

Abstract
Two new rare irregular sesquiterpenes, tricinonoic acid (1) and tricindiol (2), and the known furanopyrrolidones, NG-391 (3) and NG-393 (4), have been isolated from an EtOAc extract of Fusarium tricinctum, a fungus endophytic in the root tissue of the Sonoran desert plant, Rumex hymenosepalus. The structures of 1 and 2 were elucidated on the basis of their high-resolution mass, 1D and 2D NMR spectroscopic data. A possible biosynthetic route to 1 and 2 from farnesyl diphosphate is proposed.
Keywords: Fusarium tricinctum, endophytic fungus, Rumex hymenosepalus, irregular sesquiterpenes, tricinonoic acid, tricindiol
1. Introduction
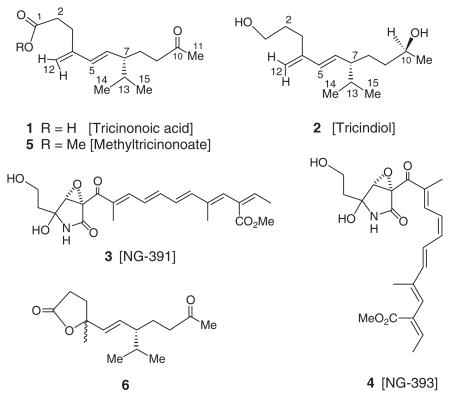
Fusarium tricinctum (Corda) Sacc., a food contaminating mould producing toxic metabolites, is found widely distributed in crops and plant products. This fungus is frequently isolated from mouldy corn and rice, and their ingestion by humans and animals cause mycotoxicoses (Bamburg, Strong, & Smalley, 1969; Hood, Kuczuk, & Szczech, 1978). Previous investigations of F. tricinctum have led to the isolation of toxin T-2, chlamidosporal (Solfrizzo & Visconti, 1996), visoltricin (Visconti & Solfrizzo, 1994), beauvericin (Rizzo, Ferracane, & Ritieni, 2002), zearalenone (Engelhardt, Schuster, Lepschy, & Wallnoefer, 1986; Vesela, Vesely, & Adamkova, 1981), enniatins (Rizzo et al., 2002), acuminatopyrone (Solfrizzo & Visconti, 1996; Visconti, Solfrizzo, Fruchier, & ApSimon, 1994) and diphenyl methanol (Gu, Ma, & Miao, 1994). In continuing our search for bioactive and/or novel metabolites from arid land plant-associated microorganisms (Zhan, Burns, Liu, Faeth, & Gunatilaka, 2007), we have investigated an EtOAc extract of a strain of F. tricinctum endophytic in the root tissue of the Sonoran desert plant, Rumex hymenosepalus Torr. Canigre (wild rhubarb; Polygonaceae), and in this article we report the isolation and characterisation of two new rare irregular sesquiterpenes, tricinonoic acid (1) and tricindiol (2), together with two known furanopyrrolidones, NG-391 (3) and NG-393 (4). This constitutes the first report of metabolites of an endophytic strain of F. tricinctum.
2. Results and discussion
Fractionation of an EtOAc extract of a liquid culture of F. tricinctum involving solvent–solvent partitioning followed by Sephadex LH–20 size-exclusion, and repeated normal and reversed phase chromatography furnished the compounds 1–4. Tricinonoic acid (1) was isolated as a colourless liquid that was analysed for C15H24O3 by a combination of HRFABMS, DEPT and 13C NMR data and indicated four degrees of unsaturation. Its IR spectrum had strong bands at 3425 and 1712 cm−1 indicating the presence of OH and ketone carbonyl groups, respectively. The 1H NMR spectrum of 1 (Table 1) consisted of two 3H doublets (J=6.5 Hz) at δ 0.82 and 0.87 due to methyl groups of an isopropyl moiety, one low-field 3H singlet at δ 2.09 assignable to a COCH3 group. The presence of 1H double doublets at δ 5.43 (J=16.0 and 9.5 Hz) and a 1H doublet at 5.97 (J=16.0 Hz) which was assignable to a –CH=CH–CH– moiety with E configuration and two olefinic 1H broad singlets at δ 4.94 and 4.90 suggested a disubstituted conjugated diene system. The cross peaks in its COSY spectrum between H-6 and H-7 and HMBC correlations of H-6 to C-7 and H-7 to C-6 indicated the connectivity of C-6 olefinic carbon of the conjugated diene to the C-7 carbon bearing the isopropyl group. The signals at δ 2.52 (br s) and 2.34 (m) were assigned to methylene protons, H-2 and H-9, on the basis of HMBC correlations of H-2 to carboxyl carbon and H-9 to C-8 and C-7. Multiplets at δ 2.52, 1.76 and 1.48 in the 1H NMR spectrum of 1 were shown to be due to methylene protons, H-3 and H-8, based on HMBC correlation of H-3 to C-4 and C-5 and H-8 to C-7, respectively (Figure 1). The 13C NMR spectrum of 1 (Table 1) when analysed with the help of DEPT and HSQC spectra indicated the presence of three CH3, five CH2 (one of which is olefinic), four CH (two due to olefinic) and three quaternary carbons (one of the each is due to carboxyl and ketone carbonyls, and the third is due to an olefinic carbon). Methylation of 1 with diazomethane afforded its methyl ester 5. The 1H NMR spectrum of 5 was almost superimposable with that of 1 except for the signal due to the OCH3 methyl which appeared as a singlet at δ 3.67. The large coupling constant (16.0 Hz) observed for signals due to H-5 and H-6 of both 1 and 5 suggested trans configuration for the C5=C6 double bond. The configuration of C-7, the carbon bearing the iso-propyl group, is assumed to be S based on the large coupling constant (9.5 Hz) observed for H-6 and H-7 similar to the structurally related compound, 5E,7S-isopropyl-4-methyl-10-oxo-undecen-4-olide (6) (Aasen, Hlubucek, & Enzell, 1975a; Coates, Ghisalberti, & Jefferies 1977; Demole & Enggist, 1975), and on biogenetic considerations (Aasen, Junker, & Enzell, 1975b; Zhang et al., 2003). Based on the above reasoning and the observed HMBC correlations (Figure 1), the structure of tricinonoic acid was elucidated as 5E,7S-isopropyl-4-methylene-10-oxo-undec-4-enoic acid (1).
Table 1.
NMR data 1H (500 MHz) in CDCl3 and 13C (125 MHz) in acetone-d6 for 1 and 2.
| Tricinonoic acid (1) |
Tricindiol (2) |
|||
|---|---|---|---|---|
| Position | δHa | δCb | δHa | δCb |
| 1 | 174.2 s | 3.67 t (6.5) | 62.2 t | |
| 2 | 2.52 br s | 33.4 t | 1.58 m | 32.8 t |
| 3 | 2.52 m | 28.1 t | 2.28 t (7.5) | 29.0 t |
| 4 | 146.0 s | 147.4 s | ||
| 5 | 5.97 d (16.0) | 134.0 d | 5.98 d (15.5) | 133.9 d |
| 6 | 5.43 dd (16.0, 9.5) | 133.0 d | 5.46 dd (16.0, 9.5) | 133.5 d |
| 7 | 1.76 m | 50.4 d | 1.75 m | 51.0 d |
| 8a | 1.76 m | 27.2 t | 1.75 m | 28.8 t |
| 8b | 1.48 m | 1.58 m | ||
| 9 | 2.34 m | 42.1 t | 1.36 m | 38.7 t |
| 10 | 208.2 s | 3.75 sextet (6.0) | 67.8 d | |
| 11 | 2.09 s | 30.1 q | 1.16 d (6.0) | 24.1 q |
| 12a | 4.94 br s | 114.4 t | 4.90 s | 113.7 t |
| 12b | 4.90 br s | 4.87 s | ||
| 13 | 1.59 oct. (6.5) | 33.1 d | 1.58 m | 33.0 d |
| 14 | 0.87 d (6.5) | 21.2 q | 0.86 d (6.5) | 21.3 q |
| 15 | 0.82 d (6.5) | 19.6 q | 0.82 d (6.5) | 19.5 q |
Notes: Multiplicities deduced from HSQC; coupling constant (J values in Hz) are in parentheses.
Multiplicities deduced from DEPT.
Figure 1.

Selected HMBC correlations for 1.
Tricindiol (2), obtained as a colourless liquid, was analysed for C15H28O2 by a combination of HRFABMS and 13C NMR, and indicated two degrees of unsaturation. IR spectrum of 2 exhibited a strong band at 3398 cm−1 suggesting the presence of an OH group. The 1H NMR and 13C NMR spectral data of 2 (Table 1) closely resembled those of tricinonoic acid (1), except for the signals in the vicinity of C-1 and C–10. The C-1 carboxyl and C-10 carbonyl groups of 1 have changed to CH2OH and CH(OH)CH3 groups in 2, consequently the chemical shifts observed for carboxyl and the carbonyl carbons were not observed in the 13C NMR spectrum of 2. The signals due to CH2-1 appeared at δ 3.67 (t, J=6.5) in 1H NMR and at δ 62.2 (t) in 13C NMR, the signals due to CH-10 appeared at δ 3.75 (sextet, J=6.0) in 1H NMR, and at δ 67.8 (d) in 13C NMR and the signals due to CH3-11 appeared at δ 1.16 (d, J=6.0) in 1H NMR and at δ 24.1 (q) in 13C NMR of 2. Because of these changes in the functionalities of C-1 and C-10, a slight up-field shifts in 1H NMR and 13C NMR signals of CH2-2 [δH 1.58 (m); δC 32.8 (t)] and CH2-9 [δH 1.36 m; δC 38.7 (t)] were observed. The 10S-configuration was deduced from a modified Mosher’s ester method (Ohtani, Kusumi, Kashman, & Kakisawa, 1991; Su et al., 2002) using the (S)- and (R)-MTPA esters of tricindiol (Figure 2). These data when combined with 2D NMR spectral analysis identified tricindiol as 5E,7S-isopropyl-4-methyleneundec-5-ene-1,10S-diol (2). None of the encountered compounds exhibited biological activity when tested in antimicrobial (up to 100 μgmL−1) assays and cytotoxicity (up to 10 mgmL−1) assay (Rubinstein et al., 1990).
Figure 2.

Δδ value [Δδ (in ppm)= δS−δR] obtained for (S)- and (R)-MTPA esters of trincindiol (2).
Irregular sesquiterpenes such as 6 with structural similarities to tricinonoic acid (1) and tricindiol (2) have previously been encountered in cured tobacco (Nicotiana tabacum L.) leaves and in tobacco smoke. In tobacco, 6 and related solanane-type irregular sesquiterpenes have been assumed to be formed during the curing process from macrocyclic thunbergane-type cembranoid diterpenes, which were found to be abundant in this plant (Aasen et al., 1975a). However, in F. tricinctum it is likely that 1 and 2 arise from E,Z-farnesyl diphosphate (FPP; 7) via germacrene D (8) (Scheme 1), commonly found as volatile constituents of some plants (Bansal, Moriarity, Takaku, & Setzer, 2007) and microorganisms (He & Cane 2004; Tsuchiya, Matsumoto, Shudo, & Okamoto, 1980). Interestingly, germacrene D (8) and a few other sesquiterpene constituents have been reported to be responsible for the peach-like aroma produced by Fusarium poae cultures (Le Quere, Semon, Latrasse, & Etievant 1987).
Scheme 1.

Possible biosynthetic origin of 1 and 2 from E,Z-farmesyl diphosphate (7).
3. Experimental
3.1. General experimental procedures
Optical rotations were measured with a Jasco DIP-370 polarimeter using CHCl3 as solvent. For IR spectral determinations, samples were dissolved in CH2Cl2 and adsorbed into KBr, dried in vacuum, disks were made and spectra were recorded on a Shimadzu FTIR–8300 spectrometer. 1D and 2D NMR spectra were recorded in CDCl3 with a Bruker DRX-500 instrument at 500MHz or DRX-600 instrument at 600MHz for 1H NMR and 125MHz for 13C NMR using residual solvent as internal standard. The chemical shift values (δ) are given in parts per million (ppm), and the coupling constants are given in Hertz. High-resolution MS were recorded in JEOL HX110A spectrometer.
3.2. Fungal isolation, identification and cultivation
Roots of R. hymenosepalus were collected in the vicinity of Sierra Ancha on Highway 188 in Arizona, in early 2005, and were processed as described previously (Bashyal, Wijeratne, Faeth, & Gunatilaka, 2005) for the isolation of endophytic fungal strains. The strain selected for further investigation was identified as F. tricinctum based on its morphological characteristics and partial LSU rRNA sequences, compared to MicroSeq library (Microbial ID, Newark, DE) and GenBank sequence database (Wijeratne et al., 2003). A culture is deposited at the Arizona State University Biology Department and the Southwest Center for Natural Products Research and Commercialization of the University of Arizona microbial culture collection under the accession numbers Rum-1RZ and CS-95-25, respectively. The strain was sub-cultured on potato dextrose agar (PDA). For the isolation of secondary metabolites, the endophyte was cultured in PDA (PDB; Difco, Plymouth, MN) in five 4 L shaker flasks at 120 RPM, each containing 2L of the medium at 26°C for 15 days.
3.3. Extraction and isolation
The liquid culture (10 L) obtained above was filtered through Whatman No. 1 filter paper and the filtrate extracted with EtOAc (3×2 L). The resulting EtOAc extract was evaporated under reduced pressure to afford a yellow oil (420 mg) which was subjected to solvent–solvent partitioning (Bashyal et al., 2005; Wijeratne et al., 2003) to afford a CHCl3 fraction as a yellow oil (273 mg). This fraction on gel permeation chromatography employing Sephadex LH-20 (10 g) and elution with hexane: CH2Cl2 (4 : 1), hexane :CH2Cl2 (1 : 4) (100 mL), CH2Cl2 : acetone (3 : 2) (50 mL), CH2Cl2 : acetone (1 : 4) (50 mL), CH2Cl2 :MeOH (1 : 4) (50 mL), and MeOH (50 mL) furnished six fractions. These were combined based on their TLC profiles to yield three major fractions, A (27.5 mg), B (42.1 mg) and C (128.0 mg). Column chromatography of fraction A (27.5 mg) on LiChroprep diol (2 g) and by elution with increasing amounts of acetone in CH2Cl2 followed by preparative TLC on RP-18 (MeCN :H2O, 50 : 50) yielded tricinonoic acid (1) (6.8 mg). Purification of fraction B (42.1 mg) by column chromatography as mentioned above followed by preparative TLC on RP-18 (MeCN : H2O, 50 : 50) yielded tricindiol (2) (1.6 mg). Chromatographic separation of fraction C (128 mg) on LiChroprep diol (12 g) followed by preparative TLC on silica gel (CH2Cl2 : acetone, 3 : 2) yielded NG-391 (3) (7.6 mg) and NG-393 (4) (4.2 mg). NG-391 (3) and NG-393 (4) were identified by direct comparison (TLC, MS and 1H NMR) with the samples obtained previously (Bashyal, Faeth, & Gunatilaka, 2007).
Tricinonoic acid (1)
Colourless oil; (c=0.23, CH3OH); UV (CH3OH) λmax 234 (sh), (5.23) nm; IR (KBr) νmax 3425, 1712, 1643, 1621, 1564, 1404, 1118, 538 cm−1; for 1H and 13C NMR data, see Table 1; HRFABMS m/z 253.1812 [M+H]+ (Calcd for C15H25O3, 253.1804).
Tricindiol (2)
Colourless oil; (c=0.11, CH3OH); UV (CH3OH) λmax 233 (sh), (5.17) nm; IR (KBr) νmax 3398, 1652, 1566, 1402, 1087, 534 cm−1; for 1H and 13C NMR data, see Table 1; HRFABMS m/z 241.2173 [M+H]+ (Calcd for C15H29O2, 241.2168).
Methyl tricinonoate (5)
Colourless oil; 1H NMR (600 MHz, CDCl3) δ 5.96 (1H, d, J=16.2 Hz, H-5), 5.39 (1H, dd, J=16.2, 9.6 Hz, H-6), 4.92 (1H, s, Ha-12), 4.87 (1H, s, Hb-12), 3.67 (3H, s, CO2CH3), 2.50 (2H, m, H-2), 2.50 (2H, m, H-3), 2.33 (2H, m, H-9), 2.09 (3H, s, CH3-11), 1.75 (2H, m, H-7), 1.75 (1H, m, Ha-8), 1.59 (1H, m, H-13), 1.47 (1H, m, Hb-8), 0.87 (3H, d, J=6.6 Hz, CH3-14), δ 0.82 (3H, d, J=6.6 Hz, CH3-15), APCIMS (+)-ve mode m/z 267 [M+H]+.
3.4. Preparation of (S)- and (R)-MTPA ester derivatives of tricindiol (2)
3.4.1. (S)-MTPA ester
Compound 2 (1.0 mg) was dissolved in pyridine (0.3 mL) and (R)-(−)- α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (20 μL) was added under a N2 gas stream and stirred for 2.5 h at room temperature. Methanol (1 mL) was added and evaporated under reduced pressure to obtain an yellowish residue which was purified by using column chromatography on LiChroprep diol (0.4 g) by elution with increasing the amount of acetone in CH2Cl2 to give (S)-MTPA ester derivative of 2. 1H NMR (500 MHz, CDCl3): δ 5.945 (1H, d, J=16.0 Hz, H-5), 5.333 (1H, dd, J=16.0, 9.5 Hz, H-6), 4.906 (1H, s, Ha-12), 4.819 (1H, s, Hb-12), 4.320 (2H, m, H-1), 4.320 (1H, m, H-10), 2.218 (2H, m, H-3), δ 1.220 (3H, d, J=6.0 Hz, H-11), δ 0.826 (3H, d, J=6.5 Hz, H-14), δ 0.772 (3H, d, J=6.5 Hz, H-15).
3.4.2. (R)-MTPA ester
Compound 2 (1.0 mg) was reacted with (S)-(+)- α-methoxy-α-(trifluoromethyl)-phenylacetyl chloride (20 μL) under similar conditions and purified as said above to give the (R)-MTPA ester derivative of 2. 1H NMR (500 MHz, CDCl3): δ 5.909 (1H, d, J=16.0 Hz, H-5), 5.295 (1H, dd, J=16.0, 9.5 Hz, H-6), 4.895 (1H, s, Ha-12), 4.807 (1H, s, Hb-12), 4.303 (2H, m, H-1), 4.303 (1H, m, H-10), 2.204 (2H, m, H-3), δ 1.290 (3H, d, J=6.0 Hz, H-11), δ 0.755 (3H, d, J=6.5 Hz, H-14), δ 0.708 (3H, d, J=6.5 Hz, H-15).
Acknowledgments
This work is supported by a grant from US National Institutes of Health/National Cancer Institute (Grant No. R01 CA90265) and this support is gratefully acknowledged. We thank 195 Dr Stanley H. Faeth (Arizona State University) for providing the fungal strain and C.J. Seliga, S. Wittlinger, C. Hamilton, L. Morse, C. Hayes and A. Das for their expert technical assistance.
Footnotes
Part 19 in the series: Studies on Arid Land Plants and Microorganisms. For part 18 see: Wijeratne, E.M.K., Paranagama, P.A., Marron, M.T.; Gunatilaka, M.K., Arnold, A.E.; Gunatilaka, A.A.L., J. Nat. Prod. 71, 218 (2008).
References
- Aasen AJ, Hlubucek JR, Enzell CR. Acta Chemica Scandinavica B. 1975a;29:677. doi: 10.3891/acta.chem.scand.29b-0677. [DOI] [PubMed] [Google Scholar]
- Aasen AJ, Junker N, Enzell CR. Tetrahedron Letters. 1975b;30:2607. [Google Scholar]
- Bamburg JR, Strong FM, Smalley EB. Journal of Agricultural and Food Chemistry. 1969;17:443. [Google Scholar]
- Bansal A, Moriarity DM, Takaku S, Setzer WN. Natural Products Communications. 2007;2:781. [Google Scholar]
- Bashyal BP, Wijeratne EMK, Faeth SH, Gunatilaka AAL. Journal of Natural Products. 2005;68:724. doi: 10.1021/np058014b. [DOI] [PubMed] [Google Scholar]
- Bashyal BP, Faeth SH, Gunatilaka AAL. Natural Products Communications. 2007;2:547. [Google Scholar]
- Coates P, Ghisalberti EL, Jefferies PR. Australian Journal of Chemistry. 1977;30:2717. [Google Scholar]
- Demole E, Enggist P. Helvetica Chimica Acta. 1975;58:1602. doi: 10.1002/hlca.19750580614. [DOI] [PubMed] [Google Scholar]
- Engelhardt G, Schuster M, Lepschy J, Wallnoefer PR. Zeitschrift für Lebensmittel-Untersuchung und – Forschung. 1986;182:123. doi: 10.1007/BF01042812. [DOI] [PubMed] [Google Scholar]
- Gu G, Ma Q, Miao Z. Zhenjun Xuebao. 1994;13:80. [Google Scholar]
- He X, Cane DE. Journal of the American Chemical Society. 2004;126:2678. doi: 10.1021/ja039929k. [DOI] [PubMed] [Google Scholar]
- Hood RD, Kuczuk HM, Szczech GM. Teratology. 1978;17:25. doi: 10.1002/tera.1420170108. [DOI] [PubMed] [Google Scholar]
- Le Quere JL, Semon E, Latrasse A, Etievant P. Sciences des Aliments. 1987;7:93. [Google Scholar]
- Ohtani I, Kusumi T, Kashman Y, Kakisawa H. Journal of the American Chemical Society. 1991;113:4092. [Google Scholar]
- Rizzo A, Ferracane R, Ritieni A. Applied and Environmental Microbiology. 2002;68:82. doi: 10.1128/AEM.68.1.82-85.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein LV, Shoemaker RH, Paul KD, Simon RM, Tosini S, Skehan P, et al. Journal of the National Cancer Institute. 1990;82:1113. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- Solfrizzo M, Visconti A. Journal of Chromatography A. 1996;730:69. doi: 10.1016/0021-9673(95)00899-3. [DOI] [PubMed] [Google Scholar]
- Solfrizzo M, Visconti A, Savard ME, Blackwell BA, Nelson PE. Mycopathologia. 1994;127:95. doi: 10.1007/BF01103065. [DOI] [PubMed] [Google Scholar]
- Su BN, Park EJ, Mbwambo ZH, Santarsiero BD, Mesecar AD, Fong HHS, et al. Journal of Natural Products. 2002;65:1278. doi: 10.1021/np0202475. [DOI] [PubMed] [Google Scholar]
- Tsuchiya Y, Matsumoto A, Shudo K, Okamoto T. Yakugaku zasshi: Journal of the Pharmaceutical Society of Japan. 1980;100:468. [PubMed] [Google Scholar]
- Vesela D, Vesely D, Adamkova A. Veterinary Medicine. 1981;26:737. [PubMed] [Google Scholar]
- Visconti A, Solfrizzo M. Journal of Chromatography A. 1994;42:195. [Google Scholar]
- Visconti A, Solfrizzo M, Fruchier A, ApSimon JW. Journal of Natural Products. 1994;57:695. [Google Scholar]
- Wijeratne EMK, Turbyville TJ, Zhang Z, Bigelow D, Pierson LS, III, VanEtten HD, et al. Journal of Natural Products. 2003;66:1567. doi: 10.1021/np030266u. [DOI] [PubMed] [Google Scholar]
- Zhan J, Burns AM, Liu MX, Faeth SH, Gunatilaka AAL. Journal of Natural Products. 2007;70:227. doi: 10.1021/np060394t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Peng L, Zhang T, Mei T, Liu H, Li Y. Synthetic Communications. 2003;33:3761. [Google Scholar]