

Abstract
Background
Nucleotide variations not changing protein sequences are considered silent mutations; accumulating data suggest that they can, however, be important in human diseases.
Design and Methods
We report an altered splicing process induced by a silent substitution (c.7056C>T) in the von Willebrand factor gene in a case of type 1 von Willebrand disease originally classified as lacking von Willebrand factor mutations.
Results
The c.7056C>T synonymous substitution introduces a new donor splice site within exon 41, leading to messenger RNA lacking nucleotides 7055-7081 (c.7055_7081del). The encoded von Willebrand factor protein is predicted to lack amino acids 2352-2360 in the B2 domain. The patient’s von Willebrand disease phenotype was characterized by reduced plasma and platelet von Willebrand factor, which was normal in function and multimer structure. In vitro expression studies demonstrated that co-transfection of equimolar c.7055_7081del and wild-type von Willebrand factor (mimicking the patient’s heterozygous state) induced a 50% lower von Willebrand factor secretion than the wild type, while almost no von Willebrand factor secretion was seen with the mutated von Willebrand factor alone. The secreted von Willebrand factor was structurally and functionally normal, suggesting that the c.7056C>T substitution behaves like a loss-of-function allele.
Conclusions
This is the first report of a synonymous von Willebrand factor substitution being responsible for von Willebrand disease. Our findings suggest the need to reconsider the role of von Willebrand factor polymorphisms in von Willebrand disease.
Keywords: von Willebrand factor, von Willebrand disease, type 1 VWD, splice site mutations, synonymous nucleotide substitution
Introduction
von Willebrand factor (VWF) is a high-molecular-weight multimeric glycoprotein with a pivotal role in primary hemostasis, mediating platelet adhesion and aggregation at the site of vascular lesions, and stabilizing blood coagulation factor VIII (FVIII). VWF is synthesized in megakaryocytes and endothelial cells, and it occurs in plasma, platelets and the subendothelial matrix.1–4
VWF deficiency causes von Willebrand disease (VWD), the most common inherited bleeding disorder, classified into three main types: type 1 accounts for more than 70% of all cases of VWD, and is a partial quantitative deficiency of structurally and functionally normal VWF; type 2 involves qualitative VWF abnormalities and comprises four subtypes (2A, 2B, 2M, 2N), according to the nature of the functional alteration; in type 3, the VWF protein is virtually absent.5,6
In addition to its heterogeneous phenotype, VWD is characterized by an incomplete penetrance due to genetic and acquired factors that influence plasma VWF levels. The ABO blood group is the main genetic modulator of VWF concentration, accounting for approximately 30% of its variability in normal individuals and VWD patients alike.7–9 Moreover, many physiological and pathological conditions up-regulate VWF, including exercise, pregnancy, surgery, hyperthyroidism, diabetes, renal failure, and cancer. VWF levels can consequently vary to such a degree that it may often be difficult to diagnose VWD, especially the mild type 1 forms, which are the most strongly influenced by inherited or acquired VWF modulators.10–12
The genetic characterization of VWD may contribute to its diagnosis, clarifying the nature of the defect and identifying the carriers within a given family. Type 2 and type 3 VWD have been relatively well characterized from the molecular perspective (see the ISTH-SSC VWF Online Database at http://www.vwf.group.shef.ac.uk/ for a review), while the genetic grounds for type 1 VWD are more elusive and approximately 30–40% of cases remain uncharacterized because no mutations in the VWF gene are found.13,14 Recent studies on large cohorts of patients with type 1 VWD have reported that a linkage between VWD and the VWF gene is only identified in a minority of families, suggesting that loci other than VWF may play a part in the pathogenesis of this disorder – and thus raising the question, once again, of how to diagnose VWD reliably.15–17
In the present study, we identified a synonymous nucleotide substitution in exon 41 of the VWF gene responsible for mild type 1 VWD. This substitution was initially thought to be a polymorphism, but has now been found to alter VWF gene splicing by generating a new donor splice site within exon 41.
Design and Methods
All the subjects involved were studied after obtaining their written informed consent in accordance with the Helsinki Declaration and the study was approved by our institutional review board.
Hemostatic tests
Basic hemostatic analyses, i.e. plasma VWF antigen (VWF:Ag), VWF collagen binding (VWF:CB), VWF ristocetin cofactor (VWF:RCo), ristocetin-induced platelet aggregation (RIPA), VWF multimers and FVIII, were done as described elsewhere.18 Platelet VWF content was measured as described previously.19 VWF multimer analysis was performed using 1.5% agarose gel containing 0.075% sodium dodecylsulfate. Multimers were detected using 125I-labeled anti-VWF antibody following autoradiography.
Genetic analysis
Exons, 5′ and 3′-UTR, and all exon/intron junctions of the VWF gene were analyzed, from genomic DNA, by direct sequencing. All polymerase chain reactions (PCR) were performed using AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA) and a thermal cycler, GeneAmp®PCR System 2700 (Applied Biosystems). For DNA sequencing, the Big Dye Terminator Sequencing v.2.5 kit (Perkin Elmer, Wellesley, MA, USA) and an ABI 3100 Genetic Analyzer (Applied Biosystems) were used.
Linkage study
Three intragenic VWF short tandem repeats (STR) were analyzed: the -2144_-2105(GT)n (GTn), the 6977-564_6977-541(ATCT)n (STR I) and the 6977-181_6977-150 (TCTA)n (STR II)20–22. STR were amplified from genomic DNA, using primer pairs in which the forward oligonucleotides were 5′-fluorescently labeled with 6-FAM or HEX dye. PCR products were analyzed in an ABI 3100 Genetic Analyzer (Applied Biosystems) and the length of each allele was determined with Peak Scanner 1.0 software (Applied Biosystems).
VWF cDNA analysis
Total RNA was extracted from platelets using Trizol reagent (Invitrogen, San Diego, CA, USA), then cDNA was synthesized using random primers and SuperScriptTM II Reverse Transcriptase (Invitrogen), and amplified by PCR. All primer sequences are available on request.
The pGEM-T Easy Vector System I (Promega Madison, WI, USA) was used to clone the cDNA11 PCR fragment found to have an altered sequence (see Results section), in order to separate and better characterize the patient’s two alleles.
VWF gene nucleotides were numbered according to the latest recommendations of the ISTH Scientific Subcommittee on VWD, assigning +1 to the “A” of the initiator codon ATG.23
Bioinformatic analysis
Alamut 1.51 software (Interactive Biosoftware, Rouen, France) was used to investigate the generation of a potential splicing site in the VWF gene as a consequence of the c.7056 C>T nucleotide substitution. Alamut 1.51 integrates data from known constitutive human splicing signals and a number of prediction methods such as the SpliceSiteFinder-like, the MaxEntScan, the GeneSplicer, the ESEFinder and the RESCUE-ESE methods.
In vitro expression of the c.7055_7081del mutation
The pSVvWFA plasmid containing normal human full-length VWF cDNA (kindly provided by Dr. C Mazurier, Lille, France) was mutated by deleting the nucleotides from 7055 to 7081 (pSV7055_7081delvWF) using the QUIKCHANGE II XL kit (Stratagene, La Jolla, CA, USA), the phosphorylated primers del-ex41(F) 5′cctgcaggaaggaggagtgcaaaagagtgtcccc3′ and del-ex41(R) 5′CAGGGTTGGTCAGTGTGGGCTG GAGGCCACGTTC3′, and T4 DNA ligase (New England Biolabs, Ipswich, MA, USA). For the expression studies the pSV7055_7081delvWF was transiently transfected into baby hamster kidney (BHK) cells stably transfected with furin (FUR4BHK) (kindly provided by Dr. JE Sadler, St Louis, MO, USA), using the Fugene transfection reagent (Roche, Mannheim, Germany). Co-transfections with pSVvWFA and pSV7055_7081delvWF constructs were used to mimic the patient’s heterozygous state. After 72 h, the transfection media containing VWF were removed, concentrated about five times with Centricon filters (Millipore, Billerica, MA, USA), and quantified using an enzyme-linked immunosorbent assay (ELISA). The results of each transfection experiment were calculated as the mean of six replicates. The intracellular VWF was also measured, lysing cells with a 2% Triton-X-100 solution according to procedures already described.24
Results
Patients
The family tree is shown in Figure 1. The proband (III-3) is a 15-year old female with a history of mild bleeding characterized by epistaxis, heavy menstrual periods, hematomas occurring after minimal trauma and bleeding gums. Her 44-year-old mother (II-3) and the 73-year-old grandmother (I-1) had similarly moderate clinical bleeding symptoms, and her mother also reported bleeding episodes after tooth extractions and tonsillectomy. The proband’s father and sister were asymptomatic.
Figure 1.

Family tree and co-segregation of VWD and the VWF gene in the family investigated. The genotype for (GT)n, STR I and STRII is given for each family member. Affected individuals are represented by filled symbols and the arrow indicates the index case. Numbers indicate allele size in base pairs. The (GT)n/STR I/STR II haplotype segregating with VWD is boxed: it was shared by all affected members, but not found in their unaffected relatives.
Hemostatic picture
The main hemostatic findings in the proband and her relatives are shown in Table 1. The proband (III-3), her mother (II-3) and her grandmother (I-1) had a mild decrease in VWF:Ag, VWF:CB and VWF:RCo. Their platelet VWF:Ag content was reduced, confirming their defective VWF synthesis. Plasma VWF multimer analysis showed that all oligomers were present, but all at lower levels than normal; there was no evidence of any accumulation of low-molecular-weight multimers (Figure 2A). A similar pattern emerged for the affected subjects’ platelet VWF multimers (Figure 2B).
Table 1.
Hemostatic picture of the proband and her relatives.
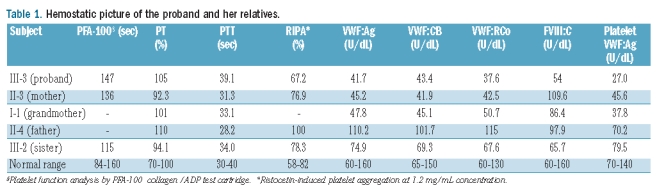
Figure 2.

Multimer composition of plasma (A) and platelet (B) VWF in the proband (1) and her mother (2), and of rwtVWF, r7055_7081delVWF and heterozygous rwtVWF/r7055_7081delVWF from expression experiments (C). NP=normal plasma. NPlat=normal platelets. Large VWF multimers are at the top, small multimers at the bottom. The patients’ VWF is fully represented in both plasma and platelet samples, although it is slightly reduced.
Sequencing of the von Willebrand factor gene
When the proband’s VWF gene was sequenced, no mutations were identified in the coding region, 5′ and 3′-UTR, and all exon/intron junctions. Some polymorphisms were, however, found: these were both synonymous (i.e. silent, they did not change the amino acid sequence encoded by the gene) and otherwise (ISTH-SSC VWF Online Database, http://vwf.group.shef.ac.uk/). The proband was consequently first classified as a case of type 1 VWD lacking mutations in the VWF gene.
Linkage analysis
Given the absence of any VWF gene mutations, we performed a linkage study to verify the co-segregation of VWD and the VWF gene, analyzing the VWF intragenic STR (GT)n, STR I and STR II. All three affected individuals shared the same (GT)n/STR I/STR II haplotype, i.e. 237/102/235, which was not found in any of their healthy relatives, thus confirming the co-segregation between VWD and the VWF gene (Figure 1).
Analysis of von Willebrand factor cDNA
The results of the linkage analysis prompted us to screen the proband’s VWF cDNA, hypothesizing a VWF gene splicing alteration. Total RNA was extracted from platelets, retrotranscribed into cDNA and amplified in 12 fragments. PCR products were run in a 2% agarose gel and the migration pattern of the proband’s cDNA fragments was compared with that of control subjects, seeking any large VWF gene splicing alteration. As no differences emerged, the patient’s cDNA was sequenced to identify any small splicing anomalies escaping detection by gel electrophoresis. In fact, sequencing of the fragment cDNA11 (including exons 38–44) revealed overlapping peaks in the chromatogram suggestive of a deletion at a heterozygous level. To separate the patient’s two alleles and precisely define the boundaries of the deletion, the cDNA11 PCR fragment was cloned into pGEM-T Easy Vector and sequenced. This analysis revealed that the proband had one allele lacking the last 27 nucleotides of exon 41, from 7055 to 7081 (c.7055_7081del) (Figure 3A). The deleted nucleotides correspond to amino acids 2352 to 2360 in the B2 domain of the VWF protein (p.Gly2352_Cys2360del). The fact that the c.7055_ 7081del mutation was found in the patient’s cDNA, but not in her genomic DNA, meant that it was the result of a VWF gene splicing error.
Figure 3.

Mutation c.7055_7081del in the proband’s VWF cDNA (A), and splicing mechanism of the normal and mutated allele of the VWF gene (B). The proband carried a wild-type allele and a mutated allele lacking the last 27 nucleotides of exon 41 (c.7055_7081del). This deletion is generated as a consequence of the c.7056C>T substitution in the VWF gene: the presence of a T at position 7056 gives rise to a new donor splicing site in exon 41 (GT at 7055-7056). When the new splice site is recognized, instead of the wild-type one, the last portion of exon 41 is deleted, together with intron 41.
The synonymous c.7056 C>T substitution generates a new donor splicing site
To characterize the prime cause of this splicing disruption, we examined the patient’s genomic sequence upstream and downstream of the deleted region in depth. The donor and acceptor splicing sites flanking exon 41 were normal, so we focused on the c.7056 C>T substitution in exon 41, adjacent to the first deleted nucleotide. Although it was characterized as a synonymous substitution14, a bioinformatic analysis performed by Alamut 1.51 software predicted that the presence of a T at position 7056 generates a new donor splice site in exon 41 (GT at position 7055-7056). This donor splice site matches the consensus better than the wild-type (SpliceSiteFinder-like score 87.1 versus 83.0) and is predicted to cause the excision of the last 27 nucleotides of exon 41, together with intron 41 of the VWF gene (Figure 3B). Sequencing exon 41 from genomic DNA of all family members confirmed that the c.7056 C>T substitution co-segregates with type 1 VWD: all affected individuals had the C/T genotype, while all healthy relatives were C/C at position 7056.
In vitro expression of the c.7055_7081del mutation
To investigate the effect of the c.7055_7081del mutation on VWF synthesis and secretion, plasmids pSVvWFA and pSV7055_7081delvWF - expressing recombinant wild-type VWF (rwtVWF) and the mutated form (rGly2352_Cys2360del VWF), respectively - were used in transfection experiments on FUR4BHK cells.
Virtually no rVWF secretion was found in the medium from cells transfected with the mutated pSV7055_ 7081delvWF plasmid alone (Figure 2C). The expression of mutation c.7055_7081del at a heterozygous level (transfection with equimolar amounts of pSVvWFA and pSV7055_7081delvWF plasmids) produced around 50% of VWF:Ag by comparison with the wild-type, and 40% of VWF:CB (Figure 4); all VWF multimers were present, albeit in decreased quantities, and there was no evidence of accumulation of small VWF oligomers (Figure 2C). Co-transfections with pSVvWFA and increasing amounts of pSV7055_7081delvWF revealed a linear decrease in the secretion of rVWF and its collagen binding activity, suggesting that the c.7055_7081del mutation does not have a dominant-negative effect (Figure 4).
Figure 4.

VWF:Ag and VWF:CB levels in conditioned media obtained from FUR4BHK cells expressing recombinant wild-type VWF (rwtVWF), mutant VWF (r7055_7081delVWF) or both rwtVWF/ r7055_7081 delVWF. Values are given as percentages of the rwtVWF expression, taken as 100. In the table, the percentages indicate the relative amounts of wild-type and mutated vectors used in the transfection experiments.
The intracellular rVWF obtained from cell lysates was also quantified: cells transfected with equimolar amounts of the pSVvWFA and pSV7055_7081delvWF plasmids revealed a rVWF production reduced to 44% by comparison with that of cells expressing the wild-type vector, while rVWF was undetectable in lysates from cells transfected with the mutated vector alone (Figure 5). VWF multimers were found to be homogeneously decreased in cells transfected with equimolar amounts of pSVvWFA and pSV7055_7081delvWF plasmids, and absent in the cells transfected with the mutated vector alone. These data confirm defective VWF synthesis as well as the absence of a dominant-negative effect of the c.7055_7081del VWF mutation.
Figure 5.

Intracellular production of r7055_7081delVWF (lysate). Values are given as percentages, assuming the production of wild-type VWF to be 100%. Means and standard errors of the mean were calculated on six measurements. The corresponding multimer pattern is shown in the lower panel, taking normal pooled plasma (NP) as the reference.
Discussion
Clarifying the mechanisms behind inherited VWD with no apparent VWF mutations may be useful for understanding the pathophysiology of this disorder. Here we report an altered splicing process induced by a synonymous nucleotide substitution (c.7056 C>T) in the coding region of the VWF gene, in a patient with type 1 VWD previously classified as not having any VWF mutations. This is the first report of a synonymous VWF mutation being responsible for VWD.
It is often difficult to characterize cases of type 1 VWD genetically because the related mutations are spread all along the VWF gene, which is 180 kb in size, including its 5′- and 3′-UTR.25 Moreover, loci other than VWF seem to have a role in the pathogenesis of VWD, as demonstrated by the small proportion of affected families showing co-segregation between VWD and the VWF gene.16,25–27 Mouse models of type 1 VWD have also been described in which the causative genetic locus is not VWF.28–30 All these reasons contribute to explaining why more than 30% of cases of type 1 VWD still remain genetically unexplained. It is important to remember, moreover, that searches for VWF mutations usually only investigate the coding regions of the gene (given its large size), so most intronic portions remain unexplored. This is a far from negligible drawback because mutations within introns are known to interfere with gene splicing or mRNA stability. Finally, genomic DNA sequencing (the most commonly used method for screening the VWF gene) represents a further intrinsic limit because it may fail to detect large deletions occurring at a heterozygous level.
All these issues are reflected in the family discussed here, diagnosed as having a mild form of type 1 VWD on the strength of the phenotype characterized by a homogeneous reduction of VWF:Ag and its associated functions, and a normal pattern of VWF multimers. Although the genetic analysis failed to reveal any mutations in the VWF gene, VWD was demonstrated to segregate with the VWF gene. This result prompted us to analyze the proband’s cDNA, which revealed a deletion of the last 27 nucleotides of exon 41 (c.7055_7081del), not present in the patient’s genomic DNA. Bioinformatic analysis demonstrated that the deletion was caused by a synonymous nucleotide substitution (c.7056C>T) that creates a new donor splice site (GT at 7055-7056) within exon 41 characterized by a presumably stronger biological potency than the wild type, and generating a new mature mRNA lacking the last 27 nucleotides of exon 41, exactly the same nucleotides as were missing in our patient.
As documented by in vitro expression experiments, the c.7055_7081del mutation interferes with the synthesis of VWF, and does not have a dominant-negative effect.31 The hemostatic profile of recombinant VWF is consistent with the patient’s phenotype, thus suggesting that the c.7056 C>T substitution acts as a loss-of-function mutation, i.e. a pure VWF quantitative defect.
The c.7056 C>T mutation is associated with the deletion of nine amino acids (Gly Glu Cys Arg Pro Asp Phe Thr Cys) from 2352 to 2360 of the VWF B2 domain (p.Gly2352_Cys2360). This domain’s function has yet to be fully defined, but it has to be intact for proper VWF synthesis and function.14,32 The B2 domain is in close proximity to the cysteine knot-like (CK) domain, a common motif involved in the dimerization of proteins, a step playing a primary role in the assembly of VWF mul-timers.33 Even though the B2 domain is not directly involved in the dimerization process, abnormalities of this domain might induce conformational changes of the CK domain capable of disrupting the proper alignment of VWF monomers during dimer formation, without interfering with the dimerization of normal VWF monomers, as suggested by the normal profile of the residually synthesized VWF.
The key finding emerging from this study concerns the role of silent nucleotide substitutions in causing type 1 VWD. Even if the c.7056 C>T substitution does not modify the VWF amino acid composition, it still has a marked impact on VWF gene expression, generating a new donor splice site leading to a mutated mRNA. While the contribution of splicing site defects is well documented as a common cause of disease, including VWD,34–36 less is known about the role of synonymous nucleotide substitutions in inherited human disorders. In the majority of the cases described, such mutations have been shown to interfere with the proper elimination of gene introns, but there are also reports of alterations to proper mRNA folding, which interfere with ribosome activity and/or increase the mRNA degradation process, resulting in a slower rate of protein translation.37
Finally, our results confirm once again how heterogeneous the penetrance of VWD may be, since the patients’ VWF defect was expressed more than we would have expected on the basis of a gene null VWF condition. Carriers of one null allele are frequently asymptomatic with regards to bleeding,37,38 but laboratory studies can often reveal lower than normal platelet and plasma VWF levels. Little is known about the modulators affecting VWF synthesis and/or a bleeding tendency, so these aspects are not taken into account in the assessment and characterization of VWD.
In conclusion, we demonstrated that the c.7056 C>T synonymous substitution is responsible for type 1 VWD, thereby showing that apparently harmless nucleotide variations may have a role in the development of VWD and help to explain genetically unsolved cases of VWD. Based on these findings, we suggest the need to investigate VWF gene sequence variations more extensively to clarify whether silent substitutions contribute to VWD or modulate its severity.
Supplementary Material
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologi-ca.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
Funding: this work was supported by a grant from the Ministero dell’Universita’ e della Ricerca Scientifica e Tecnologica (MURST, 60%, 2007)
References
- 1.Ruggeri ZM. Von Willebrand factor. Curr Opin Hematol. 2003;10(2):142–9. doi: 10.1097/00062752-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 3.Foster PA, Fulcher CA, Marti T, Titani K, Zimmerman TS. A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J Biol Chem. 1987;262 (18):8443–6. [PubMed] [Google Scholar]
- 4.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of von Willebrand factor by cultured human endothelial cells. Proc Natl Acad Sci USA. 1974;71(5):1906–9. doi: 10.1073/pnas.71.5.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruggeri ZM, Zimmerman TS. Von Willebrand factor and von Willebrand disease. Blood. 1987;70(4):895–904. [PubMed] [Google Scholar]
- 6.Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005;56:173–91. doi: 10.1146/annurev.med.56.082103.104713. [DOI] [PubMed] [Google Scholar]
- 7.Gallinaro L, Cattini MG, Sztukowska M, Padrini R, Sartorello F, Pontara E, et al. A shorter von Willebrand factor survival in O blood group subjects explains how ABO determinants influence plasma von Willebrand factor. Blood. 2008;111(7):3540–5. doi: 10.1182/blood-2007-11-122945. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins PV, O’Donnell JS. ABO blood group determines plasma von Willebrand factor levels: a biologic function after all? Transfusion. 2006;46(10):1836–44. doi: 10.1111/j.1537-2995.2006.00975.x. [DOI] [PubMed] [Google Scholar]
- 9.Gill JC, Endres-Brooks J, Bauer PJ. The effect of ABO blood group in the diagnosis of von Willebrand disease. Blood. 1987;69 (6):1691–5. [PubMed] [Google Scholar]
- 10.Stakiw J, Bowman M, Hegadorn C, Pruss C, Notley C, Groot E, et al. The effect of exercise on von Willebrand factor and ADAMTS-13 in individuals with type 1 and type 2B von Willebrand disease. J Thromb Haemost. 2008;6(1):90–6. doi: 10.1111/j.1538-7836.2007.02790.x. [DOI] [PubMed] [Google Scholar]
- 11.Pottinger BE, Read RC, Paleolog EM, Higgins PG, Pearson JD. Von Willebrand factor is an acute phase reactant in man. Thromb Res. 1989;53(4):387–94. doi: 10.1016/0049-3848(89)90317-4. [DOI] [PubMed] [Google Scholar]
- 12.Stirling Y, Woolf L, North WR, Seghatchian MJ, Meade TW. Hemostasis in normal pregnancy. Thromb Haemost. 1984;52(2):176–82. [PubMed] [Google Scholar]
- 13.Casonato A, Gallinaro L, Cattini MG, Pontara E, Padrini R, Bertomoro A, et al. Type 1 von Willebrand disease due to reduced von Willebrand factor synthesis and/or survival: observations from a case series. Transl Res. 2010;155(4):200–8. doi: 10.1016/j.trsl.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 14.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109(1):145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 15.Eikenboom J, Van Marion V, Putter H, Goodeve A, Rodeghiero F, Castaman G, et al. Linkage analysis in families diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD. J Thromb Haemost. 2006;4 (4):774–82. doi: 10.1111/j.1538-7836.2006.01823.x. [DOI] [PubMed] [Google Scholar]
- 16.James PD, Paterson AD, Notley C, Cameron C, Hegadorn C, Tinlin S, et al. Genetic linkage and association analysis in type 1 von Willebrand disease: results from the Canadian type 1 VWD study. J Thromb Haemost. 2006;4(4):783–92. doi: 10.1111/j.1538-7836.2006.01860.x. [DOI] [PubMed] [Google Scholar]
- 17.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 18.Casonato A, Pontara E, Sartorello F, Cattini MG, Sartori MT, Padrini R, et al. Reduced von Willebrand factor survival in type Vicenza von Willebrand disease. Blood. 2002;99(1):180–4. doi: 10.1182/blood.v99.1.180. [DOI] [PubMed] [Google Scholar]
- 19.Casonato A, Pontara E, Boscaro M, Sonino N, Sartorello F, Ferasin S, et al. Abnormalities of von Willebrand factor are also part of the prothrombotic state of Cushing’s syndrome. Blood Coagul Fibrinolysis. 1999;10(3):145–51. doi: 10.1097/00001721-199904000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Daidone V, Cattini MG, Pontara E, Sartorello F, Gallinaro L, Marotti A, et al. Microsatellite (GT)(n) repeats and SNPs in the von Willebrand factor gene promoter do not influence circulating von Willebrand factor levels under normal conditions. Thromb Haemost. 2009;101(2):298–304. [PubMed] [Google Scholar]
- 21.Othman M, Elbatarny HS, Byrne CD, O’Shaughnessy DF. Von Willebrand factor short sequence repeat locus 2 (intron 40) consists of three polymorphic subloci. Acta Haematol. 2007;117(3):177–80. doi: 10.1159/000097660. [DOI] [PubMed] [Google Scholar]
- 22.Vidal F, Julià A, Altisent C, Puig L, Gallardo D. Von Willebrand gene tracking by single-tube automated fluorescent analysis of four short tandem repeat polymorphisms. Thromb Haemost. 2005;93(5):976–81. doi: 10.1160/TH04-10-0699. [DOI] [PubMed] [Google Scholar]
- 23.Goodeve AC, Eikenboom JC, Ginsburg D, Hilbert L, Mazurier C, Peake IR, et al. ISTH SSC Subcommittee on von Willebrand factor. A standard nomenclature for von Willebrand factor gene mutations and polymorphisms. On behalf of the ISTH SSC Subcommittee on von Willebrand factor. Thromb Haemost. 2001;85(5):929–31. [PubMed] [Google Scholar]
- 24.Casonato A, Sartorello F, Cattini MG, Pontara E, Soldera C, Bertomoro A, et al. An Arg760Cys mutation in the consensus sequence of the von Willebrand factor propeptide cleavage site is responsible for a new von Willebrand disease. Blood. 2003;101(1):151–6. doi: 10.1182/blood-2002-04-1046. [DOI] [PubMed] [Google Scholar]
- 25.Goodeve A. Genetics of type 1 von Willebrand disease. Curr Opin Hematol. 2007;14(5):444–9. doi: 10.1097/MOH.0b013e32826f4b41. [DOI] [PubMed] [Google Scholar]
- 26.Casaña P, Martínez F, Haya S, Espinós C, Aznar JA. Significant linkage and non-linkage of type 1 von Willebrand disease to the von Willebrand factor gene. Br J Haematol. 2001;115(3):692–700. doi: 10.1046/j.1365-2141.2001.03132.x. [DOI] [PubMed] [Google Scholar]
- 27.Castaman G, Eikenboom JC, Bertina RM, Rodeghiero F. Inconsistency of association between type 1 von Willebrand disease phenotype and genotype in families identified in an epidemiological investigation. Thromb Haemost. 1999;82(3):1065–70. [PubMed] [Google Scholar]
- 28.Nichols WC, Cooney KA, Mohlke KL, Ballew JD, Yang A, Bruck ME, et al. von Willebrand disease in the RIIIS/J mouse is caused by a defect outside of the von Willebrand factor gene. Blood. 1994;83(11):3225–31. [PubMed] [Google Scholar]
- 29.Mohlke KL, Purkayastha AA, Westrick RJ, Smith PL, Petryniak B, Lowe JB, et al. Mvwf, a dominant modifier of murine von Willebrand factor, results from altered lineage-specific expression of a glycosyltransferase. Cell. 1999;96(1):111–20. doi: 10.1016/s0092-8674(00)80964-2. [DOI] [PubMed] [Google Scholar]
- 30.Shavit JA, Manichaikul A, Lemmerhirt HL, Broman KW, Ginsburg D. Modifiers of von Willebrand factor identified by natural variation in inbred strains of mice. Blood. 2009;114(26):5368–74. doi: 10.1182/blood-2009-07-233213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casari C, Pinotti M, Lancellotti S, Adinolfi E, Casonato A, De Cristofaro R, et al. The dominant-negative von Willebrand factor gene deletion p.P1105_C1926delinsR: molecular mechanism and modulation. Blood. 2010;116(24):5371–6. doi: 10.1182/blood-2010-02-268920. [DOI] [PubMed] [Google Scholar]
- 32.Casonato A, De Marco L, Gallinaro L, Sztukowska M, Mazzuccato M, Battiston M, et al. Altered von Willebrand factor subunit proteolysis and multimer processing associated with the Cys2362Phe mutation in the B2 domain. Thromb Haemost. 2007;97(4):527–33. [PubMed] [Google Scholar]
- 33.Tjernberg P, Vos HL, Spaargarenvan Riel CC, Luken BM, Voorberg J, Bertina RM, Eikenboom JCJ. Differential effects of the loss of intrachain- versus interchaindisulfide bonds in the cystineknot domain of von Willebrand factor on the clinical phenotype of von Willebrand disease. Thromb Haemost. 2006;96(6):717–24. doi: 10.1160/th06-08-0460. [DOI] [PubMed] [Google Scholar]
- 34.Gallinaro L, Sartorello F, Pontara E, Cattini MG, Bertomoro A, Bartoloni L, et al. Combined partial exon skipping and cryptic splice site activation as a new molecular mechanism for recessive type 1 von Willebrand disease. Thromb Haemost. 2006;96(6):711–6. [PubMed] [Google Scholar]
- 35.Jensen CJ, Oldfield BJ, Rubio JP. Splicing, cis genetic variation and disease. Biochem Soc Trans. 2009;37(6):1311–5. doi: 10.1042/BST0371311. [DOI] [PubMed] [Google Scholar]
- 36.Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220(2):152–63. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chamary JV, Hurst LD. The price of silent mutations. Sci Am. 2009;300(6):46–53. doi: 10.1038/scientificamerican0609-46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.