

Abstract
Background
In a study of childhood acute lymphoblastic leukemia (CoALL 06-97 study), the in vitro sensitivity of the patients’ cells to prednisolone, vincristine and asparaginase was introduced as a new additional risk parameter for treatment stratification. In parallel in vivo treatment response was assessed by determining the presence and extent of minimal residual disease in a subset of patients (n=224). Here we report the long-term impact of in vitro sensitivity-based risk stratification according to survival and compare the results of in vitro sensitivity with in vivo response.
Design and Methods
Patients with a sensitive in vitro profile were treated with a reduced intensity protocol (n=167) whereas patients defined as low risk according to conventional parameters but with a resistant in vitro profile were given intensified therapy (n=47).
Results
At a median follow-up of 6.8 years event-free survival was 0.80±0.03 for patients with a sensitive profile, 0.73±0.03 for those with an intermediate profile and 0.67±0.08 for those with a resistant profile (P=0.015). Overall, the treatment results of the cases stratified according to in vitro sensitivity were similar to those of the historical control group stratified based on conventional risk factors. Minimal residual disease at the end of induction was a strong predictor of outcome in B-precursor and T-cell acute lymphoblastic leukemia. There was no correlation between in vitro and in vivo treatment response in B-precursor leukemia (Spearman’s r=0.13; P=0.15) in contrast to T-cell acute lymphoblastic leukemia (Spearman’s r=0.63; P<0.001)
Conclusions
A moderate reduction in treatment intensity for patients with a sensitive in vitro profile was possible without jeopardizing treatment outcome. However, in vitro drug testing was affected by a decrease in risk predictive power over time and was not correlated with in vivo assessment of minimal residual disease in B-precursor acute lymphoblastic leukemia. It was, therefore, abandoned in favor of the assessment of in vivo response in subsequent CoALL trials.
Keywords: acute lymphoblastic leukemia, in vitro drug testing, risk-adapted treatment, childhood, minimal residual disease
Introduction
Traditionally, antileukemic therapy has been adjusted on the basis of risk factors such as age, white blood cell count, immunological phenotype, specific chromosomal alterations and response to therapy. In vitro1–4 and in vivo5–8 drug resistance testing, e.g. by a colorimetric-based 3-[4.5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay or a fluorescence-based fluorometric micro-culture cytotoxicity assay, was established with the aim of strengthening the power of risk prediction in childhood acute lymphoblastic leukemia (ALL).
Several groups have studied the prognostic value of drug resistance in children with newly diagnosed ALL. Resistance to single agents such as glucocorticoids, vinca alkaloids, L-asparaginase and anthracyclines was shown to be associated with unfavorable immunophenotypic and cytogenetic subtypes of ALL as well as an older age of patients.9,10 Furthermore, resistance of leukemic cells to single agents predicted unfavorable short-term (induction failure) and long-term (relapse) clinical outcome.1,11–13
A drug resistance profile, combining resistance data on three drugs, was shown to predict clinical outcome better than resistance to single agents.3,11,13,14 A profile combining in vitro cellular resistance to prednisolone, vincristine and L-asparaginase (PVA) was most discriminative for clinical outcome. In Japanese, Polish and Dutch studies, patients with a resistant profile suffered from induction failure or relapse more often than did patients with an intermediate or sensitive profile. In multivariate analyses, the predictive value of the drug resistance profile remained highly significant suggesting a potential use as a prognostic factor for newly diagnosed ALL of childhood.3,12,14
The value of a drug resistance profile as a risk predictor was confirmed in a German cohort of patients treated according to the CoALL 05-92 protocol. In this study, the MTT assay was used to determine the patients’ in vitro responsiveness towards PVA. A risk was assigned in 202 cases and very low-risk and a high-risk groups were identified using previously determined criteria for resistance.15,16
Consequently, in vitro resistance to drugs was used in the CoALL 06-97 study as an additional risk parameter for treatment stratification, allocating patients with a very sensitive profile to reduced therapy and patients with a resistant profile to more intensive treatment.
The PVA score as well as age, initial white blood cell (WBC) count and immunophenotype have the advantage that they are obtained upfront at diagnosis and therefore allow for early adjustment of therapy. However, over the past years it became increasingly clear that in the interplay between host and leukemia, not only biological characteristics of the malignant cells but also host factors might critically influence treatment response and outcome. Techniques with sufficient sensitivity and specificity to detect submicroscopic levels of leukemic cells (minimal residual disease, MRD) have been developed. Several studies showed that regular assessment of MRD by polymerase chain reaction (PCR) analysis or flow cytometry does indeed facilitate the identification of patients with a high or low risk of relapse.5–7,17,18 While both MRD and the PVA score have been used for treatment stratification in different clinical trials, there are only two small retrospective19,20 and one prospective study21 investigating a potential relationship between MRD and in vitro chemoresistance. They revealed a weak correlation between MRD and response to single drugs such as prednisolone and doxorubicin.21 Here we describe the relevance of the in vitro response to treatment (evaluated by the PVA score) and the in vivo response (assessed by the presence and degree of MRD) for predicting outcome in a large cohort of 224 children uniformly treated according to the CoALL 06-97 protocol.
Design and Methods
Assessment of the PVA score
At diagnosis leukemia samples from bone marrow and/or peripheral blood were tested for in vitro cytotoxicity based on the MTT assay which has been described elsewhere in detail.16 Briefly, leukemic cells were incubated in the absence (control) or presence of six different concentrations of drugs in duplicate (prednisolone: range, 0.06 to 250 μg/mL; vincristine: range, 0.05 to 50 μg/mL; asparaginase: range, 0.003 to 10 IU/mL). After 4 days of culture at 37ºC in humidified air containing 5% CO2, 0.45 mg/mL of MTT were added. Formazan crystals, produced by viable cells only, were dissolved in acidified isopropanol and quantified by spectrophotometry at 562 nm. The in vitro cytotoxicity of prednisolone, vincristine and L-asparaginase was determined. For each patient a three-drug resistance profile was generated that combined the results of the individually tested drugs into one score, i.e. the PVA score, as described previously.16 In brief, marked sensitivity towards a drug was scored as 1, intermediate sensitivity was scored as 2 and resistance was scored as 3. Combining the separate scores for prednisolone, vincristine and L-asparaginase of each patient resulted in an individual PVA score that varied between 3 (sensitive to all three drugs) and 9 (resistant to all three drugs).
Analysis of minimal residual disease
For MRD analysis, DNA was isolated from bone marrow mononuclear cells obtained at diagnosis, day 15 of the induction phase, at the end of induction (day 29) and at week 12 for low-risk patients and at week 15 for high-risk patients.
PCR studies for MRD analysis were performed with immunogloblulin heavy chain (IGH) and T-cell receptor (TCR) gene rearrangements as targets. DNA samples from diagnosis were screened for clonal products using the BIOMED-2 primers for the Igκ deleting element (Kde), complete and incomplete TCRδ, TCRγ and TCRβ gene rearrangements. IGH rearrangements were identified using seven VH region and one JH consensus primer. Products were analyzed for clonality on 6% acrylamide gels and positive products were further processed. Junctional regions of clonal products were sequenced directly and patient-specific junctional regions were identified for generation of allele-specific PCR primers. Biclonal or biallelic products were cloned using the TOPO-TA cloning kit (Invitrogen) and then processed adequately for generation of suitable patient-specific primers. Subsequently PCR-MRD targets were tested for specificity and sensitivity to reach a sensitivity and a quantifiable range of 1×10−4 for at least two targets. Real-time quantitative PCR was performed and interpreted according to the guidelines developed within the European Study Group for MRD detection in ALL (ESG-MRD ALL).22 In detail, sequence-specific TaqMan probes were used on a LC480 machine (Roche). Tenfold serial dilutions of diagnostic DNA were prepared in pooled peripheral blood DNA extracted from at least five healthy donors. Quantification was performed using this standard curve and triplicate follow-up samples including 500 ng DNA in each reaction.
Patients
Between August 4, 1997 and June 30, 2003 752 patients younger than 18 years with newly diagnosed ALL from 18 cooperating centers in Germany were registered in the CoALL 06-97 study. Eighty-five patients were excluded: 19 patients because they were younger than 1 year at diagnosis and were treated according to the international Interfant study, 36 patients because they had non-Hodgkin’s lymphoma and were not stratified according to the results of in vitro cytotoxicity testing and 30 patients not eligible for this study because of pre-treatment (n=14), other treatment following the parents’ or physicians’ decision (n=8), ALL being a second malignancy (n=5), or an additional major medical condition preventing therapy according to the protocol (n=3). Thus 667 patients were eligible and evaluable for the study. The study was approved by competent authorities and ethics committees. Written informed consent was obtained from all parents or guardians.
A first risk stratification comprised the traditional risk criteria: age, WBC count, immunophenotype, response to induction and chromosomal rearrangements. Patients older than 10 years, with T-lineage or pro-B ALL, an initial WBC of 25×109/L or more, no remission at day 29 after induction therapy, or translocation 4;11 or 9;22 were considered at high risk. All other patients were allocated to the low-risk group. After achieving remission a second stratification was performed according to the results of the in vitro drug resistance testing (Figure 1).
Figure 1.

Stratification in CoALL 06-97 study according to WBC count, age, immunophenotype and PVA score.
A PVA score of 8 or 9 in the high-risk group constituted an indication for stem cell transplantation provided that a matched sibling donor was available. Patients in whom the score could not be determined were allocated to low-risk or high-risk therapy on the basis of traditional risk criteria. The characteristics of the patients are summarized in Online Supplementary Table S1.
Treatment
The complete treatment is outlined in Online Supplementary Table S2. Apart from treatment modifications according to the second stratification based on the PVA score there were no other differences between the CoALL 92 and CoALL 97 therapy. All patients received the same prephase treatment with daunorubicin and one dose of intrathecal methotrexate. During induction, vincristine and daunorubicin were given weekly and methylpred-nisolone was given daily.
Low-risk patients
All low-risk patients received identical consolidation therapy, followed by 6-mercaptopurine daily for 4 weeks and four doses of intrathecal methotrexate (central nervous system phase). In reinduction for ‘low-risk reduced’ patients the doses of adriamycin and dexamethasone were halved (Figure 2). Thus ‘low-risk reduced’ patients received only one dose of adriamycin and 7 days of dexamethasone, whereas ‘low-risk standard’ patients received two doses of adriamycin and 14 days of dexamethasone. All low-risk patients received the second part of reinduction (Figure 2).
Figure 2.

Treatment in reinduction for low- and high-risk patients depending on PVA stratification according to PVA score.
High-risk and ‘low-risk intensified’ patients
‘Low-risk intensified’ and ‘high-risk standard’ patients received the same treatment. All patients were given identical consolidation therapy, followed by daily 6-mercaptopurine for 4 weeks and four doses of intrathecal methotrexate. Patients with central nervous system involvement at diagnosis received cranial irradiation (18 Gy), patients with T-ALL and patients with B-precursor ALL and an initial WBC of 100×109/L or more at diagnosis received prophylactic cranial irradiation (12 Gy). Since 2001 irradiation has been omitted for patients with B-precursor ALL and an initial WBC count between 100×109/L and 200×109/L, if they have less than 1×109/L blasts in the peripheral blood 7 days after the first dose of daunorubicin. In analogy to the management of ‘low-risk reduced’ patients with a PVA score of 3 or 4, doses of adriamycin and dexamethasone were halved for the ‘high-risk reduced’ patients during reinduction (Figure 2). As a consequence, ‘high-risk reduced’ patients received two doses of adriamycin and 14 days of dexamethasone, whereas ‘high-risk standard’ and ‘low-risk intensified’ patients received four doses of adriamycin and 28 days of dexamethasone. All high-risk patients received the second part of reinduction. In February 2003, analysis of the patients in the high-risk group as a whole showed that these patients had an inferior outcome compared to that of patients in the CoALL 05-92 study. During the remaining study period, 12 high-risk patients with a PVA score of 3 or 4 were, therefore, treated with ‘high-risk standard’ therapy. Patients who failed to achieve remission by day 29, or those with a t(4;11) or t(9;22) translocation were eligible for stem cell transplantation in first remission. No treatment intervention was made according to the result of MRD assessment.
Maintenance therapy, consisting of daily 6-mercatopurine and weekly methotrexate per os, was administered until 104 weeks after diagnosis. Three, 6 and 9 months after the start of maintenance therapy all patients who were not given central nervous system irradiation received two doses of intrathecal methotrexate.
Statistical analysis
Each year patients’ data were collected in protocol-specific forms and reviewed before data entry by the trial data monitoring board, which also performed the statistical analysis. Probability of event-free survival (pEFS) was estimated according to Kaplan and Meier and compared between subgroups using the log-rank test.23 Event-free survival was defined as the time from diagnosis to the first outcome event, which was failure to achieve remission (no remission until day 56), death during induction, relapse, death from any cause in remission or a second malignancy. Disease-free survival was defined as the time from complete remission to first relapse. The cumulative incidence of relapse was calculated by the method of Kalbfleisch and Prentice and compared using Gray’s test.24 Survival estimates are given as 10-year estimates with standard error (SE). Cox regression analysis was used for multivariate analysis of pEFS.25 The models for multiple analyses included age (cut-off 10 years), WBC (cut-off 25×109/L), immunophenotype (pro-B-ALL and T-ALL versus common/pre-B-ALL), and the additional high-risk criteria grouped into one covariate (other high-risk criteria i.e. induction failure – no remission at day 29- or presence of the chromosomal translocation t(9;22) or an MLL rearrangement), level of MRD at the end of induction and the drug resistance profile based on the PVA score as covariates. The χ2 test, Fischer’s exact test and Spearman’s rank correlation analyses were applied to compare the distribution of parameters between subgroups and correlation between parameters.
Results
Overall treatment results
The pEFS at 6 years and 10 years of follow-up for all 667 patients was 0.77±0.02 and 0.75 ±0.02, respectively. The median follow-up of patients at risk was 6.8 years (range, 5.1–11.0 years). Between LR and HR patients the prognosis of low-risk and high-risk patients differed significantly (10-year pEFS 0.81±0.02 versus 0.67±0.03; P=0.001) as did the incidence of relapse (0.16±0.02 versus 0.25±0.03; P=0.001).
There were no significant differences in outcome and incidence of relapse between patients with or without a measurable PVA score (10-year pEFS 0.73±0.02 versus 0.76±0.03; P=0.985).
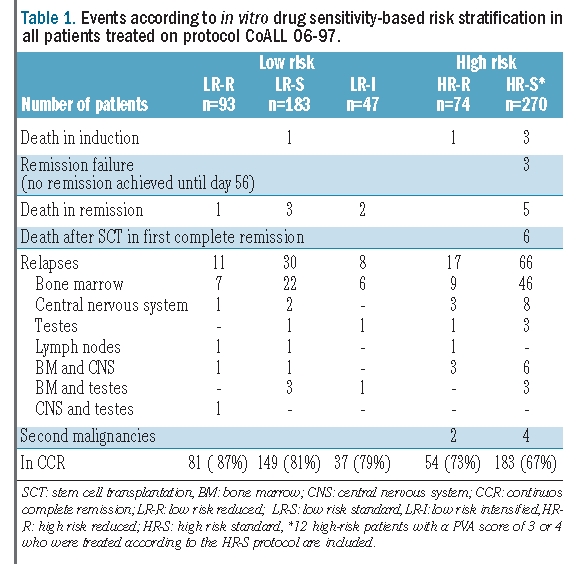
Five of 667 children (0.75%) died during induction therapy and nine patients (1.3%; 3 low-risk and 6 high-risk patients) died on therapy in remission due to infectious complications. Thirty-seven patients underwent stem cell transplantation, 12 of whom received a graft from a matched related donor and 25 of whom were transplanted from a matched unrelated donor. Only one high-risk patient received a stem cell transplant based on a PVA score of 8 without other criteria qualifying for transplantation. Six high-risk patients died after stem cell transplantation in first remission due to therapy-related complications and two patients died several years after completion of therapy due to accidents. Thirty-six patients were not in remission on day 29 but had achieved remission by day 56. All these patients were treated according to the ‘high-risk standard’ protocol. Remission failure, defined as no remission on day 56, was only seen in three high-risk patients. Six patients developed a second malignancy. The distribution of events is presented in Table 1.
Table 1.
Events according to in vitro drug sensitivity-based risk stratification in all patients treated on protocol CoALL 06-97.
Success rate and distribution of the PVA scores
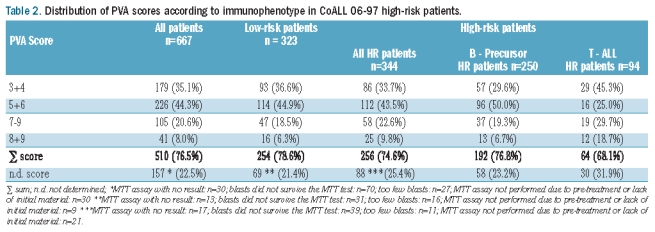
The PVA score could be determined in 510 of 667 patients (Table 2). The main reasons for failure to be able to assign a score were a low fraction of blasts in the bone marrow or peripheral blood and blasts not surviving 4 days of culture. The proportions of low-risk and high-risk patients in whom the score could be determined were similar. Patients for whom a PVA score could be determined did not differ from patients without a score with respect to gender, age, initial WBC count and immunophenotype (data not shown). The distributions of the scores between low-risk and high-risk patient with B-precursor ALL were similar (χ2 P=0.33). There was a significant difference in score distribution between patients with B-precursor ALL and T-ALL high-risk patients (P=0.0004). TALL patients presented more frequently with the very ends of the PVA score, i.e. a score of 3 or 4 was found in 45.3% of T-ALL patients compared to in 29.6% of patients with B-precursor ALL whereas a score of 8 or 9 was found in 18.7% of T-ALL patients and in 6.7% of B-precursor ALL patients (Table 2). There was a significant trend to higher PVA scores in patients who failed to achieve remission at the end of induction than in patients in remission on day 29 (χ2 P=0.0001). Fourteen of the 39 patients (35%) in whom induction failed had a PVA score of 7 or more compared to 15% within the whole group and only four out of 39 patients (10%) had a PVA score of 3 or 4 compared to 25% of the patients in the entire group.
Table 2.
Distribution of PVA scores according to immunophenotype in CoALL 06-97 high-risk patients.
Treatment outcome of patients stratified by PVA profile
Figure 3 shows the 10-year pEFS of patients with a sensitive PVA profile (score 3 or 4; pEFS 0.80±0.03), an intermediate PVA profile (score 5 or 6, pEFS 0.73±0.03) and a resistant PVA profile (score 7, 8 or 9; pEFS 0.67±0.05). The difference between patients with a PVA score of 3 or 4 and that of patients with a score of 7, 8 or 9 is statistically significant (P=0.015). This is mainly due to the worse prognosis of high-risk patients with a score of 7–9 (pEFS 0.57±0.07). In contrast, the prognosis of low-risk patients with a score of 7–9 who received intensified treatment (pEFS 0.79±0.06) was similar to that of low-risk patients with a score of 3 or 4 (pEFS 0.87±0.04) or a score of 5 or 6 (pEFS0.80±0.03). High-risk patients with a favorable score (3 or 4) had a significantly better outcome than patients with higher scores (7–9) (pEFS: 0.71±0.05 versus 0.57±0.07, respectively; P=0.05).
Figure 3.

Ten-year probability of event-free survival in CoALL 06-97 patients according to PVA score; 167 patients with PVA score 3 or 4 versus 226 patients with PVA score 5 or 6 versus 105 patients with PVA score 7–9 (in light gray 41 patients with PVA score 8 or 9) (log-rank test for patients with score 3+4 versus 8+9 P=0.01)
In vitro sensitivity testing and outcome in CoALL 05-92 compared to CoALL 06-97
Two-hundred and two patients from the CoALL 05-92 study and 510 from the CoALL 06-97 study with an informative PVA score were available for a comparative analysis.
The overall outcome in both studies, which had an identical treatment backbone, was similar with a 10-year pEFS of 0.75±0.02 in CoALL 06-97 versus 0.76±0.03 in CoALL 05-92 (P=0.95). Analyses of subgroups with different PVA scores (3+4, 5+6 and 7–9) showed no statistically significant differences in the pEFS between the two studies, either for low-risk and high-risk patients combined or in separate analyses (Online Supplementary Table S3). Notably 167 patients (32%) with a score of 3 or 4 and reduced therapy in the CoALL 06-97 study fared equally well as 39 patients with a score of 3 or 4 without therapy reduction in the CoALL 95-92 study (pEFS 0.79 ±0.03 versus 0.81±0.06; P=0.81). Therapy intensification for 47 low-risk patients with a resistant PVA profile (score 7–9) resulted in a 10-year pEFS of 0.79 (SE=0.06) compared to 0.75 (SE=0.12) for 12 patients in the CoALL 05-02 study with a resistant profile (P=0.76).
Predictive value of resistance to L-asparaginase
We also looked at the prognostic value of sensitivity to each single drug. The individual scores for asparaginase and prednisolone but not for vincristine were of prognostic value. The overall population of patients, as well as low-risk and high-risk patients separately, had a significantly better outcome with a sensitive score of 1 for asparaginase compared to patients with an asparaginase score of 2 or 3 (P= 0.0037 for score 1 versus 2, P<0.0001 for score 1 versus 3) (Figure 4). With regard to the pharmacological sensitivity towards prednisolone a score of 1 was associated with a higher pEFS than the pEFS in patients with a resistant score of 3 (P=0.0498 for score 1 versus 3) whereas score 2 did not discriminate between the groups (Online Supplementary Figure S1A,B)
Figure 4.

Ten-year probability of event-free survival in CoALL 06-97 patients according to asparaginase (ASP) score (log-rank test: PVA score 1 versus 2 P=0.0037, score 1 versus 3 P=0.0001).
Assessment of minimal residual disease
PCR-based measurements of MRD were undertaken during the study period to evaluate whether the in vitro sensitivity towards the three selected drugs was also reflected by the in vivo response to induction therapy. MRD was assessed in 224 patients (68/94 (72%) with TALL and 156/573 (27%) with B-precursor ALL. The subset of patients with B-precursor ALL in whom MRD was measured did not differ from the whole cohort with respect to gender, age, initial WBC count or chromosomal translocation.
Prognostic value of minimal residual disease
MRD at the end of induction was a strong predictor of outcome. Among the 153 with B-precursor ALL, 59 (39%) were MRD-negative at day 29 with an incidence of relapse of 0.09 (SE=0.04), whereas the risk of relapse in patients with a MRD load of 10−3 or more (39/153, 25%) was significantly higher at 0.33 (SE=0.08) (P=0.002). Patients who had a detectable MRD load of less than 10−3 had a relapse rate of 0.24 (SE=0.06) (Figure 5A). Among the patients with T–ALL no difference in outcome was seen between patients with a negative or low MRD load (<10−3) at day 29. However, patients with a high MRD load (≥10−3) on day 29 had a significantly worse outcome than that of patients with a MRD load less than 10−3 on day 29 (relapse incidence 0.41, SE=0.08 versus 0.18, SE=0.07; P=0.02) (Figure 5B).
Figure 5.

Incidence of relapse in CoALL 06-97 patients with MRD measurement (A) Incidence of relapse according to MRD level at the end of induction in patients with B-precursor ALL. (B) Incidence of relapse according to MRD level at the end of induction in T–ALL patients.
After consolidation treatment (at week 12 in the low-risk arm and week 15 in the high-risk arm), a sustained positive MRD signal was detected predominantly in TALL patients. Twelve of 46 T–ALL patients, in whom MRD was assessed at this time point, had a MRD level greater than 10−4. Among these 12 patients with a MRD signal greater than 10−4 at this late time point, five suffered from relapsing disease. Nine of 63 patients with B-precursor ALL showed a positive MRD signal, three of whom had a relapse.
Correlation between in vitro PVA score and in vivo assessment of minimal residual disease
No association was found between PVA score and MRD level at day 15 or 29 in patients with B-precursor ALL (Spearman’s correlation coefficient day 15: r=0.13, P=0.17; day 29: r=0.13, P=0.15) in contrast to T-ALL patients (correlation coefficient day 15: r=0.57, P<0.0001; day 29: r=0.63, P<0.0001) (Figure 6A,B). When comparing the in vitro sensitivity towards the single drugs with induction MRD levels, a strong correlation was found between MRD levels on days 15 and 29 and asparaginase score only in T-ALL patients (χ2 0.00004).
Figure 6.

Correlation of MRD level at the end of induction and PVA score. (A) Correlation of MRD level at the end of induction and PVA score in T–ALL patients. (B) Correlation of MRD level at the end of induction and PVA score in patients with B-precursor ALL.
Multivariate analyses
When a Cox regression analysis of event-free survival was applied taking into account the PVA score and traditional risk factors, it was found that initial WBC count and high-risk features, such as induction failure and adverse chromosomal translocations, had prognostic significance. A PVA score of 3 or 4 was of borderline significance (P=0.05). The Cox regression analysis of the scores for single agents showed a strong prognostic power for the asparaginase score (P=0.0001); initial WBC count and high-risk features retained statistical significance. When integrating MRD in this model only low MRD levels at the end of induction (day 29) remained significant (P=0.01) (Table 3A,B).
Table 3A.
Cox regression analysis of event-free survival.
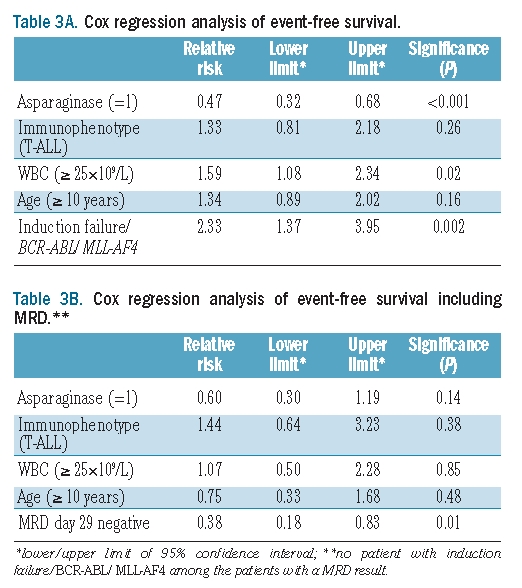
Table 3B.
Cox regression analysis of event-free survival including MRD.**
Discussion
Since the introduction of the first antileukemic agent by Sidney Farber in 1948,27 cure rates for childhood ALL have increased steadily.28–32 However, the intensity of treatment protocols has reached a high level, and there has been concern that a substantial proportion of patients is being overtreated due to the lack of a refined risk assignment which relies on traditional risk factors such as initial white blood count, age, immunophenotype and chromosomal translocations. In 1991 the group of Pieters et al.1 demonstrated the prognostic value of an in vitro assay testing the sensitivity of leukemic cells to drugs. Subsequent reports from this group2–4 and others11–13 confirmed that a simple and inexpensive assay is useful for allocating patients to different risk categories. The CoALL 05-92 study prospectively evaluated the value of the MTT assay in a cohort of 202 patients; after 5 years the probability of disease-free survival for patients with a PVA score of 3 or 4 was 0.93±0.05 versus 0.5±0.12 for patients with a score of 8 or 9 (P=0.01, unpublished data). This finding prompted us to introduce the in vitro drug resistance score as an additional parameter for risk assignment in the subsequent CoALL 06-97 trial. Given the low rate of accrual of 110 patients per year this could not be done in a randomized fashion.
Patients with a sensitive PVA profile were candidates for therapy reduction irrespective of the assigned risk arm. In contrast, therapy was intensified in patients with a resistant PVA profile, but only in low-risk patients because a further escalation of dose intensity did not seem feasible in high-risk patients.
A moderate reduction in therapy was performed in a substantial number of patients with a sensitive PVA profile, with this number being three times greater than the number of patients receiving treatment intensification. Although treatment intensity was adjusted according to the PVA score in the CoALL 06-97 study, patients with a sensitive PVA profile nevertheless had a better outcome than that of patients with an intermediate or resistant PVA profile. Patients from the CoALL 06-97 study and those from the predecessor CoALL 05-92 trial with a sensitive PVA score (3 or 4) had a similar long-term pEFS, although the former patients had received therapy reduction. The reduced intensity regimen resulted in a significant decline of the rate of infectious complications, which were more than halved in the low-risk group and diminished by 36% in the high-risk patients.26
A separate subgroup analysis of the CoALL 06-97 data revealed that low-risk patients undergoing intensified treatment according to the PVA score had an excellent outcome, which was not very different from that of the low-risk patients with sensitive or intermediate scores. The small number of patients in the previous study did not, however, allow for a test of equivalence.
With longer follow-up of the original CoALL 05-92 cohort it became apparent that the PVA score predicted early rather than late relapses leading to a decrement of the prognostic power of the score over time.16 This observation also holds true for the CoALL 06-97 study. The PVA score was of significant prognostic value exclusively in high-risk patients, who generally develop a relapse within the first 2 years after completion of therapy. However, since therapy was intensified only in low-risk patients with a high PVA score, treating them as high-risk patients, the prognostic impact of the PVA score in the low-risk group may disappear.
The prognostic impact of in vitro drug sensitivity testing per se is restricted by the selection of drugs. Analysis of the prognostic impact of sensitivity to the single drugs incorporated in the PVA score revealed that in vitro sensitivity towards asparaginase alone was still an independent predictor of relapse in both risk groups, suggesting that current therapy is not suitable for overcoming L-asparaginase resistance. The observation that the asparaginase score lost its predictive power when MRD was integrated in the multivariate analysis might be due to a selection of identical patients by both methodologies but a stronger predictive power of MRD. In vitro sensitivity towards prednisolone was associated with a superior prognosis only in a univariate analysis. A prognostic impact of in vitro sensitivity to vincristine could not be found. This suggests that the impact of resistance to these two drugs on outcome was lost upon treatment adaption. Previous studies showed that in vitro sensitivity to vincristine and prednisolone had a significant impact, but the number of patients included in these studies was substantially smaller than that in the present CoALL study, which may also account for the conflicting results.1–3,9
It is becoming increasingly clear that immunological control mechanisms play a pivotal role in the maintenance of long-term remission. Blasts that remain unrecognized by the immune system or hide in immunological niches may give rise to relapse. These host-specific factors are not adequately reflected by in vitro systems testing sensitivity to drugs, but might well contribute to the treatment response measured in vivo; MRD levels, in contrast, also account for host pharmacokinetics, pharmacodynamics and pharmacogenetics.
This comment is in line with data from a substantial number of studies showing that MRD levels within the first 4–5 weeks of treatment are highly predictive of the outcome of children with ALL.5–7,17,18 In patients with B-precursor ALL the NOPHO group observed a significant correlation between MRD level at day 15 (i.e. after induction) and in vitro sensitivity to prednisolone, vincristine and doxorubicin, drugs that were used in the induction therapy. In the CoALL 06-97 trial a correlation between in vitro drug resistance and in vivo response to chemotherapy could only be found in patients with T-ALL but not in those with B-precursor ALL. The PVA score as determined in the CoALL 05-92 and 06-97 trials comprises sensitivity against prednisolone, vincristine and asparaginase, but the last – not given in CoALL induction - was the only drug which strongly correlated with MRD in T-ALL patients.
In summary, treatment reduction in the CoALL 97 trial was feasible for patients with a sensitive in vitro drug testing profile without introducing an increased risk of relapse. Besides the immediate prevention of life-threatening infectious complications, this treatment reduction is anticipated to have a long-term benefit of reducing late adverse effects such as anthracycline-induced cardiotoxicity.
Surprisingly, no correlation was found between the in vitro response to drugs and in vivo MRD levels in patients with B-precursor ALL. Although in vitro drug testing is a simple, up-front and low cost diagnostic tool, it has been abandoned in favor of the more predictive, but somewhat sophisticated, in vivo evaluation of MRD in the ongoing CoALL trial which yields a clear cut and robust separation of risk groups.
Supplementary Material
Acknowledgments
We would like to thank all members of the COALL trial group for providing patient’s information and materials (Online Supplementary Table S4). The CoALL study group was supported by the ‘Fördergemeinschaft Kinderkrebszentrum Hamburg e.V.’
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Pieters R, Huismans DR, Loonen AH, Hahlen K, van der Does-van den Berg A, van Wering ER, et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet. 1991;338(8764):399–403. doi: 10.1016/0140-6736(91)91029-t. [DOI] [PubMed] [Google Scholar]
- 2.Kaspers GJ, Pieters R, Van Zantwijk CH, De Laat PA, De Waal FC, Van Wering ER, et al. In vitro drug sensitivity of normal peripheral blood lymphocytes and childhood leukaemic cells from bone marrow and peripheral blood. Br J Cancer. 1991;64 (3):469–74. doi: 10.1038/bjc.1991.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaspers GJ, Veerman AJ, Pieters R, Van Zantwijk CH, Smets LA, Van Wering ER, et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood. 1997;90(7):2723–9. [PubMed] [Google Scholar]
- 4.Kaspers GL, Veerman AJ, Pieters R, van Zantwijk I, Klumper E, Hählen K, et al. In vitro cytotoxicity of mitoxantrone, daunorubicin and doxorubicin in untreated childhood acute leukemia. Leukemia. 1994;8(1):24–9. [PubMed] [Google Scholar]
- 5.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352 (9154):1731–8. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Goldwasser MA, Li A, Dahlberg SE, Neuberg D, Wang H, Dalton V, et al. Dana-Farber Cancer Institute ALL Consortium. Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood. 2007;110(5):1607–11. doi: 10.1182/blood-2006-09-045369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.zur Stadt U, Harms DO, Schlüter S, Schrappe M, Goebel U, Spaar H, et al. MRD at the end of induction therapy in childhood acute lymphoblastic leukemia: outcome prediction strongly depends on the therapeutic regimen. Leukemia. 2001;15(2):283–5. doi: 10.1038/sj.leu.2402019. [DOI] [PubMed] [Google Scholar]
- 8.Silverman LB, Sallan SE. Newly diagnosed childhood acute lymphoblastic leukemia: update on prognostic factors and treatment. Curr Opin Hematol. 2003;10(4):290–6. doi: 10.1097/00062752-200307000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, et al. Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia-implications for treatment of infants. Leukemia. 1998;12(9):1344–8. doi: 10.1038/sj.leu.2401129. [DOI] [PubMed] [Google Scholar]
- 10.Ramakers-van Woerden NL, Pieters R, Loonen AH, Hubeek I, van Drunen E, Beverloo HB, et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood. 2000;96(3):1094–9. [PubMed] [Google Scholar]
- 11.Hongo T, Yajima S, Sakurai M, Horikoshi Y, Hanada R. In vitro drug sensitivity testing can predict induction failure and early relapse of childhood acute lymphoblastic leukemia. Blood. 1997;89(8):2959–65. [PubMed] [Google Scholar]
- 12.Kaspers GJ, Pieters R, Van Zantwijk CH, Van Wering ER, Van Der Does-Van Den Berg A, et al. Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood. 1998;92(1):259–66. [PubMed] [Google Scholar]
- 13.Frost BM, Nygren P, Gustafsson G, Forestier E, Jonsson OG, Kanerva J, et al. Increased in vitro cellular drug resistance is related to poor outcome in high-risk childhood acute lymphoblastic leukaemia. Br J Haematol. 2003;122(3):376–85. doi: 10.1046/j.1365-2141.2003.04442.x. [DOI] [PubMed] [Google Scholar]
- 14.Styczynski J, Wysocki M. Is the in vitro drug resistance profile the strongest prognostic factor in childhood acute lymphoblastic leukemia? J Clin Oncol. 2004;22(5):963–4. doi: 10.1200/JCO.2004.99.274. [DOI] [PubMed] [Google Scholar]
- 15.Janka-Schaub GE, Harms DO, den Boer ML, Veerman AJ, Pieters R. [In vitro drug resistance as independent prognostic factor in the study COALL-O5-92 Treatment of childhood acute lymphoblastic leukemia; two-tiered classification of treatments based on accepted risk criteria and drug sensitivity profiles in study COALL-06-97. Klin Padiatr. 1999;211(4):233–8. doi: 10.1055/s-2008-1043794. [DOI] [PubMed] [Google Scholar]
- 16.Den Boer ML, Harms DO, Pieters R, Kazemier KM, Gobel U, Korholz D, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21 (17):3262–8. doi: 10.1200/JCO.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 17.Stow P, Key L, Chen X, Pan Q, Neale GA, Coustan-Smith E, Mullighan CG, et al. Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood. 2010;115(23):4657–63. doi: 10.1182/blood-2009-11-253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–14. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 19.De Haas V, Kaspers GJ, Oosten L, Bresters D, Pieters R, Van Der Velden VH, et al. Is there a relationship between in vitro drug resistance and level of minimal residual disease as detected by polymerase chain reaction at the end of induction therapy in childhood acute lymphoblastic leukaemia? Br J Haematol. 2002;118(4):1190–200. doi: 10.1046/j.1365-2141.2002.36211.x. [DOI] [PubMed] [Google Scholar]
- 20.Schmiegelow K, Nyvold C, Seyfarth J, Pieters R, Rottier MM, Knabe N, et al. Post-induction residual leukemia in childhood acute lymphoblastic leukemia quantified by PCR correlates with in vitro prednisolone resistance. Leukemia. 2001;15 (7):1066–71. doi: 10.1038/sj.leu.2402144. [DOI] [PubMed] [Google Scholar]
- 21.Lönnerholm G, Thörn I, Sundström C, Frost BM, Abrahamsson J, Behrendtz M, et al. In vitro cellular drug sensitivity at diagnosis is correlated to minimal residual disease at end of therapy in childhood acute lymphoblastic leukemia. Leuk Res. 2009;33(1):46–53. doi: 10.1016/j.leukres.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 22.van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER, et al. European Study Group on MRD detection in ALL (ESG-MRD-ALL) Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21(4):604–11. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 24.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. New York, NY: Wiley; 2002. [Google Scholar]
- 25.Andersen PK, Borch-Johnsen K, Deckert T, Green A, Hougaard P, Keiding N, Kreiner S. A Cox regression model for the relative mortality and its application to diabetes mellitus survival data. Biometrics. 1985;41 (4):921–32. [PubMed] [Google Scholar]
- 26.Graubner UB, Porzig S, Jorch N, Kolb R, Wessalowski R, Escherich G, et al. Impact of reduction of therapy on infectious complications in childhood acute lymphoblastic leukemia. Ped Blood Cancer. 2008;50 (2):259–63. doi: 10.1002/pbc.21298. [DOI] [PubMed] [Google Scholar]
- 27.Farber S, Diamond L, Mercer R, Sylvester R, Wolff J. Temporary remissions in acute leukemia in children produced by folic and antagonist 4-aminopteroyl-glutamic acid (Aminopterin) N Engl J Med. 1948;238:787–93. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 28.Möricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H, et al. Long term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24(2):265–84. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 29.Pui CH, Pei D, Sandlund JT, Ribeiro RC, Rubnitz JE, Raimondi SC, et al. Long term results of St Judes total therapy studies 11,12,13A,13B and 14 in childhood acute lymphoblastic leukemia. Leukemia. 2010;24(2):371–82. doi: 10.1038/leu.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silverman LB, Stevenson KE, O’Brien JE, Asselin BL, Barr RD, Clavell L, et al. Long term results of Dana-Farber Cancer Institute ALL consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985–2000) Leukemia. 2010;24 (2):320–34. doi: 10.1038/leu.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaynon PS, Angiolillo AL, Carroll WL, Nachman JB, Trigg ME, Sather HN, et al. Children’s Oncology Group. Long term results of the Children’s Cancer Group studies for childhood acute lymphoblastic leukemia 1983–2002. A Childrens Oncology Group report. Leukemia. 2010;24(2):285–97. doi: 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Söderhäll S, Taskinen M Nordic Society of Paediatric Haematology and Oncology. Long term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblstic leukemia. Leukemia. 2010;24(2):345–54. doi: 10.1038/leu.2009.251. [DOI] [PubMed] [Google Scholar]
- 33.Pession A, Valsecchi MG, Masera G, Kamps WA, Magyarosy E, Rizzari C, et al. Long-Term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukaemia. J Clin Oncol. 2005;23(28):7161–7. doi: 10.1200/JCO.2005.11.411. [DOI] [PubMed] [Google Scholar]
- 34.Duval M, Suciu S, Ferster A, Rialland X, Nelken B, Lutz P, et al. Comparison of Escherichia coli asparaginase with Erwinia asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood. 2002;99(8):2734–9. doi: 10.1182/blood.v99.8.2734. [DOI] [PubMed] [Google Scholar]
- 35.Panosyan EH, Seibel NL, Martin-Aragon S, Gaynon PS, Avramis IA, Sather H, et al. Asparaginase antibody and asparaginase activity in children with higer risk acute lymphoblastic leukaemia: Children’s Cancer Group Study CCG-1961. J Pediatr Hematol Oncol. 2004;26(4):217–26. doi: 10.1097/00043426-200404000-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.