [DOI] [PubMed] [Google Scholar]
- Ferezou I, Haiss F, Gentet LJ, Aronoff R, Weber B, Petersen CC. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–23. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Fonseca M, Soriano E. Calretinin-immunoreactive neurons in the normal human temporal cortex and in Alzheimer's disease. Brain Res. 1995;691:83–91. doi: 10.1016/0006-8993(95)00622-w. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Nicholson C. Experimental optimization of current source–density technique for anuran cerebellum. J Neurophysiol. 1975;38:369–82. doi: 10.1152/jn.1975.38.2.369. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Jays PR, Bacon SJ. Calretinin neurons in human medial prefrontal cortex (areas 24a,b,c, 32', and 25) J Comp Neurol. 1997;381:389–410. doi: 10.1002/(sici)1096-9861(19970519)381:4<389::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Barrionuevo G, Lewis DA. Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. 2000;10:82–92. doi: 10.1093/cercor/10.1.82. [DOI] [PubMed] [Google Scholar]
- Grenier F, Timofeev I, Steriade M. Neocortical very fast oscillations (ripples, 80–200 Hz) during seizures: intracellular correlates. J Neurophysiol. 2003;89:841–52. doi: 10.1152/jn.00420.2002. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–45. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren E, Wang C, Schomer DL, Knake S, Marinkovic K, Wu J, et al. Processing stages underlying word recognition in the anteroventral temporal lobe. Neuroimage. 2006;30:1401–13. doi: 10.1016/j.neuroimage.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Henze DA, Csicsvari J, Hirase H, Buzsaki G. Accuracy of tetrode spike separation as determined by simultaneous intracellular and extracellular measurements. J Neurophysiol. 2000;84:401–14. doi: 10.1152/jn.2000.84.1.401. [DOI] [PubMed] [Google Scholar]
- Heitler JW. DataView v5: software for the display and analysis of digital signals in neurophysiology. http://www.st-andrews.ac.uk/∼wjh/dataview/ (13 June 2010, date last accessed) [Google Scholar]
- Herculano-Houzel S, Collins CE, Wong P, Kaas JH. Cellular scaling rules for primate brains. Proc Natl Acad Sci USA. 2007;104:3562–7. doi: 10.1073/pnas.0611396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromadka T, Deweese MR, Zador AM. Sparse representation of sounds in the unanesthetized auditory cortex. PLoS Biol. 2008;6:e16. doi: 10.1371/journal.pbio.0060016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Iber C, Ancoli-Israel S, Chesson A, Quan SF. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. Westchester: American Academy of Sleep Medicine; 2007. [Google Scholar]
- Isomura Y, Sirota A, Ozen S, Montgomery S, Mizuseki K, Henze DA, et al. Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron. 2006;52:871–82. doi: 10.1016/j.neuron.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Jacobs J, Levan P, Chatillon CE, Olivier A, Dubeau F, Gotman J. High frequency oscillations in intracranial EEGs mark epileptogenicity rather than lesion type. Brain. 2009;132:1022–37. doi: 10.1093/brain/awn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O, Kaiser J, Lachaux JP. Human gamma-frequency oscillations associated with attention and memory. Trends Neurosci. 2007;30:317–24. doi: 10.1016/j.tins.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Jirsch JD, Urrestarazu E, LeVan P, Olivier A, Dubeau F, Gotman J. High-frequency oscillations during human focal seizures. Brain. 2006;129:1593–608. doi: 10.1093/brain/awl085. [DOI] [PubMed] [Google Scholar]
- Keller CJ, Cash SS, Narayanan S, Wang C, Kuzniecky R, Carlson C, et al. Intracranial microprobe for evaluating neuro-hemodynamic coupling in unanesthetized human neocortex. J Neurosci Methods. 2009;179:208–18. doi: 10.1016/j.jneumeth.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knake S, Wang CM, Ulbert I, Schomer DL, Halgren E. Specific increase of human entorhinal population synaptic and neuronal activity during retrieval. Neuroimage. 2007;37:618–22. doi: 10.1016/j.neuroimage.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatos P, Karmos G, Mehta AD, Ulbert I, Schroeder CE. Entrainment of neuronal oscillations as a mechanism of attentional selection. Science. 2008;320:110–13. doi: 10.1126/science.1154735. [DOI] [PubMed] [Google Scholar]
- Lakatos P, Shah AS, Knuth KH, Ulbert I, Karmos G, Schroeder CE. An oscillatory hierarchy controlling neuronal excitability and stimulus processing in the auditory cortex. J Neurophysiol. 2005;94:1904–11. doi: 10.1152/jn.00263.2005. [DOI] [PubMed] [Google Scholar]
- Loomis AL, Harvey EN, Hobart GA. Cerebral states during sleep, as studied by human brain potentials. J Exp Psychol. 1937;21:127–44. [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Kulik A, Paulsen O, Fairen A, Lujan R. Expression and distribution of metabotropic GABA receptor subtypes GABABR1 and GABABR2 during rat neocortical development. Eur J Neurosci. 2002;15:1766–78. doi: 10.1046/j.1460-9568.2002.02032.x. [DOI] [PubMed] [Google Scholar]
- Luczak A, Bartho P, Marguet SL, Buzsaki G, Harris KD. Sequential structure of neocortical spontaneous activity in vivo. Proc Natl Acad Sci USA. 2007;104:347–52. doi: 10.1073/pnas.0605643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margrie TW, Brecht M, Sakmann B. In vivo, low-resistance, whole-cell recordings from neurons in the anaesthetized and awake mammalian brain. Pflugers Arch. 2002;444:491–8. doi: 10.1007/s00424-002-0831-z. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–13. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Esser SK, Riedner BA, Huber R, Murphy M, et al. Triggering sleep slow waves by transcranial magnetic stimulation. Proc Natl Acad Sci USA. 2007;104:8496–501. doi: 10.1073/pnas.0702495104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, Tononi G. Breakdown of cortical effective connectivity during sleep. Science. 2005;309:2228–32. doi: 10.1126/science.1117256. [DOI] [PubMed] [Google Scholar]
- Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. The sleep slow oscillation as a traveling wave. J Neurosci. 2004;24:6862–70. doi: 10.1523/JNEUROSCI.1318-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meskenaite V. Calretinin-immunoreactive local circuit neurons in area 17 of the cynomolgus monkey, Macaca fascicularis. J Comp Neurol. 1997;379:113–32. [PubMed] [Google Scholar]
- Mohajerani MH, McVea DA, Fingas M, Murphy TH. Mirrored bilateral slow-wave cortical activity within local circuits revealed by fast bihemispheric voltage-sensitive dye imaging in anesthetized and awake mice. J Neurosci. 2010;30:3745–51. doi: 10.1523/JNEUROSCI.6437-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molle M, Marshall L, Gais S, Born J. Grouping of spindle activity during slow oscillations in human non-rapid eye movement sleep. J Neurosci. 2002;22:10941–7. doi: 10.1523/JNEUROSCI.22-24-10941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molle M, Marshall L, Gais S, Born J. Learning increases human electroencephalographic coherence during subsequent slow sleep oscillations. Proc Natl Acad Sci USA. 2004;101:13963–8. doi: 10.1073/pnas.0402820101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Riedner BA, Huber R, Massimini M, Ferrarelli F, Tononi G. Source modeling sleep slow waves. Proc Natl Acad Sci USA. 2009;106:1608–13. doi: 10.1073/pnas.0807933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Freeman JA. Theory of current source-density analysis and determination of conductivity tensor for anuran cerebellum. J Neurophysiol. 1975;38:356–68. doi: 10.1152/jn.1975.38.2.356. [DOI] [PubMed] [Google Scholar]
- Ravagnati L, Halgren E, Babb TL, Crandall PH. Activity of human hippocampal formation and amygdala neurons during sleep. Sleep. 1979;2:161–73. doi: 10.1093/sleep/2.2.161. [DOI] [PubMed] [Google Scholar]
- Sakata S, Harris KD. Laminar structure of spontaneous and sensory-evoked population activity in auditory cortex. Neuron. 2009;64:404–18. doi: 10.1016/j.neuron.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–34. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Schevon CA, Trevelyan AJ, Schroeder CE, Goodman RR, McKhann G, Jr, Emerson RG. Spatial characterization of interictal high frequency oscillations in epileptic neocortex. Brain. 2009;132:3047–59. doi: 10.1093/brain/awp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder CE, Lakatos P. Low-frequency neuronal oscillations as instruments of sensory selection. Trends Neurosci. 2009;32:9–18. doi: 10.1016/j.tins.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwark HD, Li J. Distribution of neurons immunoreactive for calcium-binding proteins varies across areas of cat primary somatosensory cortex. Brain Res Bull. 2000;51:379–85. doi: 10.1016/s0361-9230(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–93. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Kisvarday ZF, Martin KA, Whitteridge D. Synaptic connections of morphologically identified and physiologically characterized large basket cells in the striate cortex of cat. Neuroscience. 1983;10:261–94. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Bragin A, Fried I, Engel J., Jr Sleep states differentiate single neuron activity recorded from human epileptic hippocampus, entorhinal cortex, and subiculum. J Neurosci. 2002a;22:5694–704. doi: 10.1523/JNEUROSCI.22-13-05694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Fried I, Engel J., Jr Single neuron burst firing in the human hippocampus during sleep. Hippocampus. 2002b;12:724–34. doi: 10.1002/hipo.10026. [DOI] [PubMed] [Google Scholar]
- Steinvorth S, Wang C, Ulbert I, Schomer D, Halgren E. Human entorhinal gamma and theta oscillations selective for remote autobiographical memory. Hippocampus. 2009;20(1):166–73. doi: 10.1002/hipo.20597. [DOI] [PubMed] [Google Scholar]
- Steriade M. Neuronal substrates of sleep and epilepsy. Cambridge: Cambridge University Press; 2003. [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Amzica F. Intracortical and corticothalamic coherency of fast spontaneous oscillations. Proc Natl Acad Sci USA. 1996;93:2533–8. doi: 10.1073/pnas.93.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993a;13:3266–83. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993b;13:3252–65. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron. 2003;37:563–76. doi: 10.1016/s0896-6273(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–85. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Tamas G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–5. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Tamas G, Somogyi P, Buhl EH. Differentially interconnected networks of GABAergic interneurons in the visual cortex of the cat. J Neurosci. 1998;18:4255–70. doi: 10.1523/JNEUROSCI.18-11-04255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenke CE, Schroeder CE, Arezzo JC, Vaughan HG., Jr Interpretation of high-resolution current source density profiles: a simulation of sublaminar contributions to the visual evoked potential. Exp Brain Res. 1993;94:183–92. doi: 10.1007/BF00230286. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc Natl Acad Sci USA. 2001;98:1924–9. doi: 10.1073/pnas.041430398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Li XG, Pyapali GK, Ylinen A, Buzsaki G. Morphometric and electrical properties of reconstructed hippocampal CA3 neurons recorded in vivo. J Comp Neurol. 1995;356:580–94. doi: 10.1002/cne.903560408. [DOI] [PubMed] [Google Scholar]
- Ulbert I, Halgren E, Heit G, Karmos G. Multiple microelectrode-recording system for human intracortical applications. J Neurosci Methods. 2001a;106:69–79. doi: 10.1016/s0165-0270(01)00330-2. [DOI] [PubMed] [Google Scholar]
- Ulbert I, Heit G, Madsen J, Karmos G, Halgren E. Laminar analysis of human neocortical interictal spike generation and propagation: current source density and multiunit analysis in vivo. Epilepsia. 2004a;45(Suppl 4):48–56. doi: 10.1111/j.0013-9580.2004.04011.x. [DOI] [PubMed] [Google Scholar]
- Ulbert I, Karmos G, Heit G, Halgren E. Early discrimination of coherent versus incoherent motion by multiunit and synaptic activity in human putative MT+ Hum Brain Mapp. 2001b;13:226–38. doi: 10.1002/hbm.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbert I, Magloczky Z, Eross L, Czirjak S, Vajda J, Bognar L, et al. In vivo laminar electrophysiology co-registered with histology in the hippocampus of patients with temporal lobe epilepsy. Exp Neurol. 2004b;187:310–8. doi: 10.1016/j.expneurol.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Urrestarazu E, Chander R, Dubeau F, Gotman J. Interictal high-frequency oscillations (100-500 Hz) in the intracerebral EEG of epileptic patients. Brain. 2007;130:2354–66. doi: 10.1093/brain/awm149. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Chauvette S, Mukovski M, Timofeev I. Precise long-range synchronization of activity and silence in neocortical neurons during slow-wave oscillations [corrected] J Neurosci. 2006;26:5665–72. doi: 10.1523/JNEUROSCI.0279-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Wang C, Ulbert I, Schomer DL, Marinkovic K, Halgren E. Responses of human anterior cingulate cortex microdomains to error detection, conflict monitoring, stimulus-response mapping, familiarity, and orienting. J Neurosci. 2005;25:604–13. doi: 10.1523/JNEUROSCI.4151-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J, Helmchen F. Background synaptic activity is sparse in neocortex. J Neurosci. 2006;26:8267–77. doi: 10.1523/JNEUROSCI.2152-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Jefferys JG. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature. 1995;373:612–15. doi: 10.1038/373612a0. [DOI] [PubMed] [Google Scholar]
- Wittner L, Henze DA, Zaborszky L, Buzsaki G. Hippocampal CA3 pyramidal cells selectively innervate aspiny interneurons. Eur J Neurosci. 2006;24:1286–98. doi: 10.1111/j.1460-9568.2006.04992.x. [DOI] [PubMed] [Google Scholar]
- Wittner L, Huberfeld G, Clemenceau S, Eross L, Dezamis E, Entz L, et al. The epileptic human hippocampal cornu ammonis 2 region generates spontaneous interictal-like activity in vitro. Brain. 2009;132:3032–46. doi: 10.1093/brain/awp238. [DOI] [PubMed] [Google Scholar]
- Worrell GA, Gardner AB, Stead SM, Hu S, Goerss S, Cascino GJ, et al. High-frequency oscillations in human temporal lobe: simultaneous microwire and clinical macroelectrode recordings. Brain. 2008;131:928–37. doi: 10.1093/brain/awn006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrell GA, Parish L, Cranstoun SD, Jonas R, Baltuch G, Litt B. High-frequency oscillations and seizure generation in neocortical epilepsy. Brain. 2004;127:1496–506. doi: 10.1093/brain/awh149. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Dantzker JL, Callaway EM. Excitatory cortical neurons form fine–scale functional networks. Nature. 2005;433:868–73. doi: 10.1038/nature03252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.