

Abstract
Molecular effects of obesity, a well-established risk factor for breast cancer progression, are mediated by adipocytokine leptin. Given the important role of leptin in breast cancer growth and metastasis, novel strategies to antagonize biological effects of this adipocytokine are much desired. We showed previously that benzyl isothiocyanate (BITC), a constituent of edible cruciferous vegetables (e.g. garden cress), confers significant protection against mammary carcinogenesis in a transgenic mouse model. The present study provides first evidence for the efficacy of BITC against oncogenic effects of leptin. The BITC treatment circumvented leptin-induced clonogenicity and anchorage-independent growth of MDA-MB-231 and MCF-7 human breast cancer cells. Leptin-stimulated migration and invasion of these cells was also inhibited in the presence of BITC. Analysis of the underlying molecular mechanisms revealed that BITC treatment suppressed leptin-induced Stat3 phosphorylation and cyclin D1 transactivation. The BITC-mediated inhibition of MDA-MB-231 xenograft growth correlated with a modest yet significant decrease in levels of Tyr705 phosphorylated Stat3. The BITC treatment efficiently inhibited Stat3 and SRC1 recruitment to cyclin D1 promoter in a chromatin immunoprecipitation analysis. Furthermore, overexpression of constitutively active Stat3 imparted significant protection against BITC-mediated inhibition of cyclin D1 transactivation, whereas RNA interference of Stat3 resulted in a significant increase in BITC-mediated inhibition of cyclin D1 transactivation in the presence of leptin. These results indicate that Stat3 plays an important role in BITC-mediated inhibition of leptin-induced cyclin D1 transactivation. In conclusion, BITC could potentially be a rational therapeutic strategy for breast carcinoma in obese patients with high leptin levels.
Introduction
Obesity is fast becoming a pandemic in the USA. Studies examining the relationship of obesity with mortality from breast cancer have found that obese women in the highest quintile of body mass index have double the death rate from breast cancer when compared with women in the lowest quintile (1,2). In addition, in women with body mass index in the highest quintile, an increased proportion of tumors were estrogen receptor (ER) negative, had a high S-phase fraction, histological grade, mitotic cell count, expression levels of proliferation markers and a larger tumor size. These clinical observations cannot be explained only by higher estrogen levels that are associated with obesity. Importantly, independent of their menopausal status, obese breast cancer patients exhibit a higher risk for lymph node metastasis, larger tumor burden and higher mortality when compared with non-obese breast cancer patients. Obesity is associated with an increase in number and size of adipocytes that greatly alters the local and systemic secretion of biologically active polypeptides, adipocytokines such as leptin (3). Adipocytokines affect various biological processes acting by endocrine, paracrine and autocrine mechanisms (3).
Leptin was found as an afferent satiety signal, regulating appetite and energy expenditure in both humans and rodents (4). Leptin was recognized only as a satiety hormone for many years until research over the last few years provided important clues about its apheliotropic actions, its role in the pathogenesis of atherosclerotic vascular disease (5,6) and importantly carcinogenesis (7). Leptin circulates as a 16 kD protein partially bound to plasma proteins and exerts its actions through specific cell surface receptors [leptin receptor (LR)] present in a variety of tissues localized to the cell membranes (8). Several epidemiological studies have linked high levels of plasma leptin with increased risk for breast carcinogenesis, but a direct link between leptin signaling and breast cancer was first established in a clinical study by Nagawa group in 2004 (9). They found that LRs were not detectable in normal mammary epithelial cells by immunohistochemistry, whereas 83% of the carcinoma cells showed positive staining for the LR (9). Importantly, overexpression of leptin was observed in 92% of breast tumors examined but in none of the normal breast epithelium (9). In recent years, many laboratories including our own have shown that leptin increases proliferation of different cancer cell types via multiple signaling pathways including Stat3/extracellular signal-regulated kinase/Akt signaling (10–13). The therapeutic potential of inhibition of leptin has been evaluated to some extent in diseases associated with metabolic syndrome, but the importance of inhibition of leptin signaling in carcinogenesis is still not known and is an active area of research.
Epidemiological results are compelling to show that dietary intake of cruciferous vegetables is protective against the risk of various types of malignancies including breast cancer (14–16). A case–control study involving >300 breast cancer patients and matched controls showed an inverse correlation between urinary levels of isothiocyanates (ITCs) as a biological measure of cruciferous vegetable intake and breast cancer risk (15). Similarly, another study showed that consumption of broccoli was inversely associated with premenopausal breast cancer risk (16). It is now widely accepted that anticarcinogenic effect of cruciferous vegetables is due to chemicals with an ITC functional group (N=C=S) that occur naturally as glucosinolates in a variety of edible cruciferous vegetables, including garden cress, broccoli, mustard, horseradish, watercress and cabbage (17). Benzyl isothiocyanate (BITC) is one of the best-studied members of the ITCs and is fast emerging as a novel anticancer agent. Importantly, BITC can inhibit chemically induced cancer in animal models (17,18). In addition, BITC suppresses proliferation and induces apoptosis in a variety of cancer cells, including breast cancer cells (19–22). Moreover, BITC administration inhibits growth of MDA-MB-231 human breast cancer xenograft and prevents mammary carcinogenesis in a transgenic mouse model (23,24).
The present study was designed to explore if BITC functions as an antagonist of leptin. We specifically investigated the protective effect of BITC against the oncogenic actions of leptin. Intriguingly, we found that BITC inhibits the effect of leptin on malignant properties of breast cancer cells including migration and invasion and also inhibits important downstream molecules of leptin signaling.
Materials and methods
Cell culture and reagents
The human breast cancer cell lines, MCF-7 and MDA-MB-231, were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gemini Bioproducts, Woodland, CA) and 2 μM l-glutamine (Invitrogen, Carlsbad, CA). Cell line authentication was done by analysis of known genetic markers or response (e.g. expression of estrogen receptor and p53 and estrogen responsiveness). For treatment, cells were seeded at a density of 1 × 106/100 mm tissue culture dish. After 24 h of serum starvation, the culture media were changed to serum free media containing treatments as indicated. Cultures were treated with human recombinant leptin (Sigma–Aldrich, St Louis, MO) at 100 ng/ml (12) and/or BITC (LKT Laboratories, St Paul, MN). d,L-sulforaphane (SFN) (purity 98.3%) was procured from LKT Laboratories. Antibodies for pStat3 (Phospho-Stat3), Stat3, JAK2, pJAK2 (Phospho-JAK2), SRC1, p-Src (phospho-Src-Tyr416) and pSrc were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against β-actin were purchased from Sigma–Aldrich.
Cell viability assay
Cells were seeded in 12-well plates at a density of 1 × 105 cells per well. After 12 h serum starvation, cells were pretreated with BITC (2.5 and 5 μM) or SFN (10 and 20 μM) for 2 h and then treated with leptin (100 ng/ml) for 22 h in the absence or presence of BITC. Effect of leptin and/or BITC treatments on cell viability was determined by trypan blue dye exclusion assay as described by us previously (25).
Clonogenicity assay
To perform colony formation assay (26), MCF-7 and MDA-MB-231 cells (single-cell suspension) were plated in 12-well plates at a density of 250 cells per well overnight. The following day, cells were treated with leptin and/or BITC treatments as indicated and the medium was replaced with fresh medium containing treatment every 3 days. After a 10 day treatment period, the medium was removed and cell colonies were stained with crystal violet (0.1% in 20% methanol). Colony numbers were assessed visually and colonies containing >50 normal-appearing cells were counted. Pictures were taken using a digital camera. All experiments were performed at least three times in triplicates.
Anchorage-independent growth assay
Anchorage-independent growth of MCF-7 and MDA-MB-231 cells was determined by colony formation on soft agar (26). Briefly, equal volumes of agar (1.2%) and complete medium were mixed to make 0.6% agar growth medium solution in six-well tissue culture plates. Cells (2 × 103 cells per well) were suspended in media with or without leptin or BITC treatment as indicated followed by mixing with equal volume of agar (0.6%). Cell suspension agar mix (2 ml) was then added to each well. Plates were incubated at 37°C with 5% CO2 in humidified incubator for 3 weeks, and media with or without treatment were added every 3 days. Colonies were stained with 0.005% crystal violet in phosphate-buffered saline for 1 h at room temperature and observed using Olympus IX50 inverted microscope. Colonies were counted in five randomly selected fields at ×10 magnification. Results are expressed as number of colonies counted. All experiments were performed three times in triplicate.
Migration assay
To perform migration assay (27), cells were plated into 24-well cell culture plates, pre-coated with human fibronectin (5 μg/cm2; Sigma–Aldrich). Cells were allowed to grow in 10% fetal bovine serum containing Dulbecco's modified Eagle's medium to confluence and then were washed with serum-free medium and serum starved for 16 h. A 1 mm wide scratch was made across the cell layer using a sterile pipette tip. After washing with serum-free medium twice, Dulbecco's modified Eagle's medium containing 10 μg/ml human fibronectin was added to replace matrix depleted with the cells. Plates were photographed immediately after scratching. Cells were treated with leptin and/or BITC as indicated. Plates were photographed after 24 and 48 h at the identical location of the initial image. All experiments were performed at least three times.
Electric cell-substrate impedance sensing wound-healing assay
Wound-healing assay was performed using the electric cell-substrate impedance sensing (ECIS) (Applied BioPhysics, Troy, NY) technology (27). For wound-healing assay, cells were grown to confluence on ECIS plates and serum starved for 16 h. The ECIS plates were submitted to an elevated voltage pulse of 40 kHz frequency, 3.5 V amplitude and 30 s duration, which led to the death and detachment of cells present on the small active electrode resulting in a wound normally healed by cells surrounding the small active electrode that have not been submitted to the elevated voltage pulse. Cells were immediately treated with leptin and/or BITC as indicated. Wound healing was then assessed by continuous resistance measurements for 24 h. All experiments were performed at least three times in triplicates.
Invasion assay
For an in vitro model system for metastasis, a Matrigel invasion assay (12) was performed by using a Matrigel invasion chamber from BD Biocoat Cellware (San Jose, CA). Cells were seeded at a density of 1 × 105 cells per insert and cultured overnight. After 16 h of serum starvation, the culture media were changed to serum free media containing treatments as indicated. Triplicate wells were used for each treatment. Cells were treated with leptin and/or BITC as indicated. After 24 h of incubation, cells remaining above the insert membrane were removed by gentle scraping with a sterile cotton swab. Cells that had invaded through the Matrigel to the bottom of the insert were fixed in methanol for 10 min. After washing with phosphate-buffered saline, the cells were stained with hematoxylin–eosin. The insert was subsequently washed with phosphate-buffered saline and briefly air-dried and mounted. The slides were coded to prevent counting bias, and the number of invaded cells on representative sections of each membrane were counted under light microscope. The number of invaded cells for each experimental sample represents the average of triplicate wells. All experiments were performed at least three times.
Western blotting
Whole cell lysate (12) was prepared by scraping MCF-7 and MDA-MB-231 cells in 250 μl of ice-cold modified RIPA buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1% NP-40, 0.25% Na-deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM Na3VO4 and 1 mM NaF]. The lysate was rotated 360° for 1 h at 4°C followed by centrifugation at 12 000g for 10 min at 4°C to clear the cellular debris. Protein was quantified using the Bradford protein assay kit (Bio-Rad, Hercules, CA). Equal amount of lysate protein was resolved on sodium dodecyl sulfate –polyacrylamide gel, transferred to nitrocellulose membrane and western blot analysis was performed. Immunodetection was performed using enhanced chemiluminescence (ECL system, Amersham Pharmacia Biotech, Arlington Heights, IL) according to manufacturer's instructions.
Immunohistochemical analysis for pSTAT3 in MDA-MB-231 xenografts
We have shown previously that BITC administration retards growth of MDA-MB-231 cells implanted in female athymic mice (23). We used tumor sections from the same study to determine the effect of BITC administration on expression of pSTAT3 by immunohistochemistry. Immunohistochemistry was performed essentially as described by us previously for other proteins (23,24). At least four non-overlapping representative images from each tumor section from five mice of each group were captured using ImagePro software for quantitation of pSTAT3 expression.
RNA interference and luciferase assay
Human cyclin D1 promoter reporter construct used for luciferase assay (–1745-CD1-Luc) was a kind gift from Dr Richard Pestell (Georgetown University, Washington, DC) (28). The plasmid expressing a constitutively active form of Stat3 (Stat3C-FLAG) was a kind gift from Dr James E.Darnell (The Rockefeller University, New York, NY) (29). The nuclear factor-kappaB (NF-κB)-luciferase reporter plasmid was generously provided by Dr Anning Lin (University of Chicago, Chicago, IL). Cells were transfected with expression vectors and luciferase construct and treated with leptin and/or BITC as indicated using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Forty-eight hours post-transfection, the cells were harvested and luciferase and Renilla activities were measured using a dual luciferase kit (Promega, Madison, WI). The relative firefly luciferase activities were calculated by normalizing transfection efficiency according to Renilla luciferase activities. For RNA interference, cells were seeded in six-well plates and transfected at 50% confluency with 100 nM of control siRNA or Stat3-targeted siRNA (Invitrogen) using Oligofectamine according to the manufacturer's instructions. Twenty-four hours after transfection, the cells were transfected with cyclin D1 promoter reporter construct and treated with leptin and/or BITC. Forty-eight hours post-transfection, the cells were harvested and luciferase and Renilla activities were measured using a dual luciferase kit (Promega). The relative firefly luciferase activities were calculated by normalizing transfection efficiency according to Renilla luciferase activities. The experiments were performed three times in triplicate.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) analyses were performed using our published procedure (12) with following modifications. Chromatin samples were sonicated on ice three times for 10 s each (i.e. until the average length of sheared genomic DNA was 1–1.5 kb) followed by centrifugation for 10 min. The immunoprecipitated DNA was ethanol precipitated and resuspended in 25 μl of water. Total input samples were resuspended in 100 μl of water and diluted 1:100 before polymerase chain reaction (PCR) analysis. Initially, PCR was performed with different numbers of cycles and/or dilutions of input DNA to determine the linear range of amplification; all results shown fall within this range. Following 28–30 cycles of amplification, PCR products were run on 1% agarose gel and analyzed by ethidium bromide staining. All ChIP assays were performed at least thrice.
Results
BITC-treatment inhibited oncogenic actions of leptin in human breast cancer cells
We showed previously that leptin increases proliferation and growth of breast cancer cells via activation of multiple downstream signaling pathways including Stat3 (10–12,27). Because BITC has been shown to inhibit Stat3, we postulated that the oncogenic effects of leptin may be reversed in the presence of BITC. Here, we experimentally tested this hypothesis using well-characterized human breast cancer cell lines (MCF-7 and MDA-MB-231) as a model. The effect of BITC on cell viability of breast cancer cells was determined by trypan blue exclusion assay and the results are summarized in Figure 1A. Leptin treatment significantly stimulated survival of MCF-7 and MDA-MB-231 cells, whereas the survival of both cell lines was decreased significantly after exposure to BITC in a concentration-dependent manner. Cell viability was reduced to ∼50–60% with 2.5 μM BITC treatment, whereas higher concentration (5 μM) was more inhibitory. The MCF7 cells were relatively more sensitive to the leptin-induced cell proliferation as compared with MDA-MB-231 cells. In addition, BITC conferred significant protection against leptin-stimulated cell viability of MDA-MB-231 and MCF-7 cells (Figure 1A). Breast cancer cells were treated with various concentrations of BITC and subjected to clonogenicity and anchorage-independent growth assay. Dose dependent and statistically significant inhibition of clonogenicity and soft-agar colony formation was observed in the presence of BITC (supplementary Figure S1 is available at Carcinogenesis Online). Consistent with these observations, BITC (2.5 μM) treatment not only reduced leptin-induced clonogenicity (Figure 1B) but also anchorage-independent growth in both cell lines (Figure 1C).
Fig. 1.
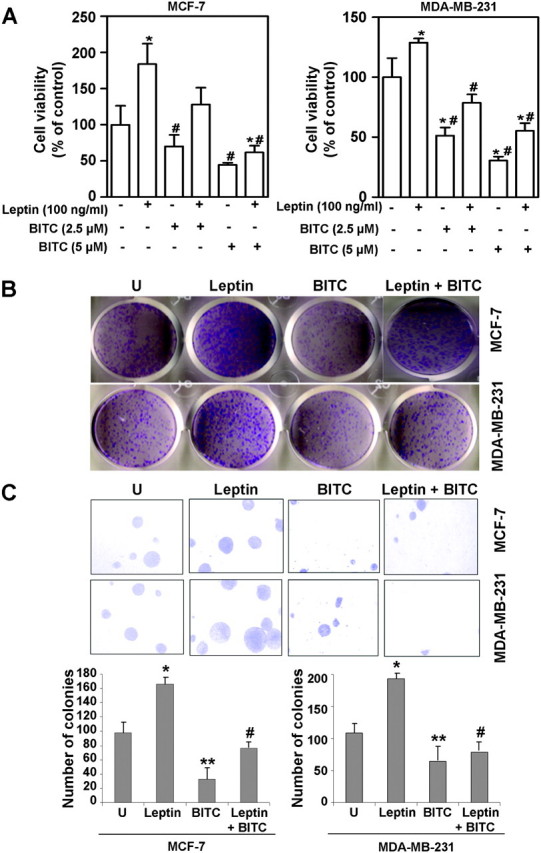
BITC reduces the stimulatory effect of leptin on clonogenicity and anchorage-independent growth of breast carcinoma cells. (A) Breast cancer cells (MCF-7 and MDA-MB-231) were treated with leptin and/or BITC and cell viability was examined by trypan blue dye exclusion assay. *P < 0.05 compared with dimethyl sulfoxide-treated control; #P < 0.05 compared with leptin treatment by one-way analysis of variance followed by Bonferroni's comparison test. (B) Breast cancer cells were treated with leptin and BITC alone and in combination and subjected to clonogenicity assay. Untreated cells are denoted with the letter ‘U’. Colonies containing >50 normal-appearing cells were counted. (C) Breast cancer cells were subjected to soft-agar colony-formation assay in the presence of leptin and/or BITC for 3 weeks. Untreated cells are denoted with the letter U. Results are expressed as average number of colonies counted (in six micro-fields). *P < 0.005 compared with controls; **P < 0.001 compared with controls; #P < 0.005 compared with leptin treatment.
Cancer progression is a multistep process that involves invasion of basement membrane by tumor cells and migration to points far from a given primary tumor mass leading to metastasis (30). We examined the effects of leptin and/or BITC treatments on invasion and migration properties of breast carcinoma cells using Matrigel invasion and scratch migration assay. BITC treatment inhibited migration and invasion potential of breast cancer cells in a concentration-dependent manner (supplementary Figure S2 is available at Carcinogenesis Online). As expected, we found that leptin increased migration of breast carcinoma cells, whereas BITC inhibited migration in a conventional scratch migration assay. Importantly, BITC treatment also inhibited migration of MCF-7 and MDA-MB-231 breast cancer cells in the presence of leptin overcoming its strong pro-migratory potential (Figure 2A). For quantitative determination of alteration in migration potential of breast cancer cells upon treatment with leptin and/or BITC, we performed a quantitative real-time impedance assay using an ECIS-based technique (27). As expected, confluent cells showed high resistance values. Confluent cells were subjected to high voltage pulse that resulted in drop in resistance indicating death and detachment of cells present on the small active electrode followed by leptin and/or BITC treatments. Leptin treatment resulted in higher resistance showing increased migration of cells surrounding the small active electrode that were not submitted to the elevated voltage pulse. BITC treatment inhibited migration resulting in lower resistance values. BITC treatment also inhibited leptin-induced migration in combination treatment (Figure 2B). Next, we performed Matrigel invasion assay to examine the effect of BITC on leptin-induced invasion potential of breast carcinoma cells. As evident from Figure 2C, leptin treatment increased invasion of cancer cells through Matrigel in comparison with untreated cells, whereas BITC treatment inhibited invasion of breast cancer (MCF-7 and MDA-MB-231) cells. Interestingly, we found that BITC treatment efficiently inhibited leptin-induced increased invasion of cancer cells (Figure 2C). Collectively, these results show that BITC treatment can effectively inhibit leptin-induced clonogenicity, anchorage-independent colony formation, migration and invasion of breast carcinoma cells.
Fig. 2.

BITC inhibits leptin-induced migration and invasion of breast carcinoma cells. (A) Breast cancer cells (MCF-7 and MDA-MB-231) were subjected to scratch migration assay. Culture media were replaced with media containing 100 ng/ml leptin, 2.5 μM BITC alone and in combination or untreated media (U). The plates were photographed at the identical location of the initial image (0 h) at 24 h. The results shown are representative of three independent experiments performed in triplicates. The histogram shows the fold change in migration. *P<0.01 compared with untreated controls; **P<0.01compared with untreated cells; ***P < 0.005 compared with leptin treatment. All the experiments were performed thrice in triplicates. (B) MCF-7 and MDA-MB-231 cells were grown to confluence in ECIS plates, serum starved for 16 h and subjected to an elevated voltage pulse of 40 kHz frequency at 3.5 V amplitude for 30 s to incite a wound. The cells were immediately treated with leptin and/or BITC as indicated. The wound was then allowed to heal from cells surrounding the small active electrode that did not undergo the elevated voltage pulse. Resistance was measured before and after the elevated voltage pulse application as described in the ‘Materials and Methods’. The measurements were stopped 24 h after the creation of wound. All the experiments were performed thrice in triplicates. (C) MCF-7 and MDA-MB-231 cells were cultured in Matrigel invasion chambers followed by treatment with leptin, BITC alone and in combination for 24 h. The number of cells that invaded through the Matrigel was counted in five different regions. The slides were blinded to remove counting bias. The results show mean of three independent experiments performed in triplicates. *P < 0.005 compared with untreated controls; **P < 0.001 compared with untreated controls; ***P < 0.001 compared with leptin-treated cells.
BITC treatment suppressed leptin-induced Stat3 phosphorylation in breast cancer cells
Binding of leptin to LR (Ob-Rb) phosphorylates conserved tyrosine residues and these phosphorylation events are important for subsequent signaling events including JAK and Stat3 activation (12). Our previous studies have shown direct involvement of JAK/Stat3, phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated kinase signaling in pro-cancerous actions of leptin (12). To investigate hierarchy of these events, we previously found that leptin signaling is transmitted mainly by the JAK/Stat3 pathway as inhibition of Stat3 activation further inhibits leptin-induced phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated kinase signaling as well as leptin-induced proliferation (10,11). These studies put forth Stat3 activation as an integral event in leptin signaling. We sought to determine the underlying molecular mechanism by which BITC treatment inhibited oncogenic actions of leptin. MCF-7 and MDA-MB-231 cells were treated with leptin for 30 min or 1 h. Consistent with previous observations (12), Tyr705 phosphorylation of Stat3 was increased markedly in leptin-treated MCF-7 and MDA-MB-231 cells, which was not due to an increase in protein level of total Stat3 (Figure 3A). The leptin-inducible Tyr705 phosphorylation of Stat3 was fully reversible in the presence of BITC. Tyrosine phosphorylation of Stat3 is an early event in the activation that is required for its dimerization and DNA-binding activity. In addition, phosphorylation of a serine residue in the C-terminal transcriptional activation domain, corresponding to Ser727 enhances its transcriptional activity (31,32). As can be seen in Figure 3B, BITC treatment reduced levels of both tyrosine and serine phosphorylated Stat3 in breast cancer cells in a concentration- and time-dependent manner. For example, the level of pStat3Tyr705 was reduced by 70% by a 24 h exposure to 2.5 and 5 μM BITC (Figure 3B). Phosphorylation of Stat3 is mediated by various kinases, including Src and JAK2 (33,34). We examined the level of pJAK2 to determine if BITC-mediated inhibition of Stat3 activation was due to suppression of JAK2 phosphorylation. The level of pJAK2 was also reduced in a concentration- and time-dependent manner upon BITC treatment (Figure 3B). BITC-mediated suppression of JAK2 phosphorylation correlated with a marked decrease in its protein level (Figure 3B). We also examined the level of pSrc to determine its involvement in BITC-mediated inhibition of Stat3 activation. BITC treatment reduced the level of pSrc in a time-dependent manner (Figure 3B). In a recent study, we showed that BITC administration retarded growth of MDA-MB-231 cells implanted in female athymic mice (23). We used tumor sections from the same study to determine effect of BITC administration on expression of pSTAT3 protein by immunohistochemistry. We found decreased phosphorylation of Stat3 in tumors from BITC-treated mice in comparison with control tumors (Figure 3C).
Fig. 3.
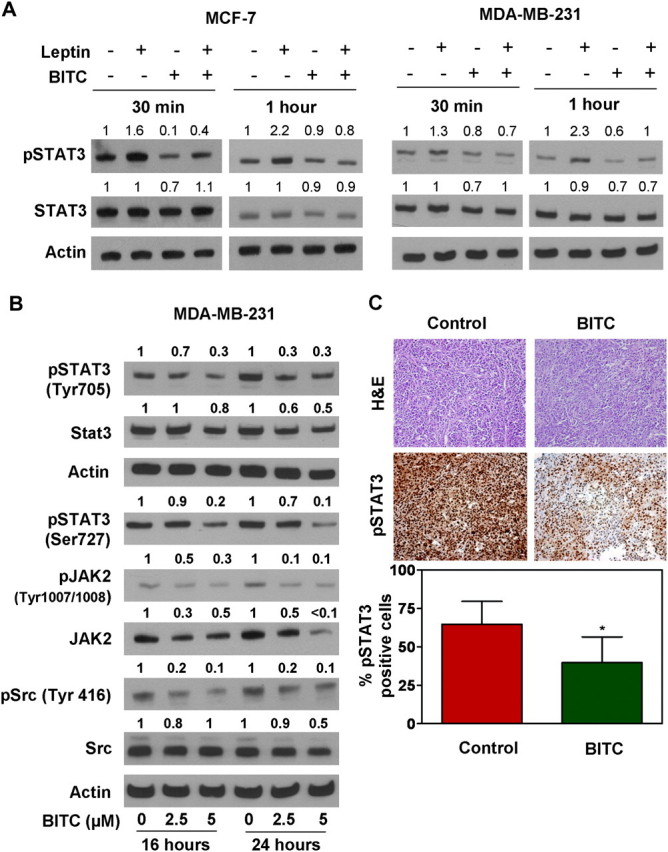
BITC inhibited leptin signaling in breast cancer cells. (A) Breast cancer cells were treated with leptin and/or BITC for 30 min or 1 h. Total protein was isolated and equal amounts of proteins were subjected to immunoblotting. The membranes were re-blotted using total Stat3 antibody. Anti-actin antibody was used as a control. The blots are representative of at least two independent experiments. (B) Immunoblotting for pSTAT3 (Tyr705 and Ser727), total Stat3, pJAK2 (Tyr1007/1008), total JAK2, pSrc and Src using lysates from MDA-MB-231 cells treated for 16 and 24 h with the indicated concentrations of BITC. The blots were stripped and re-probed with anti-actin antibody as a loading control. Numbers above bands represent densitometric quantitation relative to corresponding control. Each experiment was done twice using independently prepared lysates, and representative data from one such experiment are shown. (C) Immunohistochemical analysis for pSTAT3 (Tyr705) in representative tumor section of a control mouse and a mouse treated with 7.5 μM BITC (23) (magnification ×200). Bar diagram shows quantitation of pSTAT3 expression in tumors from control and BITC-treated mice. Columns, mean (n = 5); bar, standard deviation. *Significantly different (P < 0.05) compared with control by two-sided Student's t-test.
Stat3 was a critical target of BITC-mediated inhibition of leptin signaling
We have shown previously that one of the targets of leptin action is cyclin D1, and leptin treatment induces upregulation and transactivation of cyclin D1 (12). Next, we investigated if BITC treatment inhibits leptin-induced cyclin D1 transactivation using a cyclin D1 promoter luciferase construct, -1745-CD1-Luc (Figure 4A). The MCF-7 and MDA-MB-231 cells transfected with -1745-CD1-Luc demonstrated a 4- to –6-fold increase in luciferase activity in response to leptin exposure. BITC treatment alone significantly inhibited cyclin D1 transactivation indicating that BITC negatively regulates the activity of the cyclin D1 promoter. Interestingly, we also found that the leptin-stimulated cyclin D1 transactivation was partially but statistically significantly suppressed in the presence of BITC (Figure 4A). Thus, BITC efficiently overcame the stimulatory effects of leptin on cyclin D1 transactivation (Figure 4A). Cyclin D1 is also regulated by NF-κB (35). We investigated the effects of BITC on NF-κB in a luciferase assay. We found that BITC does not affect NF-κB activity (supplementary Figure S3 is available at Carcinogenesis Online).
Fig. 4.

BITC inhibits leptin-induced recruitment of Stat3 to cyclin D1 promoter. (A) Schematic representation of cyclin D1 promoter showing two GAS consensus sites located at -481 and -247. MCF-7 and MDA-MB-231 cells were transiently transfected with the human cyclin D1 construct (-1745-CD1-Luc) together with the internal control Renilla luciferase reporter and treated with leptin and/or BITC. Untreated cells are denoted with the letter ‘U’. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. *P < 0.005 compared with untreated controls; **P < 0.01 compared with leptin-treated cells; #P < 0.001 compared with leptin treated cells. (B) Soluble chromatin was prepared from MCF-7 and MDA-MB-231 cells, treated with leptin and/or BITC and immunoprecipitated with 5 μg of specific antibody against pStat3 and SRC1 overnight at 4°C. The immune complexes were pulled-down with protein A agarose/salmon sperm DNA beads and washed extensively and cross-linking was reversed. The purified DNA was analyzed by PCR using primers spanning the GAS sites (-481 and -247) at the cyclin D1 promoter. ChIP assay shows that BITC inhibits leptin-induced Stat3 and SRC1 recruitment to cyclin D1 promoter.
Gene expression analyses in response to cytokine signaling have shown that Stat proteins are signaling molecules with dual functions (36). Stat proteins not only transmit a signal from the cell surface to the nucleus but also directly participate in gene regulation (36). Cyclin D1 promoter contains binding sites for a number of transcription factors (37,38). The consensus Stat-binding motifs (TTCNNNGAA) at -247 and -481 were of particular interest for these studies because these consensus sites have previously been shown to be sites for cytokine action mediated by Stats in other systems. We further sought to determine if BITC treatment can inhibit direct participation of Stat3 in leptin-mediated cyclin D1 gene regulation using ChIP assay. Using specific antibodies against Stat3, formaldehyde cross-linked protein–chromatin complexes were immunoprecipitated from MCF-7 and MDA-MB-231 cells cultured with or without leptin and/or BITC. Resulting precipitated genomic DNA was then analyzed by PCR using primers spanning the Stat3-binding elements in the promoter region of the cyclin D1 gene. As shown in Figure 4B, ChIP analysis with anti-Stat3 antibody revealed that Stat3 was associated with the cyclin D1 promoter in the presence of leptin, whereas BITC treatment abrogated leptin-induced recruitment of Stat3 on cyclin D1 promoter (Figure 4B). Previously, we have shown that leptin-induced Stat3 recruits histone acetyltransferase activity containing coactivator such as SRC-1 to cyclin D1 promoter. We analyzed the recruitment of SRC1 to cyclin D1 promoter in the presence of BITC. We found that BITC inhibits recruitment of SRC1 to cyclin D1 promoter (Figure 4B).
BITC treatment inhibited leptin-induced recruitment of Stat3 to cyclin D1 promoter indicating that Stat3 may be a pivotal point in BITC-leptin cross talk. Therefore, the effect of overexpression of constitutively active Stat3 on BITC-mediated inhibition of leptin signaling was examined. Leptin-induced luciferase activity driven by the cyclin D1 promoter was augmented by co-transfection of constitutively active Stat3 (Stat3C). BITC treatment inhibited cyclin D1 transactivation with or without leptin treatment. Importantly, Stat3C overexpression interfered with BITC-mediated inhibition of cyclin D1 transactivation in the presence of leptin (Figure 5A). For further confirmation, we proceeded to determine the effect of Stat3 protein knockdown on BITC-mediated cyclin D1 transactivation in MCF-7 and MDA-MB-231 cells. The level of Stat3 and p-Stat3 protein was reduced by 80% on transient transfection of breast cancer cells with Stat3-targeted siRNA in comparison with cells transfected with a control non-specific siRNA (Figure 5B). Knockdown of Stat3 resulted in a decrease in leptin-induced cyclin D1 transactivation. On the other hand, Stat3 protein inhibition significantly potentiated inhibitory effects of BITC on cyclin D1 transactivation. We also observed a statistically significant increase in BITC-mediated inhibition of cyclin D1 transactivation in the presence of leptin (Figure 5C). Collectively, the results showed the involvement of Stat3 in BITC-leptin cross talk. BITC treatment inhibited leptin-induced stat3 activation and recruitment to cyclin D1 promoter resulting in inhibition of cyclin D1 transactivation in breast cancer cells.
Fig. 5.

Stat3 plays an important role in BITC-mediated inhibition of leptin signaling. (A) MCF-7 and MDA-MB-231 cells were transiently co-transfected with the human cyclin D1 construct (-1745-CD-Luc) and constitutively active Stat3 (Stat3C) as indicated together with the internal control Renilla luciferase reporter and treated with leptin and/or BITC. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents mean of three experiments. *P < 0.001 compared with BITC treated, vector-transfected cells; **P < 0.01 compared with leptin + BITC treated, vector-transfected cells. (B) MCF-7 and MDA-MB-231 cells were transfected with Stat3-targeted siRNA or scrambled control using Oligofectamine for 24 h. Total protein was isolated and equal amounts of proteins were subjected to immunoblotting using anti-Stat3 and anti-pStat3 antibody. Untransfected controls are denoted by U. (C) MCF-7 and MDA-MB-231 cells were transfected with Stat3-targeted siRNA as above. After 24 h, cells were co-transfected with the human cyclin D1 construct (-1745-CD-Luc) together with the internal control Renilla luciferase reporter and treated with leptin and/or BITC. Letter U signifies untreated cells. Untransfected cells were included as controls. Cell lysates were assayed for luciferase activity and activities were normalized against the internal control. Data represents the mean of three experiments. *P < 0.001 compared with leptin + BITC treated, scrambled-vector transfected cells.
Next, we questioned if inhibition of oncogenic effects of leptin was unique to BITC. We addressed this question using SFN and MDA-MB-231 cells. SFN, a synthetic racemic analogue of broccoli-derived l-isomer, inhibits proliferation and induces apoptosis in many cancer cell types (39). Similar to BITC, SFN treatment reduced cell viability in the absence or presence of leptin stimulation (Figure 6A). In addition, SFN treatment inhibited invasion potential of breast cancer cells (Figure 6B). SFN was also effective in inhibiting leptin-induced cell invasion (Figure 6C).
Fig. 6.
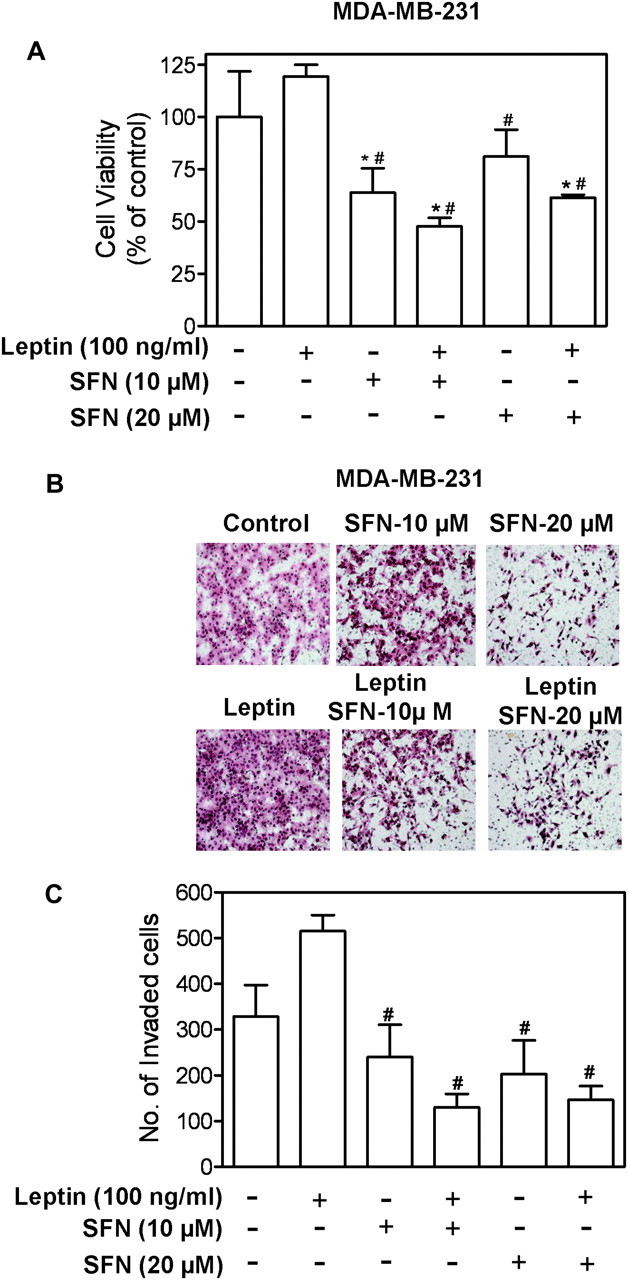
SFN inhibited leptin-stimulated proliferation and invasion of MDA-MB-231 cells. (A) MDA-MB-231 cells were serum starved for 12 h and then treated with SFN (10 or 20 μM) for 2 h followed by treatment with 100 ng/ml leptin for 22 h. Cell viability was determined by trypan blue dye exclusion assay. Columns, mean (n = 3); bar, standard deviation. *P < 0.05 compared with control; #P < 0.05 compared with leptin treated cells by one-way analysis of variance followed by Bonferroni's test. (B) Serum-starved MDA-MB-231 cells were cultured in Matrigel-coated transwell, followed by treatment by 100 ng/ml leptin and SFN (10 and 20 μM) alone or combination for 24 h. Figure shows cells that invaded through Matrigel onto the lower side of the filter. (C) Quantitation of invaded cells. Column, mean (n = 4); bar, standard deviation. Significantly different (P < 0.05) compared with dimethyl sulfoxide-treated control (*) and leptin treatment (#) by one-way analysis of variance followed by Bonferroni's comparison test.
Discussion
Adipose tissue is an active endocrine organ that secretes various biologically active adipocytokines and provides a potential molecular mechanism to link obesity with carcinogenesis (40). One of the important adipocytokine is leptin. Genetically, obese leptin-deficient MMTV-TGF-α/Lepob Lepob and leptin-receptor-deficient MMTV-TGF-α/Leprdb Leprdb female mice do not develop mammary tumors providing additional evidence for the integral role of leptin and its cognate receptor in mammary tumorigenesis (41,42). Analysis of clinical samples showed that LRs were not detectable in normal mammary epithelial cells by immunohistochemistry, whereas carcinoma cells showed positive staining for OB-R (leptin receptor) in 83% of the cases (9). Leptin has been characterized as a potent growth-stimulating factor for breast cancer that also induces breast cancer cell migration and invasion (27). Previous studies have shown that leptin induces proliferative responses both in ER-negative and ER-positive breast cancer cells (43,44). However, a recent study reported differential sensitivity to leptin treatment in breast cancer cells based on ER status (45). Discrepancy might be due to clonal variations of the cell lines used. We consistently found leptin-induced proliferation in both MCF-7 and MDA-MB-231 cells.
Reagents blocking leptin activity might prove useful for breast cancer patients with elevated leptin levels. Such neutralization can be achieved with soluble LRs that bind free leptin in the circulation, leptin antagonists that bind to, but do not activate LRs, and either specific anti-LR monoclonal antibodies (anti-LR mAbs) that bind to the receptor preventing leptin signaling or anti-leptin antibodies. Anti-LR mAbs have high molecular mass that ensures a long half-life in the circulation and good affinity for the receptor. But these mouse-generated mAbs need to be humanized to eliminate their potential immunogenicity. Nanobodies represent another approach to target LR and block the ligand-induced conformational change without interfering with the leptin-LR interaction. These recombinant, monomeric small nanobodies are stable and easy to manipulate (46). They can selectively inhibit peripheral activity of leptin as they do not cross blood–brain barrier. Importantly, recent development of leptin muteins with antagonistic properties and other proteins blocking leptin activity present new possibilities for research. To date, all these agents to counteract leptin signaling are in initial stages of development. Therefore, there is a clear and compelling need to identify novel agents that are relatively safe but could suppress leptin signaling and its biological effects.
BITC, a promising cancer chemopreventive agent, has been shown previously to noticeably suppress the incidence and/or burden of mammary hyperplasia and carcinoma in female MMTV-neu mice (24). BITC-mediated prevention of mammary carcinogenesis in MMTV-neu correlates not only with reduction of cell proliferation and increased apoptosis but also infiltration of T cells (24). BITC has also been shown to impede MDA-MB-231 breast cancer xenograft growth in association with decreased tumor cell proliferation (23). Moreover, BITC treatment reduced migratory potential of MDA-MB-231 cells (23). On the other hand, leptin has been shown to increase tumor growth in vivo and augment proliferation, invasion and migration of breast cancer cells (27,47,48) The present study shows that BITC inhibits leptin-induced breast cancer growth and metastatic properties. Specifically, BITC treatment inhibits leptin-induced clonogenicity, anchorage-independent three dimensional colony formation, invasion and migration of breast carcinoma cells. Higher levels of leptin in obese breast cancer patients have been associated with higher risk for lymph node metastasis, larger tumor burden when compared with non-obese breast cancer patients. Our studies indicate that BITC treatment may be able to inhibit tumor metastasis owing to high leptin levels.
Examination of molecular pathogenesis of cancer has shown that disruption of the cell cycle in human tumors is a critical interface between hormonal signaling and tumorigenesis. The dysregulation of cell cycle control in cancer is compressed into a model where disruption of two parallel pathways is required for the occurrence of cancer. These two parallel pathways include either the p16 (INK4a) pathway or the p19 (ARF) pathway. Our studies show that leptin led to overexpression of cyclin D1 thus inactivating one arm of the signaling pathway leading to unchecked cell cycle progression and possibly tumorigenic growth. Cyclin D1 transgenic animals show perturbed mammary gland development with increased proliferation and precocious lobuloalevolar development (49). Tumors appear in 75% of mice but after a long latency compared with oncogenes c-myc, Ha-ras and c-neu, which induce tumors relatively early (49). The ability of cyclin D1 overexpression to induce mammary carcinoma and the necessity for cyclin D1 function for cell cycle progression raises the question of whether cyclin D1 is necessary for tumor development. So far, the relationship between overexpression of cyclin D1 and breast cancer outcome has been controversial with studies reporting both positive and negative findings (50,51). We found that BITC treatment inhibited leptin-induced cyclin D1 transactivation.
Investigating the signaling pathways involved in leptin-induced cyclin D1 transactivation, we found that activation of JAK/Stat pathway regulates the transactivation of cyclin D1 gene (12). In a parallel study, Leslie at al. (52) reported that cyclin D1 is transcriptionally regulated by Stat3 and is required for transformation. We previously showed that leptin-activated-Stat3 binds to its cognate binding sites in cyclin D1 promoter, acting as a transcription factor, leading to hyperacetylation and overexpression of cyclin D1 gene. Inhibition of JAK/Stat pathway significantly reduced leptin-induced growth of breast carcinoma cells (12). Here, we show that BITC inhibits leptin-induced Stat3 activation by phosphorylation at Tyr705.Our study shows that BITC-mediated inhibition of Stat3 activation in breast cancer cells is accompanied by suppression of JAK2 activation. The kinetics of BITC-mediated inhibition of JAK2 is similar to that of pStat3 inhibition. BITC treatment also reduces protein levels of JAK2. Stat3 activation leads to dimerization, nuclear translocation and recognition of Stat3-specific DNA-binding elements and activation of target gene transcription (33,34). We found that BITC treatment inhibits leptin-induced Stat3 recruitment to cyclin D1 promoter. Furthermore, Stat3 activation represents a pivotal point in leptin-BITC cross talk as overexpression of constitutively active Stat3 interferes with BITC-mediated inhibition of leptin action, whereas Stat3 knockdown potentiates BITC-mediated inhibition of leptin action.
In summary, the present study provides concrete experimental evidence in support of a novel function of BITC as a potential antagonistic agent against oncogenic actions of leptin. Considering the high prevalence of obesity in the developed world, our study has the potential to significantly impact the vast majority of obese breast carcinoma patients by providing new possibilities for research and ultimately therapy using BITC for inhibiting invasion and migration properties of tumor cells and improving overall prognosis.
Supplementary material
Supplementary Figures S1–S3 can be found at http://carcin.oxfordjournals.org/
Funding
United States Public Health Service (NCI-RO1 CA129347-04, NCI-RO1 CA142604-01 to S.V.S.; NIDDK-K01 DK077137, NIDDK-R03 DK089130 to N.K.S.; NCI-R01 CA131294 to D.S.).
Supplementary Material
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- BITC
benzyl isothiocyanate
- ChIP
Chromatin immunoprecipitation
- ECIS
electric cell-substrate impedance sensing
- ER
estrogen receptor
- ITC
isothiocyanate
- NF-κB
nuclear factor-kappaB
- PCR
polymerase chain reaction
- SFN
d,l-sulforaphane
References
- 1.Calle EE, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 2.Rose DP, et al. Adverse effects of obesity on breast cancer prognosis, and the biological actions of leptin (review) Int. J. Oncol. 2002;21:1285–1292. [PubMed] [Google Scholar]
- 3.Vona-Davis L, et al. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer. 2007;14:189–206. doi: 10.1677/ERC-06-0068. [DOI] [PubMed] [Google Scholar]
- 4.Muoio DM, et al. Peripheral metabolic actions of leptin. Best Pract. Res. Clin. Endocrinol. Metab. 2002;16:653–666. doi: 10.1053/beem.2002.0223. [DOI] [PubMed] [Google Scholar]
- 5.Singhal A, et al. Influence of leptin on arterial distensibility: a novel link between obesity and cardiovascular disease? Circulation. 2002;106:1919–1924. doi: 10.1161/01.cir.0000033219.24717.52. [DOI] [PubMed] [Google Scholar]
- 6.Wolk R, et al. Plasma leptin and prognosis in patients with established coronary atherosclerosis. J. Am. Coll. Cardiol. 2004;44:1819–1824. doi: 10.1016/j.jacc.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 7.Somasundar P, et al. Leptin is a growth factor in cancer. J. Surg. Res. 2004;116:337–349. doi: 10.1016/j.jss.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Houseknecht KL, et al. Evidence for leptin binding to proteins in serum of rodents and humans: modulation with obesity. Diabetes. 1996;45:1638–1643. doi: 10.2337/diab.45.11.1638. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa M, et al. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 2004;10:4325–4331. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- 10.Sharma D, et al. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr. Relat. Cancer. 2006;13:629–640. doi: 10.1677/erc.1.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxena NK, et al. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saxena NK, et al. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem. 2007;282:13316–13325. doi: 10.1074/jbc.M609798200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaffler A, et al. Mechanisms of disease: adipokines and breast cancer—endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat. Clin. Pract. Endocrinol. Metab. 2007;3:345–354. doi: 10.1038/ncpendmet0456. [DOI] [PubMed] [Google Scholar]
- 14.Verhoeven DT, et al. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidemiol. Biomarkers Prev. 1996;5:733–748. [PubMed] [Google Scholar]
- 15.Fowke JH, et al. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003;63:3980–3986. [PubMed] [Google Scholar]
- 16.Ambrosone CB, et al. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 2004;134:1134–1138. doi: 10.1093/jn/134.5.1134. [DOI] [PubMed] [Google Scholar]
- 17.Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 2000;32:395–411. doi: 10.1081/dmr-100102342. [DOI] [PubMed] [Google Scholar]
- 18.Wattenberg LW. Inhibitory effects of benzyl isothiocyanate administered shortly before diethylnitrosamine or benzo[a]pyrene on pulmonary and forestomach neoplasia in A/J mice. Carcinogenesis. 1987;8:1971–1973. doi: 10.1093/carcin/8.12.1971. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura Y, et al. Involvement of the mitochondrial death pathway in chemopreventive benzyl isothiocyanate-induced apoptosis. J. Biol. Chem. 2002;277:8492–8499. doi: 10.1074/jbc.M109760200. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava SK, et al. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25:1701–1709. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 21.Xiao D, et al. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol. Cancer Ther. 2006;5:2931–2945. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 22.Xiao D, et al. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species-dependent apoptosis in human breast cancer cells. J. Biol. Chem. 2008;283:30151–30163. doi: 10.1074/jbc.M802529200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warin R, et al. Inhibition of human breast cancer xenograft growth by cruciferous vegetable constituent benzyl isothiocyanate. Mol. Carcinog. 2010;49:500–507. doi: 10.1002/mc.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warin R, et al. Prevention of mammary carcinogenesis in MMTV-neu mice by cruciferous vegetable constituent benzyl isothiocyanate. Cancer Res. 2009;69:9473–9480. doi: 10.1158/0008-5472.CAN-09-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao D, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23:5594–5606. doi: 10.1038/sj.onc.1207747. [DOI] [PubMed] [Google Scholar]
- 26.Taliaferro-Smith L, et al. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene. 2009;28:2621–2633. doi: 10.1038/onc.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saxena NK, et al. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res. 2008;68:9712–9722. doi: 10.1158/0008-5472.CAN-08-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee RJ, et al. pp.60(v-src) induction of cyclin D1 requires collaborative interactions between the extracellular signal-regulated kinase, p38, and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp.60(v-src) signaling in breast cancer cells. J. Biol. Chem. 1999;274:7341–7350. doi: 10.1074/jbc.274.11.7341. [DOI] [PubMed] [Google Scholar]
- 29.Bromberg JF, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 30.Steeg PS, et al. Metastasis: a therapeutic target for cancer. Nat. Clin. Pract. Oncol. 2008;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen Z, et al. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–2067. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen Z, et al. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 33.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 34.Yu CL, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 35.Guttridge DC, et al. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stark GR, et al. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 37.Allan AL, et al. Activating transcription factor 3 induces DNA synthesis and expression of cyclin D1 in hepatocytes. J. Biol. Chem. 2001;276:27272–27280. doi: 10.1074/jbc.M103196200. [DOI] [PubMed] [Google Scholar]
- 38.Albanese C, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 39.Stan SD, et al. Bioactive food components and cancer risk reduction. J. Cell. Biochem. 2008;104:339–356. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 40.Lorincz AM, et al. Molecular links between obesity and breast cancer. Endocr. Relat. Cancer. 2006;13:279–292. doi: 10.1677/erc.1.00729. [DOI] [PubMed] [Google Scholar]
- 41.Cleary MP, et al. Genetically obese MMTV-TGF-alpha/Lep(ob)Lep(ob) female mice do not develop mammary tumors. Breast Cancer Res. Treat. 2003;77:205–215. doi: 10.1023/a:1021891825399. [DOI] [PubMed] [Google Scholar]
- 42.Cleary MP, et al. Leptin receptor-deficient MMTV-TGF-alpha/Lepr(db)Lepr(db) female mice do not develop oncogene-induced mammary tumors. Exp. Biol. Med. (Maywood) 2004;229:182–193. doi: 10.1177/153537020422900207. [DOI] [PubMed] [Google Scholar]
- 43.Ray A, et al. Effects of leptin on human breast cancer cell lines in relationship to estrogen receptor and HER2 status. Int. J. Oncol. 2007;30:1499–1509. [PubMed] [Google Scholar]
- 44.Frankenberry KA, et al. Leptin receptor expression and cell signaling in breast cancer. Int. J. Oncol. 2006;28:985–993. [PubMed] [Google Scholar]
- 45.Fusco R, et al. Cellular and molecular crosstalk between leptin receptor and estrogen receptor-{alpha} in breast cancer: molecular basis for a novel therapeutic setting. Endocr. Relat. Cancer. 2010;17:373–382. doi: 10.1677/ERC-09-0340. [DOI] [PubMed] [Google Scholar]
- 46.Gertler A. Development of leptin antagonists and their potential use in experimental biology and medicine. Trends Endocrinol. Metab. 2006;17:372–378. doi: 10.1016/j.tem.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez RR, et al. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2) J. Biol. Chem. 2006;281:26320–26328. doi: 10.1074/jbc.M601991200. [DOI] [PubMed] [Google Scholar]
- 48.McMurtry V, et al. Leptin utilizes Jun N-terminal kinases to stimulate the invasion of MCF-7 breast cancer cells. Clin. Exp. Metastasis. 2009;26:197–204. doi: 10.1007/s10585-008-9231-x. [DOI] [PubMed] [Google Scholar]
- 49.Wang TC, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 50.Gillett C, et al. Cyclin D1 and prognosis in human breast cancer. Int. J. Cancer. 1996;69:92–99. doi: 10.1002/(SICI)1097-0215(19960422)69:2<92::AID-IJC4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 51.McIntosh GG, et al. Determination of the prognostic value of cyclin D1 overexpression in breast cancer. Oncogene. 1995;11:885–891. [PubMed] [Google Scholar]
- 52.Leslie K, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006;66:2544–2552. doi: 10.1158/0008-5472.CAN-05-2203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.