

Abstract
As a critical factor in the induction of angiogenesis, vascular endothelial growth factor (VEGF) has become an attractive target for anti-angiogenesis treatment. However, the side effects associated with most anti-VEGF agents limit their chronic use. Identification of naturally occurring VEGF inhibitors derived from diet is a potential alternative approach, with the advantage of known safety. To isolate natural inhibitors of VEGF, we established an in vitro tyrosine kinase assay to screen for diet-based agents that suppress VEGFR2 kinase activity. We found that a water-based extract from cinnamon (cinnamon extract, CE), one of the oldest and most popular spices, was a potent inhibitor of VEGFR2 kinase activity, directly inhibiting kinase activity of purified VEGFR2 as well as mitogen-activated protein kinase- and Stat3-mediated signaling pathway in endothelial cells. As a result, CE inhibited VEGF-induced endothelial cell proliferation, migration and tube formation in vitro, sprout formation from aortic ring ex vivo and tumor-induced blood vessel formation in vivo. Depletion of polyphenol from CE with polyvinylpyrrolidone abolished its anti-angiogenesis activity. While cinnamaldehyde, a component responsible for CE aroma, had little effect on VEGFR2 kinase activity, high-performance liquid chromatography-purified components of CE, procyanidin type A trimer (molecular weight, 864) and a tetramer (molecular weight, 1152) were found to inhibit kinase activity of purified VEGFR2 and VEGFR2 signaling, implicating procyanidin oligomers as active components in CE that inhibit angiogenesis. Our data revealed a novel activity in cinnamon and identified a natural VEGF inhibitor that could potentially be useful in cancer prevention and/or treatment.
Introduction
Angiogenesis, the formation of new vessels from preexisting vasculature, is an important mechanism used by tumors to promote growth and metastasis (1) and is tightly regulated by an intricate balance between stimulators and inhibitors (2). Vascular endothelial growth factor (VEGF) is one of the most critical and specific angiogenesis factors regulating normal physiological and tumor angiogenesis (3). It induces angiogenesis via binding to its two receptor tyrosine kinases expressed on endothelial cells, namely, vascular endothelial growth factor receptor 1 (VEGFR1) (Flt-1), required mainly for mitogenic and chemotactic responses, and VEGFR2 (KDR/Flk-1), which contributes to endothelial cell morphogenesis (3). By binding to and activating VEGFR, VEGF initiates a signal transduction cascade that affects numerous processes required for the formation of blood capillaries (4), including endothelial cell proliferation, migration, survival, tube formation, incorporation of endothelial cell precursors, recruitment of pericyte and smooth muscle cells and extracellular matrix remodeling.
VEGF is viewed as an attractive therapeutic target for the development of novel anticancer agents (5), and a variety of approaches to inhibit VEGF activity are currently being assessed in preclinical and clinical trials. These include monoclonal antibodies targeting VEGF ligands or VEGFRs (6), soluble receptors that sequester ligands (7) and small molecule inhibitors that inhibit kinase activity (8). Three drugs developed for their anti-angiogenic actions, bevacizumab (Avastin®), sunitinib malate (Sutent®, SU11248) and sorafenib (Nexavar®, BAY 43-9006), have been approved by the United States Food and Drug Administration for treatment of patients with specific types of cancer—all three inhibit VEGF signaling by blocking VEGF ligand or VEGFR (9).
However, serious side effects, such as hypertension, bleeding and gastrointestinal perforation, have been associated with currently available anti-VEGF agents, limiting their chronic use (9). There has, consequently, been a renewed interest in identifying natural food sources (diet-based approach) potentially rich in anti-VEGF agents, given the advantage of proven safety for human use (10–13). Additionally, consumption of a plant-based diet has been implicated in the prevention of cancer development and progression (14,15).
It has been well-established that polyphenols, especially flavonoids, are beneficial active components found in natural food products (14,15). They are abundant in a variety of foods including tea, coffee, fruits, vegetables, beans (soy), grains, seeds and spices. Recently, polyphenols extracted from various plants, including soy, berry, pomegranate, grape seed extract and green tea, have been found to be potent inhibitors of angiogenesis (16–26). However, the inhibitory activity of dietary components on VEGFR has not been fully elucidated (27–30).
To identify natural inhibitors of VEGF signaling, we have established an in vitro tyrosine kinase assay to screen for diet-based agents that suppress VEGFR2 kinase activity. We found that a water-based extract from cinnamon was a potent natural inhibitor of VEGFR2 kinase activity.
Materials and methods
Cinnamon extract
Ground cinnamon (Cinnamomum zeylanicum) powder, obtained from Frontier Natural Products Co-op in Norway, IA, was dissolved in water (70°C, 1 h). The solution was centrifuged (13 000 r.p.m., 10 min) to remove insoluble ingredients and the supernatant was then passed through a 0.22 mm filter and used for this study. A water-based cinnamon (C.zeylanicum) prepared by Dr John Lew's group (University of California, Santa Barbara), was also used to confirm some of the results obtained (Figures 1A and B and 6A and B) (31).
Fig. 1.

CE inhibits VEGFR2 kinase activity. (A) VEGFR2 was incubated with various concentrations of CE and substrate phosphorylation was monitored by enzyme-linked immunosorbent assay. Data are represented as percentage of control (not treated with CE) and are mean ± SD from four experiments. (B) Lineweaver–Burk plot and (C) Dixon plot of the inhibition of VEGFR2 by CE. Increased concentrations of adenosine triphosphate (ATP) were incubated with VEGFR2 and various concentration of CE. (D and E) CE inhibits VEGFR2 signaling. Quiescent HUVECs were incubated in the presence or absence of CE followed by stimulation with VEGF for another 5 min (for VEGFR2) or 2 h (for MAPK, Stat3, Jak2 and Src). Phosphorylation of VEGFR2, MAPK, Stat3, Jak2 and Src was assessed by western blot. β-Actin/β-tubulin level was used as a loading control. Results are representative of two to four experiments.
Fig. 6.

Oligomeric procynanidins inhibit VEGFR2 kinase activity. (A–C) PVPP treatment depletes anti-angiogenesis activity in CE. PVPP depletion of procynanidins from CE (see Materials and Methods) resulted in (A) inhibited kinase activity of purified VEGFR2, and data are mean ± SD (n = 3), (B) phosphorylation of VEGFR2 in HUVEC and (C) sprout formation from aortic ring. Scale bar, 100 μm. Experiments were repeated at least twice. (D) Oligomeric procyanidins suppressed kinase activity of purified VEGFR2. Cinnamaldehyde, trimer or tetramer of procyanidins, was incubated with VEGFR2. Data represent the percentage of kinase control (without CE treatment) and are mean ± SD (n = 3). (E) Oligomeric procyanidins inhibited VEGFR2 signaling. HUVECs were incubated in the presence or absence of trimer and tetramer of procyanidins, followed by VEGF stimulation. Phosphorylation of VEGFR-2 or MAPK was assessed by western blot using anti-phospho-VEGFR2 antibody, anti-phospho-MAPK antibody, as well as anti-total VEGFR2 and anti-total MAPK antibody. Experiments were repeated twice.
In vitro kinase assay
In vitro VEGFR2 tyrosine kinase activity was assayed using an enzyme-linked immunosorbent assay kit (Sigma, St Louis, MO) as described previously (30). Briefly, cinnamon extract (CE) was incubated with VEGFR2 (Upstate) in kinase reaction buffer in 96-well plates coated with a poly-Glu-Tyr substrate. Substrate phosphorylation was monitored using a phosphotyrosine-specific monoclonal antibody-conjugated to horseradish peroxidase. Results were expressed as percent of control (not treated with CE); IC50 values were defined as the drug concentration that resulted in 50% inhibition of enzyme activity.
Immunoblot
Human umbilical vascular endothelial cells (HUVECs; Lonza, Basel, Switzerland) were cultured (24 h) in endothelial basal medium (EBM)-2 containing 2% fetal calf serum (FCS), incubated (30 min) with various concentrations of CE and then stimulated with VEGF (100 ng/ml). Total cell extracts were prepared in Laemmli sample buffer. Proteins were resolved by electrophoresis on sodium dodecyl sulfate gels then transferred to polyvinylidene difluoride membranes. The membrane was incubated with primary antibodies anti-VEGFR2, anti-phospho-VEGFR2, anti-p44/42 MAPK, anti-phospho-p44/42 mitogen-activated protein kinase (MAPK), anti-Stat3, anti-phospho-Stat3, anti-Jak2, anti-phospho-Jak2, anti-Src and anti-phospho-Src (Cell Signaling, Boston, MA) followed by horseradish peroxidase-conjugated secondary antibody and chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Cell proliferation
HUVEC and bovine capillary endothelial cells (a generous gift from Catherine Butterfield and Dr Judah Folkman) were used for endothelial cell proliferation assay, as described previously (30). Endothelial cells were plated onto gelatinized 24-well culture plate in 0.5 ml EBM-2 (from Lonza for HUVEC) containing 10% FCS or Dulbecco's modified Eagle's medium (from Cellgro, Manassas,VA for bovine capillary endothelial cells) containing 10% bovine calf serum, incubated (24 h) and then treated with various concentrations of CE in the absence or presence of basic fibroblast growth factor (1 ng/ml) and VEGF (100 ng/ml; PeproTech, Rockyhill, NJ) or cocktail containing various growth factors (SingleQuo® kit; Lonza). After 48 h incubation, cells were trypsinized and counted with a Coulter counter.
For non-endothelial cell proliferation, cells were plated on to 24-well culture plate in 0.5 ml RPMI1640 culture medium (Cellgro; for MDA-MB-231 human breast cancer cells), McCoy 5A culture medium (Irvine Scientific, Santa Ana, CA; for human colon cancer cells) and MEM culture medium (Cellgro; for MCF-10A) containing 10% FCS, incubated (24 h) and then treated with various concentrations of CE. Cell numbers were determined by using a Coulter counter after 48 h incubation.
BrdU incorporation
DNA synthesis was measured by bromodeoxyuridine (BrdU) cell proliferation enzyme-linked immunosorbent assay (Roche, Indianapolis, IN). Briefly, 3000 HUVECs per well in 0.1 ml EBM-2 (from Lonza) containing 10% FCS were plated in 96-well tissue culture grade flat bottom plates, incubated for 24 hr and then treated (30 min) with various concentrations of CE in EBM-2 containing 2% FCS, followed by stimulation with VEGF (100 ng/ml). After 24 h, cells were incubated with BrdU labeling solution (37°C, 4 h), fixed and incubated with anti-BrdU antibody. Color intensity of substrate reaction was measured by an enzyme-linked immunosorbent assay reader at 405 nM.
Annexin V staining
Apoptosis was measured by annexin V apoptosis assay kit (Invitrogen). Briefly, HUVECs were cultured in endothelial cell growth medium-2 (Lonza) containing 10% FCS. At ∼40% confluency, cells were treated with varying concentrations (0, 16.25, 32.5 and 65 mg/ml) of CE. After 24 h, both floating and attached cells were collected and stained with annexin V-conjugated with Alexa Fluor 488 dye and propidium iodide. The signals were then detected by fluorescence-activated cell sorting analysis.
Cell migration
Endothelial cell migration was assessed using a modified Boyden chamber assay, as described previously (30). Transwells (8 mm pore; Corning, Lowell, MA) pre-coated with 200 mg/ml Matrigel (BD Bioscience, Franklin Lake, NJ) were used for the assay. HUVECs (1.5 × 105) were plated in EBM-2 containing 0.05% FCS in the upper chamber of the transwell in the presence of various concentration of CE for 30 min at 37°C. EBM-2 containing 0.05% FCS and VEGF (50 ng/ml) was then added to the lower chamber. After 5 h incubation, cells were stained and non-migrated cells (on the top side of membrane) were removed by cotton swap. The number of migrated cells was quantified by counting the cells at ×40 objectives.
Tube formation
HUVECs (50 000 cells per well) were harvested and seeded in EBM-2 (2% fetal bovine serum) containing VEGF (100 ng/ml) onto 24-well culture plates coated with 250 μl Matrigel (BD Bioscience). Various concentrations of CE were added at plating then incubated overnight (16 h). Cells were photographed using phase-contrast microscopy.
Chick aortic ring assay
The aortic arch was dissected from day 12 to 14 chick embryos, cut into rings and embedded into Matrigel in four-well plates (NUNC, Rochester, NY), as described previously (30). After incubation (37°C, 10 min), aortic rings were fed with MCDB-131 serum-free medium (GIBCO; Invitrogen) containing various concentrations of CE with or without VEGF (100 ng/ml) or endothelial cell growth supplement (ECGS; Sigma) (50 mg/ml). Growing sprouts were photographed with an Olympus inverted IX81 at ×40 magnification.
Matrigel plug assay
MDA-MB-231 cells (5 × 106 cells) were mixed with phenol red-free Matrigel and injected into both flanks of severely combined immunodeficient mice (SCID). For cinnamon-treated group, Matrigel was mixed with cells in the presence of CE (15 or 75 μg/ml). Matrigel mixed with medium alone (500 ml) was used as a negative control; 10 days after implantation, Matrigel plugs were removed and the surrounding tissues trimmed. Hemoglobin content of the gel plug was measured using a Drabkin's reagent kit (Sigma) according to the manufacturer's instructions. The concentration of hemoglobin was calculated based on a set of hemoglobin standards. Blood vessels in the Matrigel were visualized with antibody against CD31, as described previously (30).
Polyvinylpyrrolidone depletion
CE was incubated (room temperature, 30 min) with polyvinylpyrrolidone (PVPP) to deplete polyphenols. PVPP was then removed by centrifugation (13 000 r.p.m., 10 min). The absorbance of the supernatant was measured (280 nm) to ensure complete removal of polyphenol.
High-performance liquid chromatography
Trimeric and tetrameric procyanidins were isolated from CE using high-performance liquid chromatography as described previously (32).
Statistical analysis
Data were expressed as mean ± SD. Analysis of variance test followed by Dunnett's post-hoc test was used to compare the means for the multiple groups. Each assay was repeated two to four times. *P < 0.05 was considered statistically significant.
Results
Effect of CE on VEGFR2 kinase activity
In vitro tyrosine kinase assay was used to screen plant-based diets for new inhibitors of angiogenesis. CE, which is enriched in polyphenols, was found to effectively inhibit kinase activity of purified VEGFR2 with an IC50 of ∼30 ng/ml (Figure 1A). SU5416, a known inhibitor of VEGFR2, was used as a positive control and showed inhibition of kinase activity with an IC50 of 1 μM (data not shown), in agreement with previous studies (33).
To understand the biochemical mechanism of CE inhibition of VEGFR2, we assayed VEGFR2 kinase activity at various concentrations of adenosine triphosphate in the presence of increasing concentrations of CE. A Lineweaver–Burk plot analysis revealed that increasing concentrations of CE affected apparent 1/Vmax (y-axis intersection), indicating CE behaved mainly as a non-competitive inhibitor for adenosine triphosphate binding (Figure 1B). The Ki for the inhibition of CE was ∼7.5 ng/ml, as calculated by Dixon plot (Figure 1C).
Given the observed inhibition of VEGFR2 kinase activity, we further examined the effects of CE on phosphorylation of VEGFR2 to determine whether it inhibited VEGFR2-mediated signaling pathways in endothelial cells. We found that VEGFR2 was phosphorylated by the addition of exogenous VEGF to HUVECs (Figure 1D). Pretreatment of cells with CE significantly blocked VEGF-induced phosphorylation of VEGFR2, without affecting overall VEGFR2 expression levels.
In order to identify the downstream signaling pathway targeted by CE treatment, we examined the expression and phosphorylation of MAPK, one of the key signaling pathway components supporting endothelial cell proliferation (34). While treatment of HUVEC with CE inhibited VEGF-dependent phosphorylation of MAPK in a dose-dependent manner, total MAPK levels were unaffected (Figure 1D). Another critical signaling pathway activated by VEGF in endothelial cells is Stat3 (35–37). We found that the phosphorylation of Stat3, as well as Jak2 and Src kinase, two main kinases that phosphorylate Stat3, was also significantly decreased in the presence of CE (Figure 1E). Higher doses of CE appeared to be required to achieve effective inhibitory effects on VEGFR2 expressed by endothelial cells compared with the dose used to inhibit kinase activity of purified VEGFR2 in vitro. This may have been due to the possibility that the active components in CE were not stable in culture or were not able to efficiently penetrate cells to exert an effect. Taken together, our data implicated CE as a potent angiogenesis inhibitor by inhibiting VEGFR2-mediated signaling pathway.
Effect of CE on endothelial cell proliferation, migration and tube formation
To characterize the anti-angiogenesis activity of CE, we first determined whether CE inhibited growth factor-induced endothelial cell proliferation. HUVEC (Figure 2A) and bovine capillary endothelial cells (data not shown) were incubated with CE then stimulated with VEGF (100 ng/ml), basic fibroblast growth factor (1 ng/ml) or growth medium containing various growth factors; cell count was assessed 48 h later.
Fig. 2.

CE inhibits endothelial cell proliferation. (A) HUVECs were treated with various concentrations of CE, stimulated with VEGF then cell number was assessed 48 h later. Data are mean ± SD (n = 3). *P < 0.05 versus control with VEGF and #P < 0.05 versus control without VEGF. (B) MDA-MB-231, HT29 and MCF-10A cells were treated with various concentrations of CE and cells counted 48 h later. Data are represented as percentage of vehicle-treated control. (C) HUVEC were treated with various concentrations of CE and then labeled with BrdU. DNA synthesis was measured by BrdU cell proliferation enzyme-linked immunosorbent assay. Data are represented as percentage of vehicle control and are mean ± SD (n = 3). *P < 0.05 versus control. (D) CE-induced endothelial cell apoptosis. HUVECs were treated with various concentration of CE then labeled with annexin V. Data represent percentage of apoptotic cells and are mean ± SD (n = 3). *P < 0.05 versus control.
While HUVEC proliferation was significantly increased in response to VEGF treatment in the absence of CE, it was markedly suppressed in the presence of CE (Figure 2A). In contrast to endothelial cells, much higher concentrations of CE were needed to inhibit the proliferation of non-endothelial cells, such as HT29 human colon cancer cells, MDA-MB-231 human breast cancer cells and MCF-10A mammary epithelial cells (Figure 2B), suggesting that CE had greater specificity as an inhibitor for VEGF-induced endothelial cell proliferation.
To clarify whether the observed reduction in cell number in HUVEC resulted from reduced cell growth and/or increased cell death, we studied the effects of CE on DNA synthesis and apoptosis by measuring BrdU incorporation and annexin V staining, respectively. Our data suggested that CE effectively inhibited DNA synthesis and induced apoptosis in these cells (Figure 2C and D).
We next studied the effects of CE on VEGF-induced cell migration using a modified Boyden chamber assay (30,38). In the absence of CE, a large number of HUVECs migrated to the lower side of the filter in the transwell chamber following stimulation with VEGF (50 ng/ml) (Figure 3A). However, in the presence of CE, the number of migratory cells was significantly reduced in response to VEGF, in a concentration-dependent manner.
Fig. 3.
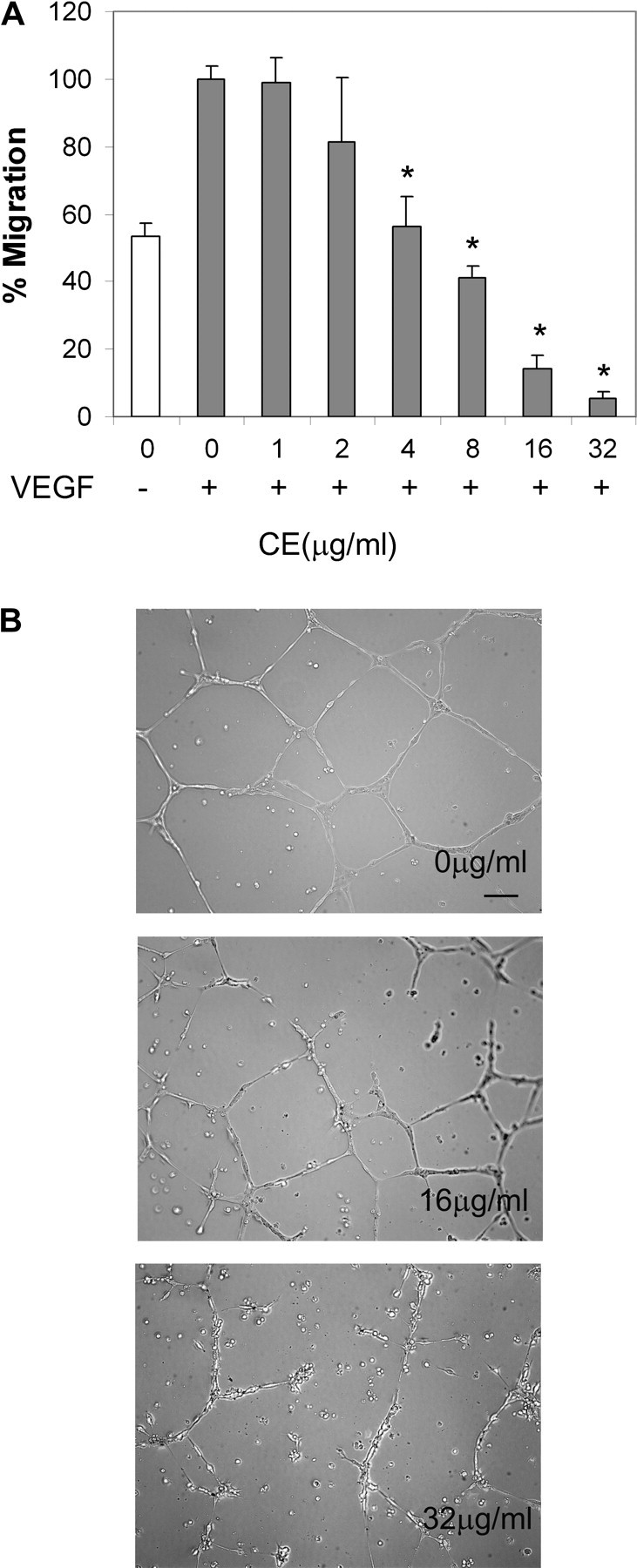
CE inhibits endothelial cell migration and tube formation. (A) CE inhibits endothelial cell migration. Migration assay of HUVEC treated with various concentrations of CE then treated with VEGF; percentage of migrating cells was examined by microscopy. Data represent percentage of control (without CE + VEGF) and are mean ± SD (n = 3). *P < 0.05 versus control (without CE treatment) with VEGF present. (B) CE inhibits endothelial tube formation. HUVECs were plated on Matrigel in the presence of VEGF with or without CE and photographed under phase-contrast after 16 h. Results are representative of three preparations. Scale bar, 100 μm.
To further characterize its anti-angiogenesis activity, we examined the effects of CE on VEGF-induced tube formation by HUVEC on Matrigel, a well-established angiogenesis assay. HUVECs formed tube-like networks within 8 h, which might, in part, reflect the process of angiogenesis. At a concentration of 32 μg/ml, CE effectively abrogated endothelial tube formation, reducing the tube-like structure both in width and in length. Endothelial cells rounded up and rendered network structures incomplete and broken in the presence of CE (Figure 3B).
Effect of CE on vessel sprout formation from aorta
The effect of CE on microvessel sprout formation (39) was determined by aortic ring sprout formation assay, a widely used ex vivo anti-angiogenesis assay that mimics several stages of angiogenesis, including endothelial cell proliferation, migration and tube formation. Chicken aortic rings were embedded in Matrigel and fed with medium containing different concentrations of CE. The rings were then stimulated with 100 ng/ml VEGF or 50 μg/ml ECGS and sprout formation was examined by microscopy.
In the absence of CE, sprouts emerged from the aortic ring and grew outward after 3 days in culture with either VEGF or ECGS. Treatment with CE resulted in a dramatic dose-dependent decrease in sprout length and density induced by VEGF (data not shown) and ECGS (Figure 4A). To test whether the inhibitory effect of CE was reversible, aortic rings were first incubated with ECGS and CE for 48 h; CE was then removed from the culture and rings were further incubated with ECGS for an additional 48 h. Removal of CE resulted in renewed vessel sprout formation (Figure 4B), suggesting that inhibition of vessel sprout formation by CE was reversible and unlikely due to cytotoxicity.
Fig. 4.
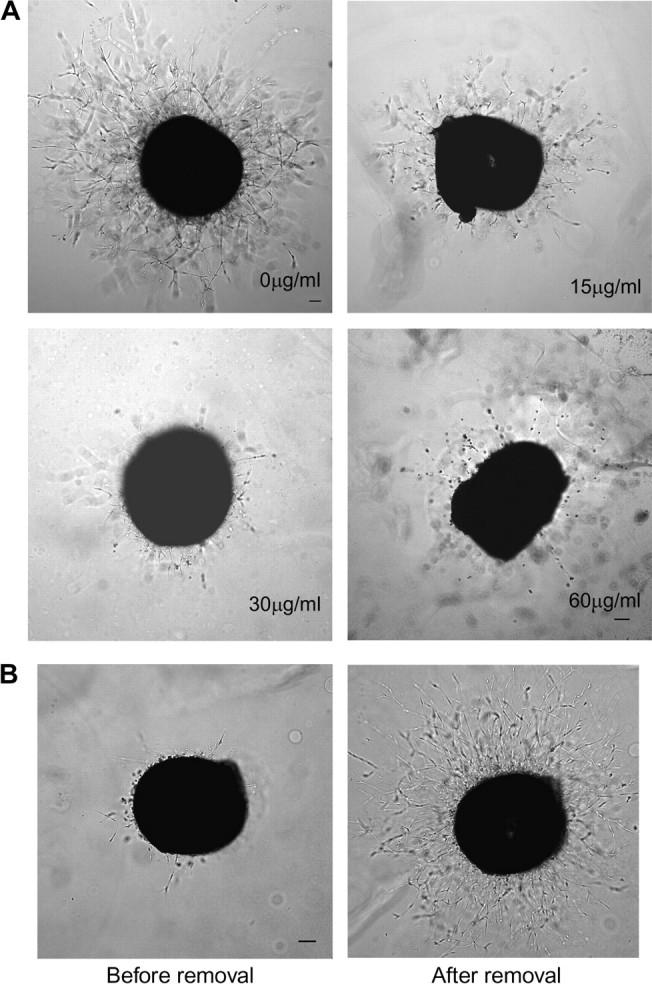
CE inhibits sprout formation from chick aorta. (A) Chick aortic rings were placed in Matrigel and treated with various concentrations of CE in the presence or absence of ECGS. The effect of CE on formation of vessel sprout from various aorta samples was examined on day 3. (B) Inhibitory effect of CE on vessel sprout formation is reversible. Aortic rings were first incubated with ECGS and CE (30 μg/ml) for 48 hr (left panel) and continued to culture with ECGS for another 48 hr after cinnamon was removed (right panel). Scale bar, 100 μm. The images shown here are representative of four experiments.
Effect of CE on blood vessel formation in mice
We next tested whether CE was able to inhibit tumor-induced angiogenesis in mice using a Matrigel plug assay. MDA-MB-231 human breast cancer cells were mixed with Matrigel and injected into the flanks of mice; gel plugs were harvested 10 days after implantation and angiogenesis was assayed by hemoglobin content of the plugs. While MDA-MB-231 cells greatly induced blood vessel formation in the plug (Figure 5A), blood vessel formation induced by MDA-MB-231 was significantly reduced in the presence of CE. To further evaluate the effects of CE, blood vessels in the plug were visualized with antibody against CD31 (Figure 5B). Compared with controls, CE treatment led to a significant reduction in blood vessel density indicated by reduced expression of CD31. These data indicated that CE is an angiogenesis inhibitor in vivo.
Fig. 5.

CE inhibits tumor-induced blood vessel formation in mice. (A) MDA-MB-231 cells were mixed with Matrigel in the presence and absence of CE and injected into both flank sites of male severely combined immunodeficient mice (SCID). Hemoglobin levels in the Matrigel plug were quantified after 10 days. Data are mean ± SD (n = 10). *P < 0.05 versus control treatment. (B) Blood vessels in the Matrigel plugs, either without CE (left panel) or with CE (75 μg/ml, right panel), were stained with CD31 antibody. The images shown here are representative of five samples for each condition. Scale bar, 20 μm.
Effect of oligomeric procyanidins derived from CE on angiogenesis
CE contains a large amount of polyphenols, including procyanidins and cinnamaldehyde, a phenylpropanoid (32,40). To test whether polyphenols are responsible for CE anti-angiogenesis activity, we used cross-linked PVPP to deplete polyphenol from CE. PVPP forms hydrogen bonds with phenolic compounds, yielding a PVPP-phenolic precipitate, which can be removed by centrifugation (31,41). Following PVPP depletion, the supernatant was subjected to a VEGFR2 kinase assay. PVPP-treated CE lost the inhibitory effect on in vitro kinase activity of VEGFR2 (Figure 6A), phosphorylation of VEGFR2 (Figure 6B) and sprout formation from aortic ring (Figure 6C), implicating the water-soluble polyphenol in CE in the inhibition of VEGFR2 kinase activity and angiogenesis.
Further testing of cinnamaldehyde, a component characteristic of the aroma of CE, found it had no inhibitory effects on VEGFR2 kinase activity (Figure 6D). The activity of high-performance liquid chromatography fractions of CE containing type A trimeric (molecular weight, 864) or tetrameric (molecular weight, 1152) procyanidins (32) was also investigated. Oligomeric procyanidin is one of the major components in CE. Our data demonstrated that these components inhibited kinase activity of purified VEGFR2 in a dose-dependent manner (Figure 6D) and were also able to suppress the phosphorylation of VEGFR2 and MAPK induced by VEGF in HUVEC. Taken together, these results identified oligomeric procyanidins as active components in CE that inhibited VEGFR2 kinase activity and angiogenesis.
Discussion
Human tumors can remain dormant for years due to the balance between cell proliferation and apoptosis. Blockade of angiogenesis is therefore an important approach for cancer treatment and prevention, given that systemic concentrations of angiogenesis inhibitors, exceeding those of stimulators, could potentially prevent tumors from growing and spreading to other organs. While VEGF, a critical factor in angiogenesis induction, has emerged as an attractive target for anti-angiogenesis treatment, the chronic therapeutic use of anti-VEGF agents is limited due to side effects. The present study investigates naturally occurring VEGF inhibitors in cinnamon as a diet-derived source of anti-VEGF agents. Diet and nutrition have been shown to play an important role in the development of cancer (14,15). Additionally, certain plant-derived dietary groups may contain phytochemicals that exert antitumor and anti-angiogenesis activity, thereby offering anticancer protection (11,13).
We found that CE and specific characterized CE components, type A procyanidin trimer and tetrameric procyanidins, effectively inhibited VEGFR2 kinase activity as well as VEGF signaling in endothelial cells. CE also inhibited various aspects of angiogenesis, including endothelial cell proliferation, migration and tube formation in vitro, sprout formation ex vivo, as well as tumor-induced blood vessel formation in mice. Our data are in agreement with other studies in which several natural products were shown to inhibit VEGFR2 signaling, including catechins from green tea extract, delphinidin, ellagic acid, as well as grape seed extract (27–30).
Cinnamon, the dry bark and twig of Cinnamomum spp., is one of the most popular and oldest spices. The bark and leaves of CE are often added to food preparations to improve taste and aroma. In addition, this herb has been found to possess potent antioxidant, antimicrobial and antipyretic properties and has been used in traditional Chinese medicine (42–44). Several recent studies have found that CE also contains anticancer activity. CE was shown to inhibit hematologic cancer cell proliferation in vitro, melanoma tumor growth in mice and VEGF expression in melanoma (45,46). In recent years, much attention has been paid to the influence of cinnamon on insulin action, which may provide benefits for diabetic patients (32,42,43,47,48). More recently, a water-based CE was found to inhibit Tau aggregation and filament formation associated with Alzheimer's disease (31).
Cinnamon contains the highest levels of procyanidins among all common foods (40). The majority of procyanidins in cinnamon is in the A-type oligomeric form. Procyanidins are of great interest in nutrition and medicine because of their potent antioxidant capacity and possible protective effects on human health (49). Although procyanidins have been suggested to account for a significant fraction of polyphenols ingested, as they are found in a variety of food sources (49), little is known about the mechanism underlying the health benefits of procyanidins.
Here we provide evidence that the high-performance liquid chromatography fraction of cinnamon containing oligomeric procyanidins inhibited VEGFR2 kinase activity, whereas no effects were observed by other constituents of CE, such as cinnamaldehyde and methylhydroxychalcone (data not shown). While a previous study suggested that cinnamaldehyde inhibited angiogenesis in a chick chorioallantoic membrane assay (50), the mechanism involved in the inhibition remained to be defined. Further research is needed to determine whether oligomeric procyanidins are the major active components responsible for CE anti-angiogenic activity as well as to identify other potential components involved.
Taken together, our data revealed a novel biological function of CE and suggested a possible molecular mechanism for its action. Further study is required to address whether CE inhibits tumor angiogenesis and tumor growth in mice through suppressing VEGFR2 signaling. As a natural inhibitor of VEGFR2, CE has the potential to be routine diet-based strategy for cancer prevention or treatment.
Funding
Stop Cancer Foundation; Concern Foundation; Markel Friedman Fund.
Acknowledgments
We thank Catherine Butterfield and Dr Judah Folkman for BCE cells, Drs John Lew, Dylan Peterson and Donald Graves for their CE and their suggestions and discussions. We thank Drs Susan Kane, Edward Newman and Tim Synold for their valuable discussions, Dr Silvia da Costa for critical reading of this manuscript and members of the Flow Cytometry Core, the Pathology Core, and the Animal Facility at the City of Hope Medical Center for their technical assistance.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- BrdU
bromodeoxyuridine
- EBM
endothelial basal medium
- ECGS
endothelial cell growth supplement
- FCS
fetal calf serum
- HUVEC
human umbilical vascular endothelial cell
- PVPP
polyvinylpyrrolidone
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
References
- 1.Kerbel R, et al. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, et al. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara N. VEGF and the quest for tumor angiogenesis factors. Nat. Rev. Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 4.Olsson A-K, et al. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell. Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, et al. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist. 2004;9:2–10. doi: 10.1634/theoncologist.9-suppl_1-2. [DOI] [PubMed] [Google Scholar]
- 7.Holash J, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc. Natl Acad. Sci. USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noble MEM, et al. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 9.Kamba T, et al. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer. 2007;96:1788–1795. doi: 10.1038/sj.bjc.6603813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Y, et al. Antiangiogenic mechanisms of diet-derived polyphenols. J. Nutr. Biochem. 2002;13:380–390. doi: 10.1016/s0955-2863(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 11.Albini A, et al. Molecular pathways for cancer angioprevention. Clin. Cancer Res. 2007;13:4320–4325. doi: 10.1158/1078-0432.CCR-07-0069. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, et al. Apigenin inhibits tumor angiogenesis through decreasing HIF-1{alpha} and VEGF expression. Carcinogenesis. 2007;28:858–864. doi: 10.1093/carcin/bgl205. [DOI] [PubMed] [Google Scholar]
- 13.Bhat TA, et al. Tumor angiogenesis—a potential target in cancer chemoprevention. Food Chem. Toxicol. 2008;46:1334–1345. doi: 10.1016/j.fct.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 14.Howe GR, et al. Dietary factors and risk of breast cancer: combined analysis of 12 case-control studies. J. Natl Cancer Inst. 1990;82:561–569. doi: 10.1093/jnci/82.7.561. [DOI] [PubMed] [Google Scholar]
- 15.Rose DP, et al. International comparisons of mortality rates for cancer of the breast, ovary, prostate, and colon, and per capita food consumption. Cancer. 1986;58:2363–2371. doi: 10.1002/1097-0142(19861201)58:11<2363::aid-cncr2820581102>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Cao Y, et al. Angiogenesis inhibited by drinking tea. Nature. 1999;398:381. doi: 10.1038/18793. [DOI] [PubMed] [Google Scholar]
- 17.Brakenhielm E, et al. Suppression of angiogenesis, tumor growth, and wound healing by resveratrol, a natural compound in red wine and grapes. FASEB J. 2001;15:1798–1800. doi: 10.1096/fj.01-0028fje. [DOI] [PubMed] [Google Scholar]
- 18.Agarwal C, et al. Anti-angiogenic efficacy of grape seed extract in endothelial cells. Oncol. Rep. 2004;11:681–685. [PubMed] [Google Scholar]
- 19.Singh RP, et al. Grape seed extract inhibits advanced human prostate tumor growth and angiogenesis and upregulates insulin-like growth factor binding protein-3. Int. J. Cancer. 2004;108:733–740. doi: 10.1002/ijc.11620. [DOI] [PubMed] [Google Scholar]
- 20.Chen T, et al. Black raspberries inhibit N-nitrosomethylbenzylamine (NMBA)-induced angiogenesis in rat esophagus parallel to the suppression of COX-2 and iNOS. Carcinogenesis. 2006;27:2301–2307. doi: 10.1093/carcin/bgl109. [DOI] [PubMed] [Google Scholar]
- 21.Singh AV, et al. Soy phytochemicals prevent orthotopic growth and metastasis of bladder cancer in mice by alterations of cancer cell proliferation and apoptosis and tumor angiogenesis. Cancer Res. 2006;66:1851–1858. doi: 10.1158/0008-5472.CAN-05-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, et al. Green tea extract and (-)-epigallocatechin-3-gallate inhibit hypoxia- and serum-induced HIF-1alpha protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Mol. Cancer Ther. 2006;5:1227–1238. doi: 10.1158/1535-7163.MCT-05-0490. [DOI] [PubMed] [Google Scholar]
- 23.Khan N, et al. Oral consumption of pomegranate fruit extract inhibits growth and progression of primary lung tumors in mice. Cancer Res. 2007;67:3475–3482. doi: 10.1158/0008-5472.CAN-06-3941. [DOI] [PubMed] [Google Scholar]
- 24.Ajaikumar BK, et al. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Lamy S, et al. Delphinidin, a dietary anthocyanidin, inhibits platelet-derived growth factor ligand/receptor (PDGF/PDGFR) signaling. Carcinogenesis. 2008;29:1033–1041. doi: 10.1093/carcin/bgn070. [DOI] [PubMed] [Google Scholar]
- 26.Sartippour MR, et al. Ellagitannin-rich pomegranate extract inhibits angiogenesis in prostate cancer in vitro and in vivo. Int. J. Oncol. 2008;32:475–480. [PubMed] [Google Scholar]
- 27.Lamy S, et al. Green tea catechins inhibit vascular endothelial growth factor receptor phosphorylation. Cancer Res. 2002;62:381–385. [PubMed] [Google Scholar]
- 28.Labrecque L, et al. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis. 2005;26:821–826. doi: 10.1093/carcin/bgi024. [DOI] [PubMed] [Google Scholar]
- 29.Lamy S, et al. Delphinidin, a dietary anthocyanidin, inhibits vascular endothelial growth factor receptor-2 phosphorylation. Carcinogenesis. 2006;27:989–996. doi: 10.1093/carcin/bgi279. [DOI] [PubMed] [Google Scholar]
- 30.Wen W, et al. Grape seed extract inhibits angiogenesis via suppression of the vascular endothelial growth factor receptor signaling pathway. Cancer Prev. Res. 2008;1:554–561. doi: 10.1158/1940-6207.CAPR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson DW, et al. Cinnamon extract inhibits tau aggregation associated with Alzheimer's disease in vitro. J. Alzheimers Dis. 2009;17:585–597. doi: 10.3233/JAD-2009-1083. [DOI] [PubMed] [Google Scholar]
- 32.Anderson RA, et al. Isolation and characterization of polyphenol type-A polymers from cinnamon with insulin-like biological activity. J. Agric. Food Chem. 2004;52:65–70. doi: 10.1021/jf034916b. [DOI] [PubMed] [Google Scholar]
- 33.Fong TA, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 34.Takahashi T, et al. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- 35.Yu H, et al. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 36.Bartoli M, et al. VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 2003;17:1562–1564. doi: 10.1096/fj.02-1084fje. [DOI] [PubMed] [Google Scholar]
- 37.Yahata Y, et al. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J. Biol. Chem. 2003;278:40026–40031. doi: 10.1074/jbc.M301866200. [DOI] [PubMed] [Google Scholar]
- 38.Miller AB. Diet and cancer. A review. Acta Oncol. 1990;29:87–95. doi: 10.3109/02841869009089996. [DOI] [PubMed] [Google Scholar]
- 39.Nicosia RF, et al. Growth of microvessels in serum-free matrix culture of rat aorta. A quantitative assay of angiogenesis in vitro. Lab Invest. 1990;63:115–122. [PubMed] [Google Scholar]
- 40.Gu L, et al. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J. Nutr. 2004;134:613–617. doi: 10.1093/jn/134.3.613. [DOI] [PubMed] [Google Scholar]
- 41.Loomis WD. Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Methods Enzymol. 1974;31:528–544. doi: 10.1016/0076-6879(74)31057-9. [DOI] [PubMed] [Google Scholar]
- 42.Jarvill-Taylor KJ, et al. A hydroxychalcone derived from cinnamon functions as a mimetic for insulin in 3T3-L1 adipocytes. J. Am. Coll. Nutr. 2001;20:327–336. doi: 10.1080/07315724.2001.10719053. [DOI] [PubMed] [Google Scholar]
- 43.Anderson RA. Chromium and polyphenols from cinnamon improve insulin sensitivity. Proc. Nutr. Soc. 2008;67:48–53. doi: 10.1017/S0029665108006010. [DOI] [PubMed] [Google Scholar]
- 44.Singh G, et al. A comparison of chemical, antioxidant and antimicrobial studies of cinnamon leaf and bark volatile oils, oleoresins and their constituents. Food Chem. Toxicol. 2007;45:1650–1661. doi: 10.1016/j.fct.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 45.Schoene NW, et al. Water-soluble polymeric polyphenols from cinnamon inhibit proliferation and alter cell cycle distribution patterns of hematologic tumor cell lines. Cancer Lett. 2005;230:134–140. doi: 10.1016/j.canlet.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 46.Kwon H-K, et al. Cinnamon extract suppresses tumor progression by modulating angiogenesis and the effector function of CD8+ T cells. Cancer Lett. 2009;278:174–182. doi: 10.1016/j.canlet.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 47.Imparl-Radosevich J, et al. Regulation of PTP-1 and insulin receptor kinase by fractions from cinnamon: implications for cinnamon regulation of insulin signalling. Horm. Res. 1998;50:177–182. doi: 10.1159/000023270. [DOI] [PubMed] [Google Scholar]
- 48.Cao H, et al. Cinnamon extract and polyphenols affect the expression of tristetraprolin, insulin receptor, and glucose transporter 4 in mouse 3T3-L1 adipocytes. Arch. Biochem. Biophys. 2007;459:214–222. doi: 10.1016/j.abb.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 49.Santos-Buelga C, et al. Proanthocyanidins and tannin-like compounds-nature, occurrence, dietary intake and effects on nutrition and health. J. Sci. Food Agric. 2000;80:1094–1117. [Google Scholar]
- 50.Kwon B-M, et al. Synthesis and biological activity of cinnamaldehydes as angiogenesis inhibitors. Bioorg. Med. Chem. Lett. 1997;7:2473–2476. [Google Scholar]