

Abstract
Cholesterol-engorged macrophage foam cells are a critical component of the atherosclerotic lesion. Reducing the sterol deposits in lesions reduces clinical events. Sterol accumulations within lysosomes have proven to be particularly hard to mobilize out of foam cells. Moreover, excess sterol accumulation in lysosomes has untoward effects, including a complete disruption of lysosome function. Recently, we demonstrated that treatment of sterol-engorged macrophages in culture with triglyceride-containing particles can reverse many of the effects of cholesterol on lysosomes and dramatically reduce the sterol burden in these cells. This article describes what is known about lysosomal sterol engorgement, discusses the possible mechanisms by which triglyceride could produce its effects, and evaluates the possible positive and negative effects of reducing the lysosomal cholesterol levels in foam cells.
Keywords: atherosclerosis, cholesterol, lysosomes, macrophage, triglyceride
Atherosclerosis is a progressive disease that eventually leads to the impairment vascular function in ways that can promote heart attacks and strokes, usually as a result of atheromatous plaque rupture. Plaque development is a multi-factoral process and understanding the various mechanisms that produce structural instability and rupture of the lesion are key areas of research towards more effective treatments. A hallmark feature of the atherosclerosis lesion, particularly those areas prone to rupture, is the presence of sterol-engorged macrophages [1]. Thus, understanding the factors that influence macrophage accumulation of cholesterol remains an important line of clinical study.
Importance of lysosomes in atherosclerotic lesion development
The macrophages in the artery wall arise from monocytes that leave the circulation, enter the artery wall and differentiate into macrophages. Simultaneously, lipids also enter the artery wall from the blood stream. Most, but certainly not all, of the lipids enter the artery as components of lipoproteins. LDLs, HDLs, VLDLs, and their metabolic remnants, have all been identified within atherosclerotic lesions. These particles are the source of most of the excess lipids that accumulate within macrophages [2]. The lipid content in macrophages can occupy a large proportion of the cell volume and give the cell a foamy appearance (Figure 1). For this reason, the cells are often referred to as foam cells.
Figure 1. Light micrograph of early atherosclerosis lesion in the aorta of a White Carneau pigeon.
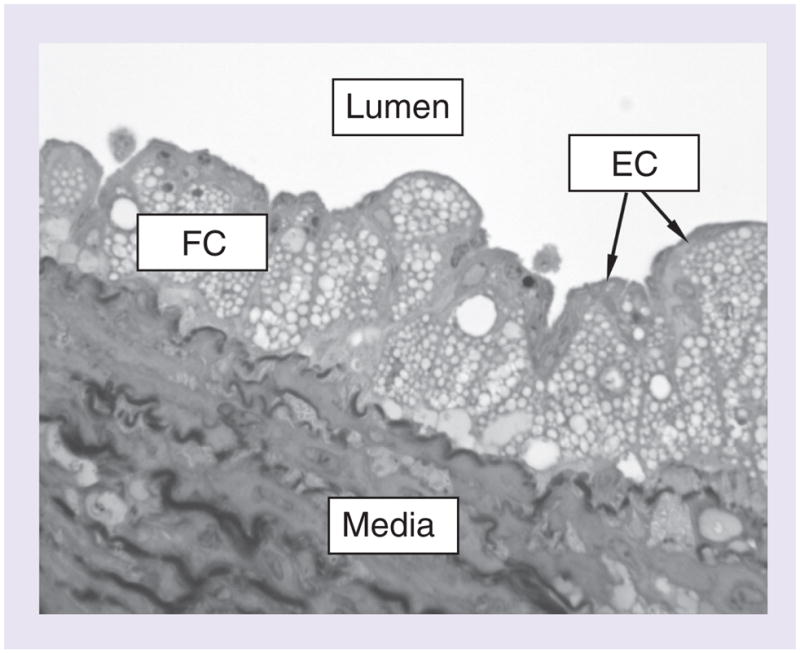
The atherosclerotic plaque fills the intima of the artery. The intima is the space between the thin layer of ECs facing the lumen of the artery and the underlying media layer. Normally, the intima is very thin. The presence of the atherosclerotic plaque has thickened the artery wall by expanding the intimal space. The plaque consists primarily of cholesterol-engorged macrophage FCs that are identifiable by the numerous round lipid droplets inside the cells. Cross-sections through approximately 14 such cells are visible in the image. The plaque sits on top of the arterial media which is composed of smooth muscle cells and elastic connective tissue.
EC: Endothelial cell; FC: Foam cell.
Although the lipid particles that enter the artery wall carry a variety of lipids, it is predominantly sterol that is accumulated in macrophages, with cholesteryl esters and native cholesterol being the most common. During the initiation phase of atherosclerosis, the sterol is found mainly within lipid droplets in the cell cytoplasm. However, as lesions progress into more clinically important stages, substantial amounts of sterol accumulate within the lysosomes of the foam cells [3]. Normal macrophages contain between 20 and 40 mg of cholesterol (mostly free cholesterol [FC]) per milligram of cell protein. Foam cells can have in excess of 300 mg of cholesterol per milligram of cell protein. The majority of this occurs as cholesteryl esters. In late-stage lesions, up to 80% of the excess sterol can be found within large, lipid-swollen lysosomes. This article summarizes what we know about the causes of this lysosomal accumulation, examines some schemes for reducing this and discusses whether such lysosomal accumulation is beneficial or detrimental to arterial health.
Normal cellular lipoprotein cholesterol metabolism
The sterol found in foam cells in atherosclerotic lesions is primarily derived from plasma LDL [2]. Much of our understanding of macrophage metabolism of sterol derived from lipoproteins has come from tissue culture experiments [4]. As the normal LDL receptor is highly regulated, uptake by this receptor does not produce massive sterol accumulation [5]. However, as professional phagocytic cells, macrophages have a number of alternate receptors that are not highly regulated. It is now generally believed that the uptake of the majority of the cholesterol found within foam cells occurs by these unregulated receptors [6,7]. As these receptors are not downregulated, they have the potential to mediate the accumulation of large amounts of cholesterol. The chemical or physical modifications associated with the induction of massive cholesterol accumulation in culture and animal models include acetylation, oxidation and aggregation [8–10]. Although acetylation is a purely artificial alteration, there is strong evidence that both oxidation and aggregation of LDL particles occurs in atherosclerotic lesions [11,12]. In the lesion, both oxidation and aggregation can, and probably do, occur by a variety of mechanisms. This has led to the hypothesis that a key factor in the uptake of lipoproteins by foam cells is the retention of native LDL particles, within the lesion, long enough to become modified [13]. Given the prevalence of modifying factors in the extracellular regions, it is highly probable that in late-stage lesions very little LDL remains in its native state. Moreover, cholesterol esters associated with phospholipids, such as those found in the extracellular spaces of lesions, also produce cholesterol accumulation in macrophages [14–16]. In addition to retained lipids, in late-stage lesions, the death of foam cells also contributes to the extracellular lipid pool [17]. Importantly, intracellular metabolism of this lipid could modify the lipid to make it more atherogenic after its release during the cells death [18].
Thus, there are numerous modified particle types that could promote macrophage cholesterol accumulation. However, no matter the uptake mechanism, the cholesteryl esters (CEs) contained in the modified particle are eventually delivered to a hydrolytic compartment for degradation. This is critical because the cell does not have the capability of ridding itself of CEs. For removal, the CEs must first be hydrolyzed to an unesterified cholesterol. Although a few novel compartments have been identified for degradation of some specialized particles [19,20], most sterol-containing particles are thought to be internalized via the canonical, endocytic pathway, involving uptake into an early endosome and delivery to a late endosome–lysosome compartment (Figure 2). The late endosome–lysosome compartment is a specialized digestive organelle. Within this compartment, the action of lysosomal acid lipase (LAL) converts CE to FC. The liberated FC then partitions into the lysosomal membrane. Removal of the majority of FC from lysosomes occurs via vesicular transport with most of the released cholesterol going to the plasma membrane [21]. Cholesterol is an important component of the plasma membrane and may become highly concentrated here. However, there is a finite solubility of cholesterol in the plasma membrane. When the plasma membrane content exceeds this limit, excess sterol is shunted to other locations, such as the endoplasmic reticulum (ER). In the ER, the cholesterol can be esterified to a fatty acid (FA) by the action of acyl-CoA:cholesterol acyltransferase (ACAT). The CEs thus formed accumulate within the cytoplasm as neutral lipid-containing droplets surrounded by other lipids and proteins. Therefore, the extralysosomal component of foam cells is an important regulator of the extralysosomal FC content. The action of another enzyme, neutral cholesterol–ester hydrolase, can convert the stored CEs back to FC for use by the cell [22]. Alternatively, if there are sterol acceptor particles, such as HDLs, in the fluid surrounding the cell, the plasma membrane FC can be extracted by these acceptor particles in a process known as efflux [23,24]. This is an alternative way of reducing the plasma membrane FC concentration.
Figure 2. Normal cellular lipoprotein-derived cholesterol metabolism.

CE-containing lipoproteins, such as modified forms of LDL, are taken up by receptor-mediated endocytosis and delivered to the late endosome–lysosome compartment for digestion. In the lysososme, LAL hydrolyzes the CE to FC and FA. Most of the FC partitions into, and is removed as vesicles form, from the lysosome membrane. The bulk of the lysosomal FC is delivered to the plasma membrane where it plays a critical role in modulating plasma membrane function. FC can be removed from the plasma membrane by transfer to acceptor particles in a process known as efflux. Excess FC that cannot be effluxed can be stored as CE droplets in the cytoplasm. The re-esterification of FC to CE occurs in the ER by the ACAT enzyme. CE within these cytoplasmic lipid droplets can be hydrolyzed back to FC by the enzyme NCEH.
ACAT: Acyl-CoA:cholesterol acyltransferase; CE: Cholesteryl ester; ER: Endoplasmic reticulum; FA: Fatty acid; FC: Free cholesterol; LAL: Lysosomal acid lipase; NCEH: Neutral cholesterol ester hydrolase.
Implication of lysosomes as a factor in atherosclerosis
The presence of large, lipid-swollen lysosomes in late lesions indicates an inhibition of normal lysosomal hydrolysis and clearance of sterol. Beginning in the mid-1960s, evidence began accumulating suggesting that specific aspects of atherosclerosis have features consistent with an acquired lysosomal storage disease [25]. Lysosomal storage disorders are characterized by excess accumulation of material in lysosomes due to innate or acquired defects in the process of lysosomal hydrolysis. In the best characterized lysosomal disorders the link between defect and dysfunction is clear and is related to defective lysosomal enzymes [26]. However, in others, the accumulation appears to be the result of secondary effects which create imbalances in the cell homeostatic mechanisms or metabolism [27]. As detailed below, the lysosomal sterol accumulation accompanying atherosclerosis appears to represent this latter type of defect.
As previously described, macrophages have two key sites for regulating intracellular sterol stores; the lysosome and extralysosomal lipid droplets. Despite the consistent evidence of sterol accumulation in lysosomes, the relative importance of lysosomal cholesterol accumulation in the disease process has not been fully established. Considerable circumstantial evidence, however, suggests it is a key event. For example, we know that during the initial fatty streak phase of lesion development, CEs accumulate mainly in cytoplasmic inclusions, indicating that lysosomal hydrolysis and sterol clearance is effective and suggesting an important role for ACAT-1 (iso-form of ACAT found in macrophages)-derived lipid droplets at this stage [28]. However, as lesions progress to fibrous plaques, substantial accumulation of CE and FC occurs in lysosomes in lesions from humans, nonhuman primates, rabbits and pigeons [28–30], indicating a failure of lysosomal hydrolysis of CE and clearance of sterol, and suggesting that lysosomal dysfunction is a participant in late-stage disease. Consistent with this, the Avasimibe and Progression of Coronary Lesions Assessed by Intravascular Ultrasound (A-PLUS) and Acylcoenzyme A:cholesterol Acyltransferase Inhibition on the Progression of Coronary Atherosclerosis (ACTIVATE) clinical trials demonstrated that ACAT inhibition, which would reduce the lipid droplet sterol pool, did not reduce advanced atherosclerosis [31,32]. Together, these studies suggest that ACAT-1 may not be the most critical player in late-stage lesions and indicate that lysosomal sterol may be an important pool of sterol during the later disease stages. Similarly, in pigeons, an animal model that closely mimics human atherosclerosis, medial smooth muscle cell proliferation and migration into the intima does not occur until after the lysosomal accumulation in macrophages [33]. Smooth muscle cell involvement in the lesion is a key transition point from reasonably benign lesions to clinically important ones. In addition, cholesterol trapped in lesion foam cell lysosomes remains trapped even when total plasma cholesterol returns to normal [34]. By contrast, cytoplasmic CE droplets are cleared rapidly. These animal studies parallel experiments on cultured macrophages, which demonstrate that lysosomal cholesterol (FC and CEs) is trapped and not available for efflux even under conditions that rapidly remove cytoplasmic and plasma membrane cholesterol stores [35]. Indicating that the lysosomal sterol is particularly difficult to remove and is resistant to treatment.
In addition to their effects on LAL, several genes for other lysosomal enzymes, including those for cathepsin D and acid sphingomyelinase, are altered in atherosclerosis, further suggesting a link between lysosomes and atherosclerosis, and indicating that sterol accumulation may produce other nonsterol-associated effects. Interestingly, exogenous administration of LAL to mice reduces atherosclerosis [36]. Several questions remain about how exogenous LAL exerts its effect but the studies are provocative and further highlight the potential for lysosomes to influence atherogenesis.
Unesterified cholesterol can partition into the lysosomal membrane & influence lysosomal function
The FC generated by lysosomal hydrolysis partitions into the lysosome membrane for clearance. Lipids in membranes are ordered into functional microdomains that greatly influence membrane function and cellular metabolism [37]. Changes in the distribution of cholesterol within membranes can have important consequences. Cholesterol-rich regions show an association with, and modulation of, specific protein and lipid functions [37,38]. Membrane proteins can also modulate cholesterol organization in the bilayer and regulate intracellular cholesterol movement [39–41]. In addition to modulating protein function, the lipid content of membranes affects the physical properties of membranes [42] and cholesterol is one of the most important regulators of lipid organization [43]. Altering the cholesterol content of membranes disrupts vesicle formation in the endocytic pathway [44] and the induction of intracellular signals [45]. Thus, there is reason to suspect that excess FC in the lysosome membrane could affect lysosomal membrane function.
Recently, we demonstrated that the generation of FC from the uptake of modified forms of LDL by human macrophages in culture produced a rise in the lysosomal pH [46] to levels above the functional range of LAL [47]. This led to an inhibition of hydrolysis and accumulation of undigested CE within lysosomes producing large, sterol-engorged lysosomes similar to those observed in atherosclerotic lesions [46]. The cause of the increased pH was an FC-induced inhibition of the vacuolar-ATPase pumps (v-ATPase) in the lysosomal membrane [46]. The v-ATPases are membrane-bound protein complexes that pump hydrogen ions into the lysosome lumen in order to maintain the necessary acidic pH of the lysosome. Pump inhibition appeared to be produced by the partitioning of excess FC into the lysosome membrane since it was possible to mimic this inhibition pharmacologically by inserting excess FC into the membranes of isolated lysosomes [46]. Moreover, the pumps could be reactivated by removing excess sterol [46]. This is not surprising, since it is known that this type of extreme alteration of the lipids within membrane domains can affect many membrane properties [48]. However, a failure of lysosomal hydrolysis caused by increased pH would explain the CE accumulation that accompanies late-stage atherosclerosis. It is not clear how the initial accumulation of cholesterol in the membrane occurs but preliminary, unpublished evidence implicates the rate of delivery of cholesterol to lysosomes as one determinant [Jerome WG, Unpublished Data]. When uptake and delivery is slow, the lysosome can efficiently clear the FC generated by hydrolysis. It is only when delivery of cholesterol to lysosomes is rapid that the dysfunction is stimulated and v-ATPase activity inhibited.
Besides v-ATPase activity, a major determinant of lysosomal pH is leakiness of the lysosomal membrane [49]. Tissue culture experiments have shown that a number of factors, including sterol, can affect lysosomal membrane permeability. Membrane leakiness can be decreased through stabilization by FC and increased by sterol oxidation [43,50]. In the case of oxysterols, the leakiness generally leads to apoptosis [50]. Increased apoptosis is associated with the areas of the plaque most prone to rupture [51].
Although there are probably multiple factors involved in the initiation of cholesterol-induced lysosome malfunction, our current data suggest a scenario where unregulated uptake of cholesteryl ester-containing particles leads to a massive accumulation of FC in lysosomes which alters lysosome function leading to pathologic changes, including inhibition of CE hydrolysis and the subsequent accumulation of CE in lysosomes, as particles continue to be delivered to the malfunctioning lysosomes. Besides the described direct effects on lysosome function, the increased lysosomal FC also has the potential for indirect effects. The inhibition of sterol removal from lysosomes, either due to the inability to hydrolyze CEs or trafficking defects, is related to several pathologies, including Niemann-Pick type C and Wolman disease [52,53]. Kruth reports that inhibiting lysosomal function in cultured cells produces a reduction in cholesterol microdomains on macrophage plasma membranes, suggesting that alterations in lysosome function can inhibit the trafficking of lysosomal membrane sterol to other locations [54]. In addition, alterations in membrane cholesterol can influence membrane sphingomyelin content and changes in membrane sphingomyelin can influence membrane and lysosome function. Under normal circumstances, there is coordinate regulation of membrane FC and sphingomyelin content and dysregulation of one can produce dysregulation of the other [55]. Sphingomyelin is an important structural component of membranes but more importantly, products of sphingomyelin metabolism are potent signaling molecules affecting cell viability, cytokine production, intracellular vesicular trafficking and cholesterol homeostasis [55–58].
Perhaps even more importantly, the trapping of sterol within lysosomes removes it from the normal metabolic pools. In addition to re-esterification, some extralysosomal cholesterol moves into alternative metabolic pools that can act to signal specific changes in the macrophage. Among these is activation of apoptosis pathways or processing to oxysterols [59–63]. Several oxysterols have been identified as liver X receptor (LXR) agonists which can upregulate the expression of genes involved in sterol efflux, amongst other functions [59,61].
Effect of triglycerides on lysosomal sterol trapping & lysosome function
Reducing the lesion cholesterol burden by stimulating removal of arterial cholesterol via the reverse cholesterol transport pathway is a major area of atherosclerosis research [64]. The first step in this pathway is the mobilization of intracellular cholesterol stores to the plasma membrane for efflux. Clearly, the trapping of sterol within lysosomes would prevent its movement into efflux pools. Therefore, the resistance to clearance of lysosomal sterol presents a barrier to treatment. Thus, it would appear that it is important to identify mechanisms to stimulate the mobilization of sterol out of the lysosome.
Atherosclerosis studies have tended to focus on cholesterol because it is the principal lipid which accumulates within the atherosclerotic lesion. Most tissue culture studies utilize LDL or modified LDL as the sole lipid source. However, circulating triglyceride (TG) levels are also associated with increased atherosclerosis risk. The mechanism(s) by which TG affects atherosclerosis is unclear and whether it is an independent risk factor remains controversial [65]. It is also not clear whether increased circulating TG levels directly affect arterial wall metabolism, since larger TG-rich particles (TRPs) do not appear to enter the artery wall. However, TGs are found in circulation as a component of TRP; including chylomicrons and VLDL and their corresponding remnants and some smaller TRPs have been identified within atherosclerotic plaques [66]. In primary cultures of human monocyte-derived macrophages and within the artery wall, the interaction of macrophages with smaller TRP can induce TG accumulation within the macrophage [67–69].
There are good reasons to suspect that cellular TG accumulation in macrophage foam cells could influence macrophage cholesterol metabolism. TGs are more metabolically active than CE and thus represent a more dynamic lipid pool than cholesterol which provides more opportunities to influence cellular lipid metabolism [70]. It is also known that macrophage lysosomes hydrolyze CE faster when it is introduced as a mixed CE and TG particle compared with CE without TG [71]. This is indicated by TGs altering the physical state of the CE and keeping it more fluid [71,72]. This physical state effect is not limited to lysosomal hydrolysis. The association of TGs with CEs in cytoplasmic CE droplets makes the CEs more susceptible to hydrolysis by neutral cholesteryl ester hydrolase [73,74]. This is important because the mobilization of FC from CE stores, either within lysosomes or from droplets, is a necessary first step for clearance [74].
Physical effects are not the only potential mediators of cholesterol homeostasis. The free FAs hydrolytically released during TG metabolism are also potential mediators of cholesterol homeostasis. FAs are key signaling molecules that greatly affect the expression of critical genes controlling cellular cholesterol mobilization. FAs can act at the level of nuclear receptors to affect the transcription of a number of genes important in cholesterol homeostasis. For example, the individual or cooperative upregulation of PPAR and LXR expression by FA has been shown to regulate the expression of a number of cholesterol homeostatic genes including the ATP-binding cassette (ABC) gene family members, A1 and G1, which are influential in intracellular sterol transport and efflux [75]. Activation of ABCA1 and ABCG1 genes enhances cholesterol movement and efflux. LXRs also influence inflammatory genes [76]. Macrophage inflammatory responses and sterol metabolism are intimately linked in the atherosclerosis environment and are important regulators of lesion progression [77–79].
Owing to the potential for interaction between TG and sterol metabolism, we explored the effect of TRP on macrophage lysosomal cholesterol sequestration [80]. These studies demonstrated that TGs delivered to cultured macrophages as part of TRPs dramatically reduced lysosomal CE accumulation and almost completely eliminated the CEs stored in cytoplasmic droplets. The effect of TRPs on cholesterol accumulation was proportional to the TG content of the cells. The reduction in lysosomal CEs was observed when cholesterol-containing particles were delivered simultaneously with TRP but, more importantly, the incubation of cells with TRPs subsequent to lysosomal sterol engorgement stimulated a greater than 50% reduction in pre-existing lysosomal sterol stores. This is a very exciting observation given that we have previously demonstrated that, without the presence of TG, the sterol in lysosomes is trapped and unresponsive to stimuli that normally enhance efflux, even when the stimulation removed 90% of the nonlysosomal cholesterol stores [35].
With TRP treatment, not only were the lysosomal sterol stores of cultured cells depleted, but the bulk of the generated cholesterol also exited the cell, presumably by sterol efflux pathways. In addition to naturally occurring TRPs, TG-phospholipid micelles also stimulated lysosomal sterol release, implicating the TG component of the particles as a causative agent [80]. The mechanism by which TG produced its effect(s) is still not clear. However, we were able to demonstrate that treatment with TG-containing particles returned the lysosomal pH to its normal acidic levels and restored lysosomal CE hydrolysis [80]. Thus, the TRP restored v-ATPase activity, which would, at least in part, contribute to the clearance of lysosomal sterol. Other than these clues it is unclear whether this was a direct effect of TG or the result of metabolites from cellular TG metabolism. However, what is clear is that in some manner, TRP treatment dramatically affected lysosome function.
At present, we do not know exactly how TRP treatment influences lysosome function. Figure 3 summarizes macrophage TG metabolism and highlights the multiple pathways involved; each of which could potentially influence some aspect of lysosomal function. The predominant mechanism for normal macrophage degradation of TG involves lipases on the surface of the macrophage [81], which can hydrolyze the TG to generate free FAs [82,83]. These FAs can be internalized into the macrophage cytosol where they can be used to form the acyl chains of newly synthesized lipids, such as di- and tri-glycerides, phospholipids, and CEs. Such an influx of FAs could alter macrophage metabolism via a number of mechanisms. For example, altering the make-up of phospholipid FAs could alter the properties of membranes such as those of the lysosome. Moreover, in addition to altering the character of cellular lipids, the FAs generated from increased TG hydrolysis are potential signaling molecules for PPAR- and LXR-regulated pathways. Of course, many of the FAs end up being converted back to TGs. It is possible that these cytoplasmic TGs could influence cellular lipid metabolism.
Figure 3. Normal cellular lipoprotein-derived triglyceride metabolism.

The TG in lipoproteins can be hydrolyzed on the surface of macrophages by lipase enzymes. The FAs generated by this hydrolysis can be internalized by cells. In the cytoplasm, the FAs can be activated and incorporated into newly synthesized TGs or other molecules, such as phospholipids or CEs. In macrophages, the TGs and CEs are stored within cytoplasmic lipid droplets. Although, under normal conditions, this is the predominant method for degradation of lipoprotein TGs, macrophages can also process TGs through the uptake of particles into endosomes and the delivery of the particles to lysosomes. In the lysosome, lysosomal acid lipase (and possibly other lipases) can hydrolyze the TGs and liberate the FAs. Liberated FAs can then be transported out of lysosomes and join the pool of FAs that are utilized for the synthesis of new lipids.
CE: Cholesteryl ester; FA: Fatty acid; TG: Triglyceride.
Furthermore, the FA flux within cells is highly dynamic, with acyl changes liberated from TG hydrolysis ending up not only as part of new TG but also as components of phospholipids and even esterified to cholesterol. A particularly interesting possibility is that the uptake of large amounts of TG could alter the phospholipid acyl chain distribution in ways that influence some aspect of lysosome membrane function. Our VLDL preparations are typically enriched in both linoleic and oleic acids; a finding consistent with those in the literature [84]. Given the membrane-stiffening effects of saturated FAs, it is possible that displacing saturated FA-containing phospholipids with unsaturated FA-enriched phospholipids could increase membrane fluidity and restore v-ATPase activity and lysosome function [85,86]. Finally, under certain conditions, the cellular TGs can be incorporated into lysosomes through autophagocytosis [87]. This presents the intriguing possibility that either TGs in the lysosomes or FAs generated from hydrolysis of autophagocytically delivered TGs could interact with the cholesterol stores in the lysosome lumen.
Surface hydrolysis is not the only method by which TGs could influence macrophage cholesterol metabolism. An alternative pathway exists for the cellular metabolism of TG-containing particles. Similar to CE-containing particles, TRPs can be internalized by receptor-mediated endocytosis [88,89]. Under normal circumstances, this pathway makes a minor contribution to cellular lipid accumulation. However, modification of TRPs can enhance their endocytic potential [88,89]. These enhancements include changes in size and composition and in the concentration of specific apolipoproteins on the particles [19,90,91]. Several of these changes have been associated with atherogenesis. The uptake of TRP via endocytosis leads to the direct delivery of TG to lysosomes. Within the lysosome, TG itself can affect the physical state of cholesterol-containing emulsions in ways that increase CE hydrolysis [92–94]. Presumably, a major effect is altering CE fluidity, making it more available for attack by lipases. In addition, lysosomal TGs can be hydrolyzed resulting in the liberation of FAs within the lysosome lumen, which could potentially interact in yet unspecified ways with other lysosomal lipids. However, under normal circumstances, these FAs rapidly exit the lysosome and join the cytosolic pool of FAs [95] with the same fate as those generated by surface hydrolysis of TG. Thus, cellular TG metabolism could potentially influence lysosomes through the following:
Endocytic delivery of TGs to lysosomes;
Generation of FAs from either lysosomal or surface hydrolysis;
Altering the pool of FAs available for cytoplasmic assembly of lipids;
Influencing signaling pathways involved in lipid metabolism.
Which, if any, of these contribute to TG-induced cholesterol release remains to be determined.
Is release of lysosomal sterol into the extralysosomal compartment a good thing?
At first glance, the removal of cholesterol from the lysosome would appear to be a positive occurrence with respect to atherosclerosis. As the first step in cholesterol clearance, lysosomal CE hydrolysis and FC clearance are obligate steps in reducing the cholesterol burden of foam cells. However, this positive effect would appear to be at odds with findings from some clinical studies in which hypertriglyceridemia has been associated with increased atherosclerosis [96]. Two important points need to be made here regarding this apparent paradox. First, the epidemiologic evidence is controversial and does not determine whether hypertriglyceridemia has a direct or indirect effect on coronary disease [65]. Thus, it is possible that high TG levels have a positive effect on foam cells while promoting other negative systemic effects.
Second, it remains to be determined whether the increased lysosomal cholesterol clearance induced by TG has a positive or negative impact on macrophages and lesion development. With regard to this point, there is growing evidence that excess cellular FC, if delivered into the wrong intracellular pools, can have adverse effects on macrophages. Although the removal of foam cell cholesterol is an important step in lesion regression [97], the triggered release of large FC stores from lysosomes would be expected to generate high extralysosomal pools of FC. If these over-whelmed the normal homeostatic mechanisms and were not efficiently directed into efflux or storage pathways, the FC could potentially accumulate in alternate cellular pools. Increases in some of these pools could have adverse effects. For example, several studies have demonstrated that disrupting normal intracellular cholesterol trafficking can induce cell death [60,98]. One effect of disrupting cholesterol trafficking is to redirect extralysosomal FC into the ER regulatory pool [99]. Excess accumulation in this pool is cytotoxic [59,100]. Thus, although proper membrane FC concentrations are required for normal cell growth and membrane stability, cellular health is regulated not only by the level but also the cellular location of sterol. Therefore, it is possible that the sequestration of cholesterol within the lysosomal compartment may be a protective measure to save the cell from the potentially toxic effects of FC accumulation in these regulatory pools. If this is true, then the rapid release of these protective pools could potentially flood the normal homeostatic mechanisms and produce excess accumulation within cytotoxic pools. This possibility highlights a critical area of macrophage biology and atherogenesis that requires further study.
Dynamics of late-stage atherosclerotic plaques
Our experiments on the ability of TG to reverse the process of cholesterol-induced lysosome malfunction are based primarily on cell culture experiments (although our results are consistent with evidence from animal and human studies). The strength of cell culture is that it provides greater control over variables. However, the late-stage atherosclerotic plaque is a complicated and highly dynamic milieu and it is almost impossible to mimic in culture all that occurs in the artery wall. Thus, it will be necessary to explore these same processes within the context of the artery wall before conclusions about their importance to atherosclerosis are confirmed. Nonetheless, our studies to date do provide important information regarding the interaction of cellular TG and cholesterol metabolism and provoke interesting questions regarding the role of their interaction in the disease process.
The necrotic core of the late-stage atherosclerotic lesion is a key area in which the interaction of lysosomes, cholesterol and TGs would be expected to play a role. The necrotic cores are areas rich in foam cells but also containing large amounts of cellular debris, extracellular lipid and other free molecules. In fact, the core often contains as much or more cholesterol and CEs as that within foam cells. The term necrotic core highlights the hypothesis that much of the extracellular material in these areas is derived from the death and decay of cells in the plaque. There is clear evidence that necrotic cores do contain the remains of dead cells but the exact mechanisms of core formation are hard to pinpoint and, similar to much of the atherogenesis, are probably the result of a variety of factors interacting [51,101–103]. However, there is strong evidence that the death of cells in the plaque occurs by both apoptosis and oncosis [102]. Undoubtedly, the various contributions of each process are dependent upon the state of the lesion at different points in time. The highly dynamic nature of the core makes it difficult to study in a controlled manner. However, macrophages are usually found surrounding the core and also exist within the core [104]. Some of these may be dying cells but many are undoubtedly recruited to the area to aid in removing the debris [105]. Unfortunately, the debris includes excess cholesterol, which can potentially be toxic to the macrophages. This sets up a scenario in which uptake of debris leads to cell death, which leads to further uptake of debris. Thus, macrophages play a role both as contributors to the pathologic features of lesion and as a major mechanism for resolving the pathology. Not discussed here, but equally important, is the fact that macrophages are important inflammatory mediators within the lesion and this function also influences atherogenesis and, potentially, the necrotic core.
There is still a lot to be learnt with regard the dynamics of the necrotic core but two recent themes have emerged that have some importance to the discussion of the role of cholesterol and TG in lysosome function. The first theme began with elegant cell culture experiments indicating that the lipid from the uptake of apoptotic cells is less toxic to the macrophage than that derived from lipoproteins [106]. The exact reasons why this is so are still being investigated but it has been shown that a partial explanation is that cholesterol from apoptotic cell uptake is more available for efflux [106]; a process that rids cells of excess cholesterol. In addition, cholesterol-trafficking patterns are altered so that cholesterol is directed away from toxic pools [106]. Finally, uptake of apoptotic cells stimulates upregulation of a number of signaling pathways that render the cells resistant to death signals [106]. These studies raise the exciting possibility that the dynamics of metabolism within the necrotic core might be manipulated towards regression of the atherosclerotic lesion. Therefore, an important question would be what role TG accumulation might play in either enhancing or disrupting these beneficial effects and the extent to which lysosomes are involved.
The second exciting observation regarding the necrotic core is the evidence of autophagy within some cells in the core region of the plaque [107,108]. Although the evidence in plaques is still circumstantial, studies in cultured cells reveal that cholesterol accumulation and other factors present within the lesion are capable of stimulating autophagy [109–111]. Autophagy is a mechanism by which cells partition their own intracellular components and deliver them to hydrolytic compartments for digestion. This is now recognized as a key homeostatic mechanism in cells allowing for the removal and recycling of worn out components and the removal of toxic material. However, when autophagy is excessive and prolonged it can lead to cell death [103]. If this occurred within the atherosclerotic lesion, it would contribute (along with apoptosis and oncosis) to further increase the necrotic core. To date, the evidence of plaque autophagy only relates to endothelial cells and smooth muscle cells in the lesion. However, this may well be because autophagy is much harder to distinguish in macrophages. At present, there is no reason to rule out autophagy as a key process occurring within lesion macrophages.
Since the lysosome is a central player in autophagic digestion, understanding how cholesterol and TG influence autophagy should provide important information towards a better comprehension of the dynamics of the necrotic core. This, in turn, would promote a better appreciation of the multiple factors that drive the changes occurring in late-stage atherosclerotic lesions.
Conclusion
The foam cell is a major regulator of atherosclerotic plaque development and it is clear that the increased cholesterol accumulation observed in foam cells can impact macrophage function and thus lesion development. For this reason, understanding what mediates foam cell intracellular cholesterol homeostasis is a key goal of atherosclerosis research. Lysosomal cholesterol accumulation is a major constituent of clinically important atherosclerotic macrophage foam cells. Most importantly, the lysosomally sequestered free and esterified cholesterol has been shown to be highly resistant to removal, even under conditions that promote extralysosomal cholesterol efflux. However, we have recently demonstated that TGs delivered to macrophage foam cells as part of lipoproteins or phospholipid micelles can stimulate a large reduction in the lysosome sterol levels. Moreover, much of the cholesterol liberated from lysosomes became available for exit from the cell via efflux pathways. The combined effect was to dramatically reduce the macrophage lipid burden.
The proximate cause of the lysosomal release of sterol appears to be a TG-induced restoration of normal lysosome function, especially the restoration of lysosome membrane proton pump activity. This indicates that TG treatment would not only enhance lysosome clearance of sterol but it could also improve overall lysosome function and help re-establish normal macrophage homeostasis. If these in vitro effects of TG-containing particles can be reproduced in vivo, it suggests that TG-induced removal of cholesterol from foam cell lysosomes, if properly managed, might be harnessed as an aid in atherosclerosis regression. However, more work must still be carried out in order to ascertain whether this TG effect can be harnessed therapeutically.
Future perspective
Reduction of circulating LDL-C and increasing reverse cholesterol transport-mediated removal of lesion sterol have shown some efficacy in reducing the overall burden of cholesterol in lesions. However, there is growing evidence that this has little effect on lysosomal cholesterol stores. Although this article presents circumstantial evidence that the build-up of sterol within lysosomes is detrimental, it is still unclear how much the lysosomal stores of sterol play in the resistance of atherosclerotic lesions to regression therapies. This remains an important area for investigation. Nonetheless, given the potential for these stores to damage macrophages and effect overall vascular health, understanding the mechanism(s) for the lysosomal sterol engorgement and determining strategies for liberating the sterol remain important areas of investigation for understanding the nuances of atherosclerosis lesion development and determining novel treatment options. The realization that TG-containing particles, at least in tissue culture, can liberate lysosomally sequestered sterol presents an exciting opportunity to investigate the causes of the lysosome engorgement and to design treatment methods. However, to reach this goal, several important questions remain to be answered. These include:
Can TG-containing particles liberate the lysosome sterol in macrophages within the artery wall?
What is the mechanism by which TG produces its effects on macrophage lysosomes?
Can the liberation of cholesterol from lysosomes be carried out in a manner that shunts the liberated cholesterol directly into storage pools or to efflux and bypasses delivery to potentially pathologic pools of sterol?
Can methods be designed to deliver TG to macrophages in the artery wall without the potential complications produced by hypertriglyceridemia? For example, can TG be delivered as part of inert particles rather than lipoproteins?
Footnotes
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 2▪.Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: part I. J Lipid Res. 2004;45(9):1583–1593. doi: 10.1194/jlr.R400003-JLR200. Review of the evolution of the cholesterol hypothesis of atherosclerosis. [DOI] [PubMed] [Google Scholar]
- 3.Jerome WG. Advanced atherosclerotic foam cell formation has features of an acquired lysosomal storage disorder. Rej Res. 2006;9:245–255. doi: 10.1089/rej.2006.9.245. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding sites on macrophages that mediate uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein J, Debose-Boyd R, Brown M. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 6.Greaves D, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. 2009;50(Suppl):S282–S286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woollard K, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7(2):77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage. Ann Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 9.Steinbrecher U, Lougheed M, Kwan W, Dirks M. Recognition of oxidized low density lipoprotein by the scavenger receptor of macrophages results from derivatization of apolipoprotein B by products of fatty acid peroxidation. J Biol Chem. 1989;264:15216–15223. [PubMed] [Google Scholar]
- 10.Buton X, Mamdouh Z, Ghosh R, et al. Unique cellular events occurring during the initial interaction of macrophages with matrix-retained or methylated aggregated LDL. J Biol Chem. 1999;274:32112–32121. doi: 10.1074/jbc.274.45.32112. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg D, Parthasarathy S, Carew T, Khoo J, Witztum J. Beyond cholesterol, modification of low-density lipoproteins that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 12.Hoff H, Hoppe G. Structure of cholesterol-containing particles accumulating in atherosclerotic lesions and the mechanisms of their derivation. Curr Opin Lipidol. 1995;6(5):317–325. doi: 10.1097/00041433-199510000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Williams K, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9:471–474. doi: 10.1097/00041433-199810000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Rothblat GH, Rosen JM, Insull W, Yau AO, Small DM. Production of cholesteryl ester-rich anisotropic inclusions by mammalian cells in culture. Exp Mol Pathol. 1977;26:318–324. doi: 10.1016/0014-4800(77)90059-4. [DOI] [PubMed] [Google Scholar]
- 15.Kruth H. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin Lipidol. 2002;13:483–488. doi: 10.1097/00041433-200210000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Tabas I. Nonoxidative modifications of lipoproteins in atherogenesis. Ann Rev Nutr. 1999;19:123–139. doi: 10.1146/annurev.nutr.19.1.123. [DOI] [PubMed] [Google Scholar]
- 17.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfbauer G, Glick JM, Minor LK, Rothblat GH. Development of the smooth muscle foam cell: uptake of macrophage lipid inclusions. Proc Natl Acad Sci USA. 1986;83(20):7760–7764. doi: 10.1073/pnas.83.20.7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabas I, Myers J, Innerarity T, Xu X, Arnold J, Maxfield F. The influence of particle size and multiple apoprotein E-receptor interactions on the endocytic targeting of β-VLDL in mouse. J Cell Biol. 1991;115:1547–1560. doi: 10.1083/jcb.115.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruth H, Chang J, Ifrim I, Zhang W-Y. Characterization of patocytosis: endocytosis into macrophage surface-connected compartments. Eur J Cell Biol. 1999;78:91–99. doi: 10.1016/S0171-9335(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 21.Soccio R, Breslow J. Intracellular cholesterol transport. Arterioscl Thromb Vasc Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S, St Clair R, Rudel L. Mobilization of cytoplasmic CE droplets by overexpression of human macrophage cholesteryl ester hydrolase. J Lipid Res. 2003;44:1883–1840. doi: 10.1194/jlr.M300162-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Barter PJ, Rye KA. Molecular mechanisms of reverse cholesterol transport. Curr Opin Lipidol. 1996;7(2):82–87. doi: 10.1097/00041433-199604000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Yancey P, Bortnick A, Kellner-Weibel G, De la Liera-Moya M, Phillips M, Rothblat G. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 25.De Duve C. The participation of lysosomes in the transformation of smooth muscle cells to foamy cells in the aorta of cholesterol fed rabbits. Acta Cardiol Suppl. 1974;20:9–25. [PubMed] [Google Scholar]
- 26.Hers HG. Lysosomes and Storage Diseases. Academic Press; NY, USA: 1973. The concept of inborn lysosomal disease; pp. 141–171. [Google Scholar]
- 27.Vellodi A. Lysosomal storage disorders. Br J Haemotol. 2004;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- 28.Jerome WG, Lewis JC. Early atherogenesis in White Carneau pigeons II ultrastructural and cytochemical observations. Am J Pathol. 1985;119:210–222. [PMC free article] [PubMed] [Google Scholar]
- 29.Fowler S, Berberian P, Shio H, Goldfischer S, Wolinsky H. Characterization of cell populations isolated from aortas of rhesus monkeys with experimental atherosclerosis. Circ Res. 1980;46:520–530. doi: 10.1161/01.res.46.4.520. [DOI] [PubMed] [Google Scholar]
- 30.Miller B, Kothari H. Increased activity of lysosomal enzymes in human atherosclerotic aortas. Exp Mol Pathol. 1969;10:288–294. doi: 10.1016/0014-4800(69)90058-6. [DOI] [PubMed] [Google Scholar]
- 31.Tardif J, Gregoire J, L’Allier P, et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–3377. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
- 32.Nissen S, Tuzcu E, Brewer H, et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354:1253–1263. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- 33.Jerome WG, Lewis JC. Cellular dynamics in early atherosclerotic lesion progression in White Carneau pigeons. Spatial and temporal analysis of monocyte and smooth muscle invasion of the intima. Arterioscler Thromb Vasc Biol. 1997;17:654–664. doi: 10.1161/01.atv.17.4.654. [DOI] [PubMed] [Google Scholar]
- 34.Jerome WG, Lewis JC. Early atherogenesis in White Carneau pigeons. Effect of a short-term regression diet. Exp Mol Pathol. 1990;53:223–238. doi: 10.1016/0014-4800(90)90046-g. [DOI] [PubMed] [Google Scholar]
- 35.Yancey PG, Jerome WG. Lysosomal cholesterol derived from mildly oxidized low density lipoprotein is resistant to efflux. J Lipid Res. 2001;42:317–327. [PubMed] [Google Scholar]
- 36▪▪.Du H, Schiavi S, Wan N, Levine M, Witte D, Grabowski G. Reduction of atherosclerotic plaques by lysosomal acid lipase supplementation. Arterioscl Thromb Vasc Biol. 2004;24:147–154. doi: 10.1161/01.ATV.0000107030.22053.1e. Highlights the role of lysosomes in atherosclerosis by demonstrating that exogenous LAL can reduce plaque size. [DOI] [PubMed] [Google Scholar]
- 37.Lingwood D, Simons K. Lipid rafts as a membrane organizing principal. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 38.Smart EJ, Anderson RG. Alterations in membrane cholesterol that affect structure and function of caveolae. Methods Enzymol. 2002;353:131–139. doi: 10.1016/s0076-6879(02)53043-3. [DOI] [PubMed] [Google Scholar]
- 39.Bagnat M, Keranen S, Shevchenko A, Shevchenko A, Simons K. Lipid rafts function in biosynthetic delivery of proteins to the cell surface of yeast. Proc Natl Acad Sci USA. 2000;97:3254–3259. doi: 10.1073/pnas.060034697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroeder F, Gallegos A, Atshves BP, et al. Recent advances in membrane microdomains: rafts, caveolae, and intracellular cholesterol traficking. Exp Biol Med. 2001;226:873–890. doi: 10.1177/153537020122601002. [DOI] [PubMed] [Google Scholar]
- 41.Liscum L. Niemann–Pick type C mutations cause lipid traffic jam. Traffic. 2000;1:218–225. doi: 10.1034/j.1600-0854.2000.010304.x. [DOI] [PubMed] [Google Scholar]
- 42.Mukherjee S, Maxfield FR. Role of membrane organization and membrane domains in endocytic lipid trafficking. Traffic. 2000;1:203–211. doi: 10.1034/j.1600-0854.2000.010302.x. [DOI] [PubMed] [Google Scholar]
- 43.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 44.Pichler H, Riezman H. Where sterols are required for endocytosis. Biochim Biophys Acta. 2004;1666:51–61. doi: 10.1016/j.bbamem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini L. Elevated plasma membrane cholesterol content alters macrophage signaling function. Arterioscler Thromb Vasc Biol. 2006;26 doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- 46▪▪.Cox B, Griffin E, Ullery J, Jerome W. Effects of cellular cholesterol loading on macrophage foam cell lysosome acidification. J Lipid Res. 2007;48:1012–1021. doi: 10.1194/jlr.M600390-JLR200. Establishes that excess lysosome membrane cholesterol inhibits lysosome proton pump activity and disturbs lysosome function. [DOI] [PubMed] [Google Scholar]
- 47.Sando G, Rosenbaum L. Human lysosomal acid lipase/cholesterol ester hydrolase. J Biol Chem. 1985;260:15186–15193. [PubMed] [Google Scholar]
- 48.Zhang G-J, Liu H-W, Yang L, Zhong Y-G, Zheng Y-Z. Influence of membrane physical state on the lysosomal proton permeability. J Membrane Biol. 2000;175:53–62. doi: 10.1007/s002320001054. [DOI] [PubMed] [Google Scholar]
- 49.Grabe M, Oster G. Regulation of organelle acidity. J Gen Physiol. 2001;117:329–343. doi: 10.1085/jgp.117.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50▪.Yuan X, Li W, Olsson A, Brunk U. The toxicity to macrophages of oxidized LDL is mediated through lysosomal damage. Atherosclerosis. 1997;133:153–161. doi: 10.1016/s0021-9150(97)00094-4. Lysosome membrane lipid alterations can produce leakiness leading to cell death. [DOI] [PubMed] [Google Scholar]
- 51.Tabas I, Seimon T, Timmins J, Li G, Lim W. Macrophage apoptosis in advanced atherosclerosis. Ann NY Acad Sci. 2009;1173(Suppl 1):E40–E45. doi: 10.1111/j.1749-6632.2009.04957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liscum L, Sturley S. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta. 2004;1685:22–27. doi: 10.1016/j.bbalip.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 53.Assmann G, Seedorf U. Acid lipase deficiency. Wolman disease and cholesteryl ester storage disease. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw Hill Inc; NY, USA: 1995. [Google Scholar]
- 54.Kruth HS, Ifrim I, Chang J, Addadi L, Peri-Treves D, Zhang W-Y. Monoclonal antibody detection of plasma membrane cholesterol microdomains responsive to cholesterol trafficking. J Lipid Res. 2001;42:1492–1500. [PubMed] [Google Scholar]
- 55.Ridgway N. Interactions between metabolism and intracellular distribution of cholesterol and sphingomyelin. Biochim Biophys Acta. 2000;1484:129–141. doi: 10.1016/s1388-1981(00)00006-8. [DOI] [PubMed] [Google Scholar]
- 56.Witting S, Maiorano J, Davidson W. Ceramide enhances cholesterol efflux to apolipoprotein A-I by increasing the cell surface presence of ATP-binding cassette transporter A1. J Biol Chem. 2003;278:40121–40127. doi: 10.1074/jbc.M305193200. [DOI] [PubMed] [Google Scholar]
- 57.Gomez-Munoz A, Kong J, Sahl B, Steinbrecher U. Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages. J Lipid Res. 2004;45:99–105. doi: 10.1194/jlr.M300158-JLR200. [DOI] [PubMed] [Google Scholar]
- 58.Mathias S, Pena L, Kolesnick R. Signal transduction of stress via ceramide. Biochem J. 1998;335:465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59▪▪.Feng B, Yao P, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. First demonstration that excess sterol in the endoplasmic reticulum can induce cell death. [DOI] [PubMed] [Google Scholar]
- 60.Kellner-Weibel G, Jerome W, Small D, et al. Effects of intracellular free cholesterol accumulation on macrophage viability: a model for foam cell death. Arterioscler Thromb Vasc Biol. 1998;18:423–431. doi: 10.1161/01.atv.18.3.423. [DOI] [PubMed] [Google Scholar]
- 61.Bjorkhem I, Andersson O, Diczfalusy U, et al. Atherosclerosis and sterol 27-hydroxylase: evidence for a role this enzyme in elimination of cholesterol from macrophages. Proc Natl Acad Sci USA. 1994;91:8592–8596. doi: 10.1073/pnas.91.18.8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu X, Menke J, Chen Y, et al. 27-Hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded macrophages. J Biol Chem. 2001;276:38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- 63.Szanto A, Benko S, Szatmari I, et al. Transcriptional regulation of human CYP27 integrates retinoid, peroxisome proliferation-activated receptor, and liver X receptor signaling in macrophages. Molec Biol Cell. 2004;24:8154–8166. doi: 10.1128/MCB.24.18.8154-8166.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ooi T, Ooi D. The atherogenic significance of an elevated plasma triglyceride level. Critical Rev Clin Lab Sci. 1998;35:489–516. doi: 10.1080/10408369891234255. [DOI] [PubMed] [Google Scholar]
- 66.Rapp J, Lespine A, Hamilton R, et al. Triglyceride-rich lipoproteins isolated by selected-affinity anti-apolipoprotein B immunosorption from human atherosclerotic plaque. Arterioscler Thromb. 1994;14:1767–1774. doi: 10.1161/01.atv.14.11.1767. [DOI] [PubMed] [Google Scholar]
- 67.Mattsson H, Johansson H, Ottosson M, Bondjers G, Wiklund O. Expression of lipoprotein lipase mRNA and secretion in macrophages isolated from human atherosclerotic aorta. JCI. 1993;92(4):1759–1765. doi: 10.1172/JCI116764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mukhin D, Orekhov A, Andreeva E, Schindeler E, Smirnov V. Lipids in cells of atherosclerotic and uninvolved human aorta. III. Lipid distribution in intimal sublayers. Exp Mol Pathol. 1991;54(1):22–30. doi: 10.1016/0014-4800(91)90040-5. [DOI] [PubMed] [Google Scholar]
- 69.Garner B, Baoutina A, Dean R, Jessup W. Regulation of serum-induced lipid accumulation in human monocyte-derived macrophages by interferon-gamma. Correlations with apolipoprotein E production, lipoprotein lipase activity and LDL receptor-related protein expression. Atherosclerosis. 1997;128(1):47–58. doi: 10.1016/s0021-9150(96)05979-5. [DOI] [PubMed] [Google Scholar]
- 70.Kritharides L, Christian A, Stoudt G, Morel D, Rothblat G. Cholesterol metabolism and efflux in human THP-1 macrophages. Arterioscl Thromb Vasc Biol. 1998;18:1589–1599. doi: 10.1161/01.atv.18.10.1589. [DOI] [PubMed] [Google Scholar]
- 71.Mahlberg F, Glick JM, Jerome WG, Rothblat GH. Metabolism of cholesteryl ester lipid droplets in a J774 macrophage foam cell model. Biochim Biophys Acta. 1990;1045:291–298. doi: 10.1016/0005-2760(90)90133-i. [DOI] [PubMed] [Google Scholar]
- 72.Snow J, Glick J, Phillips M. The phase behavior of cholesteryl esters in intracellular inclusions. J Biol Chem. 1992;267(26):18564–18572. [PubMed] [Google Scholar]
- 73.Adelman SJ, Glick JM, Phillips MC, Rothblat GH. Lipid composition and physical state effects on cellular cholesteryl ester clearance. J Biol Chem. 1984;259:13844–13850. [PubMed] [Google Scholar]
- 74.Zhao B, Fisher B, St Clair R, Rudel L, Ghosh S. Redistribution of macrophage cholesteryl ester hydrolase from cytoplasm to lipid droplets upon lipid loading. J Lipid Res. 2005;46:2114–2121. doi: 10.1194/jlr.M500207-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Chawla A, Boisvert WA, Lee C-H, et al. A PPARγ–LXR–ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7(1):161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 76.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saha P, Modarai B, Humphries J, et al. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol. 2009;9(2):109–118. doi: 10.1016/j.coph.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 78.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74(2):213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 79.Miller Y, Choi S, Fang L, Harkewicz R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. 2009;19(7):227–232. doi: 10.1016/j.tcm.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80▪▪.Ullery-Ricewick J, Cox B, Griffin E, Jerome W. Triglyceride alters lysosomal cholesteryl ester metabolism in cholesteryl-ester laden macrophage foam cells. J Lipid Res. 2009;50:2014–2026. doi: 10.1194/jlr.M800659-JLR200. Demonstrates that triglycerides can re-establish lysosome function in sterol-engorged macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81▪.Skarlatos SI, Dichek HL, Fojo SS, Brewer HB, Kruth HS. Absence of triglyceride c-deficient human monocyte-macrophages incubated with human very low density lipoprotein. J Clin Endocrinol Metab. 1993;76(3):793–796. doi: 10.1210/jcem.76.3.8383147. First conclusive demonstration to suggest that surface hydrolysis of triglyceride and fatty acid uptake are major routes for intracellular triglyceride accumulation in normal cells. [DOI] [PubMed] [Google Scholar]
- 82.Brown R, Rader D. Lipases as modulators of atherosclerosis in murine models. Curr Drug Targets. 2007;8(12):1307–1319. doi: 10.2174/138945007783220614. [DOI] [PubMed] [Google Scholar]
- 83.Olivecrona G, Olivecrona T. Triglyceride lipases and atherosclerosis. Curr Opin Lipidol. 2010;21(5):409–415. doi: 10.1097/MOL.0b013e32833ded83. [DOI] [PubMed] [Google Scholar]
- 84.Ruiz-Gutierrez V, Morgado N, Prada JL, Perez-Jimenez F, Muriana F. Composition of human VLDL triacylglycerols after ingestion of olive oil and high oleic sunflower oil. J Nutr. 1998;128(3):570–576. doi: 10.1093/jn/128.3.570. [DOI] [PubMed] [Google Scholar]
- 85.Jaureguiberry M, Tricerri M, Sanchez S, et al. Membrane organization and regulation of cellular cholesterol homeostasis. J Membr Biol. 2010;234(3):183–194. doi: 10.1007/s00232-010-9245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leekumjorn S, Cho H, Wu Y, Wright N, Sum A, Chan C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim Biophys Acta. 2009;1788(7):1508–1516. doi: 10.1016/j.bbamem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1137. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gianturco S, Bradley W. Pathophysiology of triglyceride-rich lipoproteins in atherothrombosis. cellular aspects. Clin Cardiol. 1999;22(Suppl 6):II7–II14. doi: 10.1002/clc.4960221403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gianturco S, Ramprasad M, Lin A, Song R, Bradley W. Cellular binding site and membrane binding proteins for triglyceride-rich lipoproteins in human monocyte-macrophages and THP-1 monocytic cells. J Lipid Res. 1994;35(9):1674–1687. [PubMed] [Google Scholar]
- 90.Jones N, Allen NS, Lewis JC. β-VLDL uptake by pigeon monocyte-derived macrophages: correlation of binding dynamics with three-dimensional ultrastructure. Cell Motil Cytoskel. 1991;19:139–151. doi: 10.1002/cm.970190302. [DOI] [PubMed] [Google Scholar]
- 91.Tabas I, Lim S, Xu XX, Maxfield FR. Endocytosed β-VLDL and LDL are delivered to different intracellular vesicles in mouse peritoneal macrophages. J Cell Biol. 1990;111:929–940. doi: 10.1083/jcb.111.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92▪▪.Glick JM, Adelman SJ, Phillips MC, Rothblat GH. Cellular cholesteryl ester clearance. Relationship to the physical state of cholesteryl ester inclusions. J Biol Chem. 1983;258:13425–13430. Demonstrates that triglycerides can alter the cholesteryl ester physical state, making it more susceptible to hydrolysis. [PubMed] [Google Scholar]
- 93.Minor LK, Mahlberg FH, Jerome WG, Lewis JC, Rothblat GH, Glick JM. Lysosomal hydrolysis of lipids in a cell culture model of smooth muscle foam cells. Exp Mol Pathol. 1991;54:159–171. doi: 10.1016/0014-4800(91)90028-v. [DOI] [PubMed] [Google Scholar]
- 94.Lada AT, Willingham MC, St Clair R. Triglyceride depletion in THP-1 cells alters cholesteryl ester physical state and cholesterol efflux. J Lipid Res. 2002;43:618–628. [PubMed] [Google Scholar]
- 95.Minor LK, Rothblat GH, Glick JM. Triglyceride and cholesteryl ester hydrolysis in a cell culture model of smooth muscle foam cells. J Lipid Res. 1989;30:189–197. [PubMed] [Google Scholar]
- 96.Hokanson J, Austin M. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3(2):213–219. [PubMed] [Google Scholar]
- 97.Williams KJ, Feig JE, Fisher EA. Cellular and molecular mechanisms for rapid regression of atherosclerosis: from bench top to potentially achievable clinical goal. Curr Opin Lipidol. 2007;18(4):443–450. doi: 10.1097/MOL.0b013e32823bcb15. [DOI] [PubMed] [Google Scholar]
- 98.Tabas I. Apoptosis and plaque destabilization in atherosclerosis: the role of macrophage apoptosis induced by cholesterol. Cell Death Differ. 2004;11:S12–S16. doi: 10.1038/sj.cdd.4401444. [DOI] [PubMed] [Google Scholar]
- 99.Li Y, Ge M, Ciani L, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids. J Biol Chem. 2004;279:37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- 100.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50(Suppl):S382–S387. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thim T, Hagensen M, Bentzon J, Falk E. From vulnerable plaque to atherothrombosis. J Intern Med. 2008;263(5):506–516. doi: 10.1111/j.1365-2796.2008.01947.x. [DOI] [PubMed] [Google Scholar]
- 102.Crisby M, Kallin B, Thyberg J, et al. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis. 1997;130(1–2):17–27. doi: 10.1016/s0021-9150(96)06037-6. [DOI] [PubMed] [Google Scholar]
- 103.Debnath J, Baehrecke E, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1(2):66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- 104.Stary H, Chandler A, Dinsmore R, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1995;15(9):1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 105.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86(5):1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cui D, Thorp E, Li Y, et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J Leukoc Biol. 2007;82(5):1040–1050. doi: 10.1189/jlb.0307192. [DOI] [PubMed] [Google Scholar]
- 107.Martinet W, De Bie M, Schrijvers D, De Meyer G, Herman A, Kockx M. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24(12):2296–2301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
- 108.Kockx M, De Meyer G, Muhring J, Jacob W, Bult H, Herman A. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97(23):2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 109.Xu K, Yang Y, Yan M, Zhan J, Fu X, Zheng X. Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. J Lipid Res. 2010;51(9):2581–2590. doi: 10.1194/jlr.M005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martinet W, De Meyer G. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104(3):304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Y, Cao Y, Zhang X, et al. The autophagy-lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;394(2):377–382. doi: 10.1016/j.bbrc.2010.03.026. [DOI] [PubMed] [Google Scholar]