

Abstract
We investigated the impact of carrying more than one BCR-ABL1 mutation in 207 patients with chronic myeloid leukemia (102 chronic, 61 accelerated, and 44 blast phase) post-imatinib failure. Seven (8%) of 92 patients carrying mutations had more than one mutation: 4 (4%) in chronic phase, 2 (2%) in accelerated phase, and one (1 %) in blast phase. The cytogenetic response rate to second generation TKIs for patients with no, one, or more than one mutation was 88%, 69%, 50% (P=0.03) in chronic phase, 54%, 42%, 50% in accelerated phase (P=0.67), and 35%, 25%, 0% (P=0.63) in blast phase, respectively. No differences were observed in event free survival or overall survival in accelerated or blast phase according to their mutational status. However, the 4-year event free survival rates among patients in chronic phase with no, one, or more than one BCR-ABL1 mutation were 56%, 49%, and 0%, respectively (P=0.02), with overall survival rates of 91%, 69%, and 75%, respectively (P=0.13). In conclusion, patients with more than one BCR-ABL1 mutation fare worse than those with no or one mutation.
Keywords: CML, tyrosine kinase inhibitor, ABL1 mutation, multiple mutations
Introduction
BCR-ABL1 kinase domain mutations represent the most frequent mechanism of resistance to tyrosine kinase inhibitor (TKI) therapy, being detected in 40%–50% of imatinib-resistant patients with chronic myeloid leukemia in chronic phase (CML-CP).1–6 Over 100 BCR-ABL1 single-point mutations have been reported in patients with imatinib-resistant CML, which confer different levels of TKI resistance.7 A threonine to isoleucine substitution at the 315 residue (T315I), which forms a key H-bond interaction with TKIs, results in clinical insensitivity to imatinib, nilotinib, and dasatinib.3,8–13 Other mutations, however, can be inhibited at least to some extent by 2nd generation TKIs (SG-TKIs). The sensitivity of single-point mutations to both nilotinib and dasatinib has been determined in vitro and may inform the selection of TKI upon imatinib failure.14 Patients with CML can acquire more than one BCR-ABL1 mutation during sequential TKI therapy, which may result in increased oncogenicity compared with each individual mutation.15 We analyzed the response rates and survival of patients carrying more than one BCR-ABL1 mutation during sequential TKI therapy.
Design and Methods
Among 293 patients treated at the MD Anderson Cancer Center with SG-TKI after imatinib failure between November 6, 2003 and December 21, 2008, mutation analysis was available in 207 (71%): 102 in chronic phase (CP), 61 in accelerated phase (AP), and 44 in blast phase (BP) (Table 1). The complete kinase domain of the BCR-ABL1 oncogene was analyzed using seminested reverse transcriptase PCR followed by direct sequencing.16 The study was conducted in compliance with the MD Anderson Cancer Center Institutional Review Board.
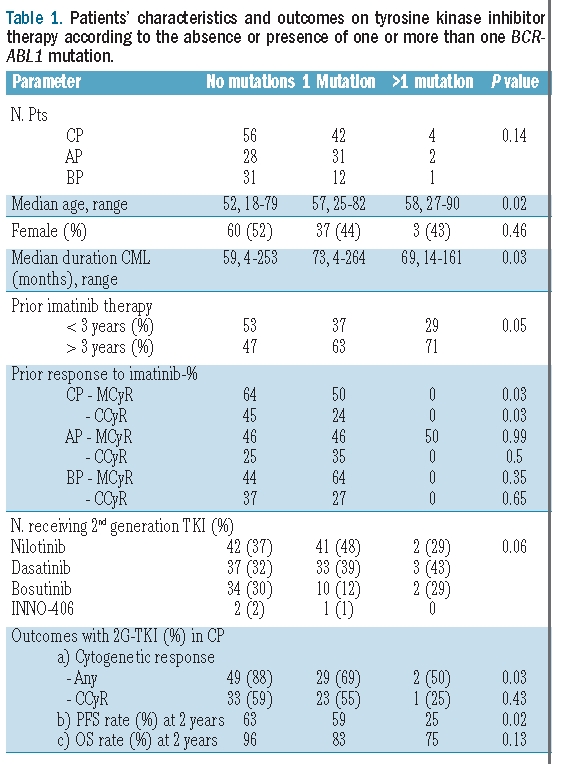
Table 1.
Patients’ characteristics and outcomes on tyrosine kinase inhibitor therapy according to the absence or presence of one or more than one BCR-ABL1 mutation.
Results and Discussion
The complete cytogenetic response (CCyR) rate with imatinib among patients in chronic, accelerated or blast phase was 35%, 19%, and 30%, respectively. BCR-ABL1 mutations upon imatinib failure were detected in 92 (44%) patients: 45% in chronic phase, 54% in accelerated phase, and 30% in blast phase. Median time from diagnosis to detection of one mutation in chronic, accelerated, and blast phase was 76 (range 21–264), 67 (range 4–191), and 39 months (range 3–189), respectively. Median time on imatinib for patients with one mutation in chronic, accelerated, and blast phase was 51 (range 14–80), 38 (range 2–63), and 34 months (range 2–56), respectively. Median time from diagnosis to detection of more than one mutation in chronic, accelerated, and blast phase was 81 (range 23–126), 37 (range 14–60), and 161 months, respectively. Median time on imatinib for patients with more than one mutation in chronic, accelerated, and blast phase was 51 (range 22–77), 25 (range 14–37), and 44 months, respectively. The most frequent mutations mapped to residues M351 (n=12), G250 (n=12), E255 (n=9), F359 (n=9), and T315I (n=7), comprising 70% of all mutations. Seven (8%) patients had more than one BCR-ABL1 mutation (Online Supplementary Table S1): 4 (4%) in chronic phase (G250E/Y253H, M351T/F317L, T315I/M351T, M351T/F359V), 2 (3%) in accelerated phase (E255K/E459K, G250E/M351T), and one (2%) in blast phase (E255V/M351T).
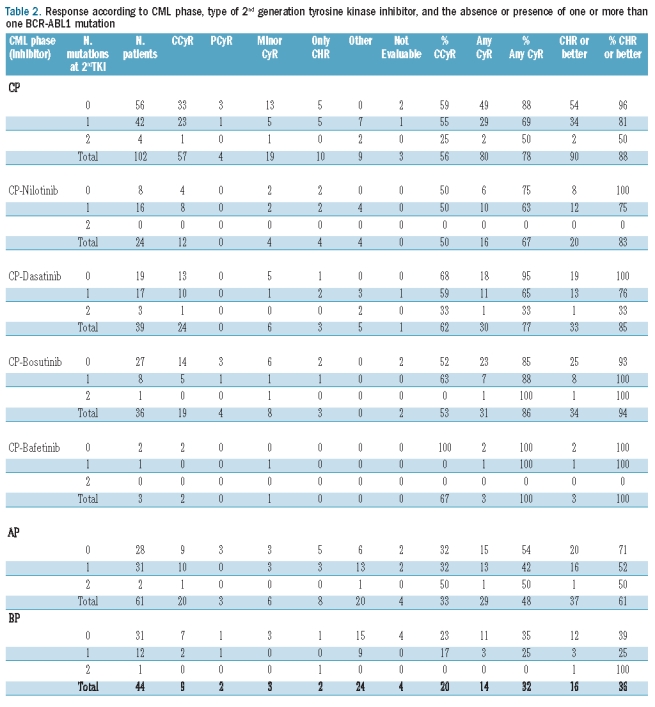
After a median follow up of 48 months, the complete cytogenetic response (CcyR) rate with imatinib among patients in chronic, accelerated, or blast phase was 35%, 19%, and 30%, respectively. The cytogenetic response rates with SG-TKIs differed among patients with no, one, or more than one mutation (88%, 69%, 50%, respectively, P=0.03) in chronic phase, but not in accelerated phase (54%, 42%, 50%, respectively, P=0.67), or in blast phase (35%, 25%, 0%, respectively, P=0.63) (Table 1). There was a similar but not statistically significant trend for CCyR rates in chronic phase (59%, 55%, 25%, P=0.41) and blast phase (23%, 17%, 0%, respectively, P=0.80), but not accelerated phase (32%, 32%, 50%, respectively, P=0.87) (Table 2). The median duration of cytogenetic response for patients in chronic phase with no, one, or more than one mutation was 22, 36, and 11 months, respectively (P=0.22). These data suggest that carrying more than one BCR-ABL1 mutation may hamper the achievement of cytogenetic response and shorten its duration compared to patients with no or only one mutation.
Table 2.
Response according to CML phase, type of 2nd generation tyrosine kinase inhibitor, and the absence or presence of one or more than one BCR-ABL1 mutation
We next evaluated the survival rates of the cohort, both overall (Online Supplementary Figure S1) and according to CML phase and mutational status (Table 1). In chronic phase, the number of BCR-ABL1 mutations segregated prognostically distinct groups. The 4-year event free survival rates among patients in chronic phase with no, one, or more than one BCR-ABL1 mutation were 56%, 49%, and 0%, respectively (P=0.02) whereas the overall survival rates were 91%, 69%, and 75%, respectively (P=0.13), indicating that patients with more than one mutation have a significantly worse event free survival compared to those with no or only one BCR-ABL1 mutation. However, no significant differences were observed regarding event free or overall survival in accelerated phase (4-year: 21% vs. 30% vs. 0%, P=0.81; 54% vs. 47% vs. 0%, P=0.47) or in blast phase (1-year: 19% vs. 13% vs. not applicable [patient with longest follow up censored at 5.4 months] P=0.24; 1-year: 52% vs. 33% vs. 100%, P=0.39) according to whether they carried no, one, or more than one BCR-ABL1 mutation, indicating that CML phase bears a higher prognostic weight than mutational status.
M351T was the most frequently detected mutation both alone and concomitantly with other mutations, being present in 5 (71%) of 7 patients carrying more than one BCR-ABL1 mutation. M351T maps to the activation loop hinge and has lower kinase activity, growth competition potential in low serum, B-lymphoid transformation potency and colony formation capacity compared with native BCR-ABL1 kinase.17 Importantly, progression free survival and overall survival were similar among patients carrying 2 mutations and those carrying highly resistant single point-mutations (any TKI: T315I; nilotinib: G250E/V, Y253F/H/K, E255K/V, F359C/V; dasatinib: F317L, V299L; bosutinib: E255K/V, F317L, V299L, F486S) (Figure 1A–B). These data suggest that acquiring more than one mutation portends a poor prognosis similar to that associated with the acquisition of a single-point mutation highly resistant to SG-TKIs. Teleologically, the association with a second mutation would allow M351T, a mutation highly sensitive to SG-TKIs, to induce a fully TKI resistant phenotype.
Figure 1.

Event-free survival (A) and overall survival (B) of patients with CML-CP carrying none or highly sensitive BCR-ABL1 mutations (“No Resistant”) mutations, a single resistant mutation (“Resistant”), or two mutations (“Double”).
The selective pressure exerted by TKI therapy facilitates the growth of mutant resistant clones. Moreover, acquiring one BCR-ABL1 mutation is associated with an increased risk of acquiring a second mutation.18 During SG-TKI therapy, imatinib-resistant mutations may associate with newly acquired ones either within the same or in different clones. As patients fail sequential TKI therapy (imatinib followed by an SG-TKI), new mutant clones are selected and may coexist with other previously selected mutant clones. Acquiring new mutations is frequently associated with loss of response.18 Since patients with CML-CP carrying more than one BCR-ABL1 mutation appear to have a worse outcome compared with those carrying no or only one mutation, they might benefit from anti-CML agents with a BCR-ABL1-independent mechanism of action. Alternatively, novel TKIs active against mutations resistant to SG-TKIs, such as ponatinib (AP24534) are highly active in vitro against CML cells carrying more than one BCR-ABL1 mutation and may be particularly suited for this particular clinical scenario.19
A limitation of our analysis is that mutational status was assessed by direct sequencing, a technique routinely employed in clinical laboratories that only detects mutations reliably when present in at least 20% of the BCR-ABL1-expressing cells.20 More sensitive techniques might have allowed the detection of additional mutant clones at incipient stages of expansion. For instance, a recent mutational analysis using sensitive cloning techniques showed that among 61 patients with imatinib-resistant CML-CP, 57% of them harbored more than one BCR-ABL1 kinase domain mutation and that the latter were prone to cluster within the same leukemic clone.21 However, the clinical impact of these clones representing a small proportion of the overall leukemic population remains unknown.
In conclusion, patients with CML-CP carrying more than one BCR-ABL1 kinase domain mutation exhibit worse response rates and long-term outcomes with TKI therapy compared with those with no or only one BCR-ABL1 mutation. A longer follow up of a larger series of patients is warranted to confirm these intriguing observations.
Supplementary Material
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Hochhaus A, La Rosee P. Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia. 2004;18(8):1321–31. doi: 10.1038/sj.leu.2403426. [DOI] [PubMed] [Google Scholar]
- 2.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 3.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 4.Lowenberg B. Minimal residual disease in chronic myeloid leukemia. N Engl J Med. 2003;349(15):1399–401. doi: 10.1056/NEJMp038130. [DOI] [PubMed] [Google Scholar]
- 5.Corbin AS, La Rosee P, Stoffregen EP, Druker BJ, Deininger MW. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood. 2003;101(11):4611–4. doi: 10.1182/blood-2002-12-3659. [DOI] [PubMed] [Google Scholar]
- 6.Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003;4(2):75–85. doi: 10.1016/s1470-2045(03)00979-3. [DOI] [PubMed] [Google Scholar]
- 7.Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–30. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 9.Weisberg E, Manley PW, Breitenstein W, Brügger J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Kantarjian H, Giles F, Wunderle L, Bhalia K, O'Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–51. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 11.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoli J, Paquette J, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354 (24):2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 12.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methyl-pyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–61. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 13.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 14.Branford S, Melo JV, Hughes TP. Selecting optimal second-line tyrosine kinase inhibitor therapy for chronic myeloid leukemia patients after imatinib failure: does the BCR-ABL mutation status really matter? Blood. 2009;114(27):5426–35. doi: 10.1182/blood-2009-08-215939. [DOI] [PubMed] [Google Scholar]
- 15.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoli JM, Paquette RL, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117(9):2562–9. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102(1):276–83. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 17.Griswold IJ, MacPartlin M, Bumm T, Goss VL, O'Hare T, Lee KA, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 2006;26(16):6082–93. doi: 10.1128/MCB.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soverini S, Gnani A, Colarossi S, Castagnetti F, Abruzzese E, Paolini S, et al. Philadelphia-positive patients who already harbor imatinib-resistant Bcr-Abl kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114(10):2168–71. doi: 10.1182/blood-2009-01-197186. [DOI] [PubMed] [Google Scholar]
- 19.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintas-Cardama AG, Gibbons DL, Kantarjian H, et al. Mutational analysis of chronic myeloid leukemia (CML) clones reveals heightened BCR-ABL1 genetic instability and wild-type BCR-ABL1 exhaustion in patients failing sequential imatinib and dasatinib therapy. Blood. 2007:110. abstract 1938. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.