

The sideroblastic anemias are a heterogeneous group of inherited and acquired disorders characterized by anemia of varying severity and the presence of ring sideroblasts in the bone marrow.1
Ring sideroblasts
Ring sideroblasts are erythroblasts with iron-loaded mitochondria visualized by Prussian blue staining (Perls’ reaction) as a perinuclear ring of blue granules (Figures 1D and 2C). The International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS) recommended that ring sideroblasts be defined as erythroblasts in which there are a minimum of five siderotic granules covering at least one third of the circumference of the nucleus.2 Ring sideroblasts are found exclusively in pathological conditions, and should not be confused with ferritin sideroblasts, which are present in normal bone marrow. These latter are normal erythroblasts that, after Prussian blue staining, show a few blue granules scattered in the cytoplasm, representing endosomes filled with excess iron not utilized for heme synthesis (siderosomes). While the iron of ferritin sideroblasts is stored in cytosolic ferritin, whose subunits are encoded by the FTH1 and FTL genes, the iron of ring sideroblasts is stored in mitochondrial ferritin, encoded by the FTMT gene.3 Indeed, mitochondrial ferritin is specifically detected in ring sideroblasts, as illustrated in Figure 2D.
Figure 1.

Representative peripheral blood and bone marrow smears from a patient with X-linked sideroblastic anemia. (A) Peripheral blood smear showing many hypochromic and microcytic cells. May-Grünwald-Giemsa (MGG), x1,000. (B) Bone marrow smear showing erythroid hyper plasia: erythroblasts are small with abnormal condensation of nuclear chromatin and ragged cytoplasm with ill-defined edges. MGG, x1,000. (C) Bone marrow smear showing erythroblasts with defective hemoglobinization (left) and erythroblasts containing multiple Pappenheimer bodies (right). MGG, x1,250. (D) Bone marrow smear. Perls’ stain shows that most erythroid precursors are ring sideroblasts with at least five positive granules disposed in a ring surrounding a third or more of the circumference of the nucleus. x1,250.
Figure 2.

Representative peripheral blood and bone marrow smears from a patient with refractory anemia with ring sideroblasts. (A). Peripheral blood smear showing dimorphic red cells with a population of macrocytes and a population of hypochromic microcytes. MGG, x1,000. (B) Bone marrow smear showing a marked erythroid hyperplasia with megaloblastoid features. The rare granulocytic cells look normal. Upper right, a late erythroblast with defective hemoglobinization; lower right, an early erythroblast with vacuolated cytoplasm and a late erythroblast with Pappenheimer bodies. MGG, x1,000. (C) Bone marrow smear stained by Perls’ reaction showing several ring sideroblasts. MGG x1,250. (D) Bone marrow smear. Mitochondrial ferritin is detected in granules surrounding the nucleus. Immunoalkaline phosphatase reaction, MGG x1250.
Classification of sideroblastic anemias
The sideroblastic anemias include both hereditary and acquired conditions, and the main disorders are reported in Table 1. Representative peripheral blood and bone marrow smears from a patient with X-linked sideroblastic anemia (XLSA) and a patient with refractory anemia with ring sideroblasts (RARS) are shown in Figures 1 and 2, respectively.
Table 1.
Classification of congenital and acquired sideroblastic anemias.
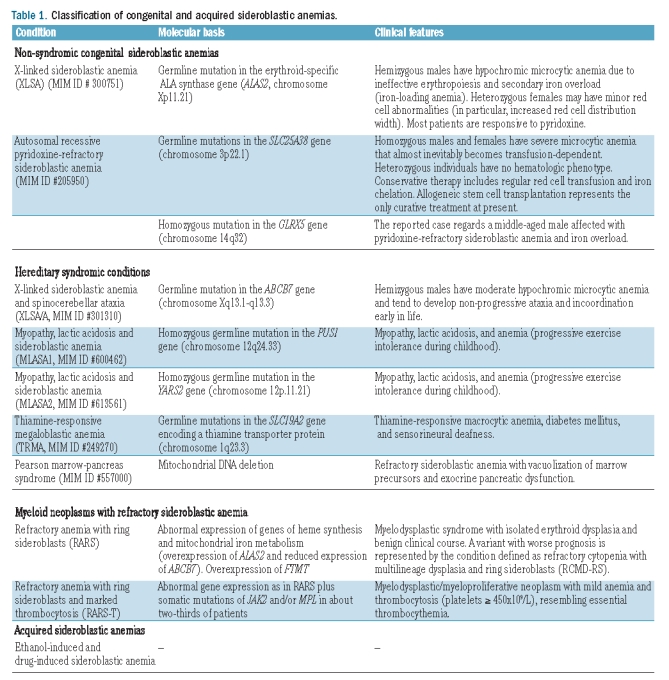
X-linked versus autosomal recessive congenital sideroblastic anemias
XLSA is caused by germline mutations in the erythroid-specific ALA synthase gene (ALAS2). Males with XLSA may present in the first two decades of life with symptoms of anemia or later with manifestations of anemia and/or those of parenchymal iron overload.4 However, the phenotypic expression of XLSA is highly variable,5 and occasional patients, both males and females, may present late in life.6,7 Distinctive laboratory features are microcytic anemia with hypochromic red cells, increased red cell distribution width and evidence of parenchymal iron overload: for a conclusive diagnosis of XLSA, however, identification of the ALAS2 mutation is required. The management of XLSA involves not only treatment of anemia, but also prevention and treatment of iron overload, family studies to identify additional at-risk individuals, and genetic counseling.1 Most patients with XLSA are responsive, to some extent, to pyridoxine, and subjects with iron overload can safely undergo mild phlebotomy programs under pyridoxine supplementation.
Patients affected with other inherited forms of sideroblastic anemia are not responsive to pyridoxine, and the molecular basis of these autosomal recessive disorders has been clarified only recently. Camaschella et al.8 studied a middle-aged anemic patient with ring sideroblasts and iron overload whose anemia was partially reversed by iron chelation therapy. The phenotype of this patient resembled that of the shiraz zebrafish, a mutant resulting from a large deletion encompassing the GLRX5 gene.9 In fact, sequencing of GLRX5 showed that the patient had a homozygous mutation of this gene. GLRX5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts.10
Two years ago, Guernsey et al.11 studied three Canadian families, each with one child affected by congenital sideroblastic anemia. The data available were inconsistent with an X-linked recessive inheritance, while the families derived from a local subpopulation isolate, consistent with a possible genetic founder effect. A single nucleotide polymorphism-based genome-wide scan performed in individuals belonging to these families led to the identification of SLC25A38 as the mutant gene responsible for this type of autosomal recessive pyridoxine-refractory sideroblastic anemia.11 Studies on additional subjects with familial or sporadic congenital sideroblastic anemia without ALAS2 mutations showed multiple additional biallelic SLC25A38 mutations. SLC25A38 encodes the erythroid specific mitochondrial carrier protein, which is important for the biosynthesis of heme in eukaryotes.
Following the identification of mutant SLC25A38 as a novel cause of inherited sideroblastic anemia, Bergmann et al.12 systematically analyzed a cohort of 60 previously unreported patients with congenital sideroblastic anemia, looking for ALAS2, SLC25A38, PUS1, GLRX5, and ABCB7 mutations. Twelve probands had biallelic mutations in SLC25A38, while 7 had ALAS2 mutations and one had a novel homozygous null PUS1 mutation.
In this issue of the journal, Kannengiesser et al.13 report on a study of 24 patients with congenital sideroblastic anemia who did not have ALAS2 mutations. Eleven patients of several different ancestral origins carried SLC25A38 mutations: 9 patients were homozygous and 2 were compound heterozygotes. All patients required blood transfusions that inevitably became regular within the first few years of life. Two patients underwent allogeneic stem cell transplantation with complete correction of anemia. Since the clinical course of congenital sideroblastic anemia associated with SLC25A38 mutations is very similar to that of thalassemia major, conservative therapy includes regular red cell transfusion and iron chelation. However, as in thalassemia major, allogeneic stem cell transplantation represents the only curative therapy at present, and should, therefore, be considered for young patients with this congenital sideroblastic anemia.
Refractory anemia with ring sideroblasts
What are the implications of recent advances in our understanding of the molecular basis of congenital sideroblastic anemia for the acquired forms, i.e. refractory anemia with ring sideroblasts (RARS) and its variant with marked thrombocytosis (RARS-T)?
RARS is a myelodysplastic syndrome characterized by isolated anemia, erythroid dysplasia only, less than 5% blasts, and 15% or more ring sideroblasts in the bone marrow.14 The natural history of RARS is characterized by an initial phase of erythroid hyperplasia and ineffective erythropoiesis, which is usually stable for many years but in a proportion of patients may be followed by a phase of marrow failure, with or without the later emergence of leukemic blasts.15,16 Since the vast majority of patients with this syndrome have no cytogenetic abnormalities, the clonal nature of RARS has been questioned. However, a few studies of X-chromosome inactivation patterns performed in female patients have suggested that RARS derives from the clonal proliferation of a multipotent hematopoietic stem cell with the potential for myeloid and lymphoid differentiation.17
Unfortunately none of the candidate genes, i.e. genes mutated in the different types of congenital sideroblastic anemia, has been found to be mutated in RARS. Of note, CD34+ cells from patients with RARS have a particular gene expression profile characterized by upregulation of mitochondria-related genes and, in particular, genes involved in heme synthesis (e.g., ALAS2),18 and reduced expression of ABCB7, a gene encoding a protein involved in the transport of iron/sulfur clusters from mitochondria to the cytoplasm.19 In addition, RARS is characterized by over-expression of mitochondrial ferritin (Figure 2D), encoded by the FTMT gene.3,20–22
RARS-T is a myelodysplastic/myeloproliferative neoplasm characterized by anemia with ring sideroblasts and marked thrombocytosis.23 Our recent studies suggest that RARS-T is indeed a myeloid neoplasm with both myelodysplastic and myeloproliferative features at the molecular and clinical levels, and that it may develop from RARS through the acquisition of somatic mutations of JAK2, MPL, or other as-yet-unknown genes.24
Thus, the available evidence suggests that the clonal hematopoiesis of RARS and RARS-T is associated with abnormal expression of several genes of heme synthesis and mitochondrial iron processing. Identifying the somatic mutation(s) that can be responsible for these abnormalities represents the current challenge in this field.
Conclusions
The two most common forms of congenital sideroblastic anemia, i.e. the X-linked form due to an ALAS2 mutation and the autosomal recessive form due to SLC25A38 mutations, have similar hematologic pictures but completely different clinical courses. Overall, XLSA is a benign disorder that generally responds to pyridoxine with substantial amelioration of anemia; prevention and treatment of iron overload is also important and can be generally achieved through phlebotomy. By contrast, the congenital autosomal recessive congenital sideroblastic anemia due to SLC25A38 mutations is a severe disease, not responsive to pyridoxine and with a clinical course very similar to that of thalassemia major: allogeneic stem cell transplantation should, therefore, be considered in young patients with this disease.
Supplementary Material
Footnotes
(Related Original Article on page 808)
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Cazzola M, Invernizzi R. Sideroblastic anemias. In: Young NS, Gerson SL, High KA, editors. Clinical Hematology. Philadelphia: Mosby Elsevier; 2005. pp. 721–32. [Google Scholar]
- 2.Mufti GJ, Bennett JM, Goasguen J, Bain BJ, Baumann I, Brunning R, et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93(11):1712–7. doi: 10.3324/haematol.13405. [DOI] [PubMed] [Google Scholar]
- 3.Cazzola M, Invernizzi R, Bergamaschi G, Levi S, Corsi B, Travaglino E, et al. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood. 2003;101(5):1996–2000. doi: 10.1182/blood-2002-07-2006. [DOI] [PubMed] [Google Scholar]
- 4.Cotter PD, May A, Li L, Al-Sabah AI, Fitzsimons EJ, Cazzola M, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood. 1999;93(5):1757–69. [PubMed] [Google Scholar]
- 5.Cazzola M, May A, Bergamaschi G, Cerani P, Ferrillo S, Bishop DF. Absent phenotypic expression of X-linked sideroblastic anemia in one of 2 brothers with a novel ALAS2 mutation. Blood. 2002;100(12):4236–8. doi: 10.1182/blood-2002-03-0685. [DOI] [PubMed] [Google Scholar]
- 6.Cotter PD, May A, Fitzsimons EJ, Houston T, Woodcock BE, al-Sabah AI, et al. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J Clin Invest. 1995;96(4):2090–6. doi: 10.1172/JCI118258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cazzola M, May A, Bergamaschi G, Cerani P, Rosti V, Bishop DF. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96(13):4363–5. [PubMed] [Google Scholar]
- 8.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110(4):1353–8. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 9.Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436(7053):1035–9. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 10.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120(5):1749–61. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41(6):651–3. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 12.Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54(2):273–8. doi: 10.1002/pbc.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannengiesser C, Sanchez M, Sweeney M, Hetet G, Kerr B, Moran E, et al. Missense SLC25A38 variations play an important role in autosomal recessive inherited sideroblastic anemia. Haematologica. 2011;96(6):808–13. doi: 10.3324/haematol.2010.039164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunning RD, Orazi A, Germing U, Le Beau MM, Porwit A, Bauman I, et al. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 88–93. [Google Scholar]
- 15.Cazzola M, Barosi G, Gobbi PG, Invernizzi R, Riccardi A, Ascari E. Natural history of idiopathic refractory sideroblastic anemia. Blood. 1988;71(2):305–12. [PubMed] [Google Scholar]
- 16.Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005;23(30):7594–603. doi: 10.1200/JCO.2005.01.7038. [DOI] [PubMed] [Google Scholar]
- 17.Malcovati L, Brisci A, Gallì A, Bruno F, Travaglino E, Pellagatti A, et al. Mutation analysis of TET2 reveals the clonal nature of refractory anemia with ring sideroblasts. Blood. 2010;116(21):776a. Abstract 1862. [Google Scholar]
- 18.Pellagatti A, Cazzola M, Giagounidis AA, Malcovati L, Porta MG, Killick S, et al. Gene expression profiles of CD34+ cells in myelodysplastic syndromes: involvement of interferon-stimulated genes and correlation to FAB subtype and karyotype. Blood. 2006;108(1):337–45. doi: 10.1182/blood-2005-12-4769. [DOI] [PubMed] [Google Scholar]
- 19.Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS ONE. 2008;3(4):e1970. doi: 10.1371/journal.pone.0001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tehranchi R, Invernizzi R, Grandien A, Zhivotovsky B, Fadeel B, Forsblom AM, et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood. 2005;106(1):247–53. doi: 10.1182/blood-2004-12-4649. [DOI] [PubMed] [Google Scholar]
- 21.Della Porta MG, Malcovati L, Invernizzi R, Travaglino E, Pascutto C, Maffioli M, et al. Flow cytometry evaluation of erythroid dysplasia in patients with myelodysplastic syndrome. Leukemia. 2006;20(4):549–55. doi: 10.1038/sj.leu.2404142. [DOI] [PubMed] [Google Scholar]
- 22.Nikpour M, Pellagatti A, Liu A, Karimi M, Malcovati L, Gogvadze V, et al. Gene expression profiling of erythroblasts from refractory anaemia with ring sideroblasts (RARS) and effects of G-CSF. Br J Haematol. 2010;149(6):844–54. doi: 10.1111/j.1365-2141.2010.08174.x. [DOI] [PubMed] [Google Scholar]
- 23.Vardiman JW, Bennett JM, Bain BJ, Baumann I, Thiele J, Orazi A. Myelodysplastic/myeloproliferative neoplasms, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 85–6. [Google Scholar]
- 24.Malcovati L, Della Porta MG, Pietra D, Boveri E, Pellagatti A, Galli A, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114(17):3538–45. doi: 10.1182/blood-2009-05-222331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.