

Abstract
HIV-1-associated neurocognitive disorders (HAND) remains a significant source of morbidity in the era of wide spread use of highly active antiretroviral therapy. Disease is precipitated by low levels of viral growth and glial immune activation within the central nervous system. Blood borne macrophage and microglia affect a proinflammatory response and release viral proteins that affects neuronal viability and leads to death of nerve cells. Increasing evidence supports the notion that HAND is functional channelopathy, but proof of this concept remains incomplete. Based on their role in learning and memory processes, we now posit that voltage-gated potassium (Kv) channels could be a functional substrate for disease. This was tested in the severe combined immunodeficient (SCID) mouse model of HIV-1 encephalitis (HIVE) by examining whether the Kv channel blocker, 4-aminopyridine (4-AP), could affect behavioral, electrophysiological, and morphological measures of learning and memory. HIVE SCID mice showed impaired spatial memory in radial arm water maze tests. Electrophysiology studies revealed a reduction of long-term potentiation (LTP) in the CA1 region of the hippocampus. Importantly, systemic administration of 4-AP blocked HIV-1-associated reduction of LTP and improved animal performance in the radial arm water maze. These results support the importance of Kv channel dysfunction in disease but, more importantly, provide a potential target for adjunctive therapies for HAND.
Keywords: voltage-gated K+ channels, 4-aminopyridine, hippocampal slice, EPSP, learning and memory, HIV-1 encephalitis
Introduction
Human immunodeficiency virus (HIV)-associated neurocognitive disorder (HAND) is a common complication of progressive viral infection in the era of highly active antiretroviral therapy (HAART). Disease commonly progresses from asymptomatic neurocognitive impairment to HIV-associated mild neurocognitive disorder and, ultimately, to HIV-associated dementia (HAD; Gonzalez-Scarano and Martin-Garcia 2005; McArthur et al. 2005; Antinori et al. 2007). While the incidence of HAND has fallen by HAART, extended life spans and growing resistance to antiviral therapy due to viral strain mutation have led to its increased prevalence (Sacktor et al. 2001, 2002; Neuenburg et al. 2002; Kandanearatchi et al. 2003; McArthur et al. 2003). In regards to disease pathogenesis, common high levels of viral replication in brain have been supplanted by more subtle viral growth and mild clinical disease. Indeed, asymptomatic neurocognitive impairment and mild cognitive motor disorder are now most common disease manifestations (Ellis et al. 1997; Sacktor et al. 2002; Antinori et al. 2007). As HAART can neither abrogate viral growth in its central nervous system sanctuaries nor provide complete protection from neurological dysfunction associated with HIV-1 infection, research into the mechanisms and treatments, including adjunctive therapies, are imperative (Bouwman et al. 1998; Sacktor et al. 2002; Langford et al. 2003; Zakharenko et al. 2003).
Despite more than two decades of investigation, the mechanisms for HAND pathogenesis remain incompletely understood. It is widely accepted that virus-linked cognitive impairments are strongly associated with the presence of immune competent and virus-infected mononuclear phagocytes (MP; blood borne macrophages and microglia; Glass et al. 1995; Nath 1999; Kaul et al. 2001). MP secretory products affect disease endpoints specifically neuronal injuries that include dendritic pruning and the reduction of synaptic contacts (Masliah et al. 1997; Everall et al. 1999), apoptosis (Adle-Biassette et al. 1995; Gelbard et al. 1995; Petito and Roberts 1995), and selective neuronal loss (Masliah et al. 1992; Fox et al. 1997). However, reduced neuropathology is seen in recent years for HAND and improvements in cognitive function have been observed by HAART (Gendelman et al. 1998; Tozzi et al. 1999; Parsons et al. 2006; Sacktor et al. 2006; McCutchan et al. 2007). This suggests that the cognitive decline seen during HAND may result more from neuronal dysfunction than from frank cell death.
Recent studies provide compelling evidence that neuronal voltage-gated potassium (K+) channels (Kv) play an important role in memory processes. Neuronal K+ currents decrease during learning and animal mutants with Kv channel dysfunction exhibit learning and memory deficits (Giese et al. 1998, 2001; Solntseva et al. 2003). Furthermore, Kv channel antagonists have been shown to improve learning and memory, while Kv channel “openers” diminish it (Messier et al. 1991; Ghelardini et al. 1998; Solntseva et al. 2003). Thus, alteration of Kv channel activity may affect learning and memory processes. Studies from our laboratory revealed conditioned media recovered from HIV-1gp120- and/or lipopolysaccharide-stimulated human monocyte-derived macrophages (MDM) produces significant increase of 4-aminopyridine (4-AP)-sensitive transient K+ current and resultant neuronal injury in cultured rat hippocampal neurons (manuscript under preparation). We hypothesize HIV-1-infected MP induce neuronal dysfunction by alteration of neuronal Kv channel physiology, leading to cognitive impairment. To test this hypothesis, we studied the protective effects of 4-AP, a nonspecific Kv channel blocker, on neurocognitive processes in a severe combined immunodeficient (SCID) mouse model of HIV-1 encephalitis (HIVE). Our results showed that 4-AP ameliorates HIV-1-induced inhibition of long-term potentiation (LTP) and improves animal performance in the radial arm water maze system. These results indicate that Kv channels are linked to HAND. Our data also suggest that Kv channels could be the potential targets for the development of adjunctive therapeutic modalities in disease.
Materials and methods
Isolation and culture of primary human monocytes
Human monocytes were obtained from HIV-1, HIV-2, and hepatitis B seronegative adults by leukophoresis and purified by countercurrent centrifugal elutriation. Monocytes were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Sigma, St. Louis, MO, USA) with 10% heat-inactivated pooled human serum, 1% glutamine, 50 μg/ml gentamicin, 10 μg/ml ciprofloxacin, and 1,000 U/ml highly purified recombinant human macrophage colony-stimulating factor (a generous gift from Wyeth Pharmaceuticals, Inc., Cambridge, MA, USA). Monocytes were plated at a density of 2×107 cells/ml for 7 days in Teflon flasks to permit cellular differentiation. Tissue culture reagents were screened before use and found negative for endotoxin (<10 pg/ml; Associates of Cape Cod, Inc., Woods Hole, MA, USA) and mycoplasma contamination (Gen-probe II; Gen-probe Inc., San Diego, CA, USA).
HIVE SCID mice and 4-AP administration
SCID mice (male C.B-17/IcrCrl-scidBR, 4 weeks old) purchased from Charles River Laboratories (Wilmington, MA, USA) were inoculated into the basal ganglia (caudate and putamen) with 10 μl of either an HIV-1ADA-infected or uninfected human MDM suspension containing 3×105 cells. Sham-operated animals received 10 μl of serum-free DMEM. 4-AP (5 mg/kg) was administered daily by intraperitoneal injection beginning on the day of MDM inoculation and continuing until sacrifice for electrophysiological studies. The Institutional Animal Use and Care Committee (IACUC) of University of Nebraska Medical Center (IACUC # 05-020-05) approved all animal use procedures.
Histopathology
Brain tissue was collected at necropsy, fixed in 4% phosphate-buffered paraformaldehyde, and embedded in paraffin. Blocks were cut to identify the injection site. For each mouse, 30–100 serial (5 μm in thickness) coronal sections were cut from the injection site at the level of the hippocampus. Immunohistochemical staining followed a basic indirect protocol. Antibodies to CD68 and HIV-1 p24 antigen (Dako) were applied to determine the number of human MDMs and HIV-1-infected cells. Astrocytes were identified using antibodies specific for glial fibrillary acidic protein (GFAP). All paraffin-embedded sections were counterstained with Mayer’s hematoxylin.
Radial arm water maze
Behavioral testing was conducted using a six-arm radial arm water maze apparatus (diameter 91 cm, height 110 cm) containing a submerged platform (13×13 cm, 2 cm below water level) in one arm. A small fixed light source along with intramaze visual cues was used for spatial orientation. Room and water temperature were maintained at 22°C and 17–22°C, respectively. A 3-day protocol, consisting of five blocks of three trials each, for a total of 15 trials per day, was performed 1 week prior to, 1 week after, and 2 weeks after inoculation. Two measurements were recorded during each 60-s trial: the number of incorrect arm choices (errors) and the amount of time required to discover the hidden platform (escape latency). Upon successfully locating the platform, mice remained on the platform for 10–15 s before being removed from water maze, dried, and given food and water. For each block, mice were run in cohorts of three, allowing for extended periods of rest between blocks. The goal arm for each individual animal was randomly assigned and fixed for a particular 3-day protocol, while the start arm varied randomly for each trial. In subsequent testing, each individual mouse was randomly assigned a different goal arm in order to assess new learning and memory. To assess whether the groups differed in the motor or proximal visual skills necessary to locate and escape to the platform, animals were tested before and after injection of infected or uninfected MDMs using a visible platform. The test runs were captured and analyzed for swim speed using Noldus EthoVision video tracking system (Leesburg, VA, USA).
Hippocampal slices and electrophysiology
Hippocampal slices were prepared as described previously (Anderson et al. 2003). Briefly, animals were anesthetized with isoflurane and decapitated. The brains were quickly removed to ice-cold (2–4°C) artificial cerebral spinal fluid (ACSF) containing (in millimolars): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1 NaH2PO3, 26 NaCO3, and 10 glucose, equilibrated with 95% O2/5% CO2 to a pH of 7.35–7.5. Transverse hippocampal slices (400 μm in thickness) were then cut using a tissue chopper and incubated in an oxygenated holding chamber for at least 1 h before being used for experiments. For recording, a single slice was submerged in the recording chamber and perfused continuously with ACSF (30°C, 2 ml/min). Field excitatory postsynaptic potentials (fEPSPs) were elicited by constant current (50–300 μA), low frequency (0.05 Hz) orthodromic stimulation of Schaffer collateral–commissural axons with the use of an insulated bipolar tungsten electrode (except for the tip). The stimulation intensity was adjusted to generate approximately 40–50% of a maximal response. The evoked fEPSPs were recorded with an Axopatch-1D amplifier (Axon Instruments, Inc., Union City, CA, USA) in the CA1 dendrite field (stratum radium). The recording microelectrodes were made from borosilicated glass capillaries. The tip diameter of the microelectrode was 3–5 μm and had a resistance of 1–5 MΩ when filled with ACSF. High-frequency stimulation (HFS; 100 Hz, 500 ms×2) was delivered after a 20-min control (baseline) recording. The initial slopes of the fEPSPs were measured and expressed as a percentage of the basal level.
Electron microscopy
After completion of water maze testing, animals were sacrificed and hippocampal slices were cut and immediately fixed in 2% glutaraldehyde, 2% paraformaldehyde, and 0.5% acrolein. Vials were left at room temperature for 1–2 h, then stored at 4°C when necessary. Tissue slices were then washed with 0.1 M Sorenson’s phosphate buffer and postfixed in 1% osmium tetroxide for 1 h before dehydration processes. After dehydration, tissue slices were embedded in blocks of media (39.1 g Araldite, 23.0 g dodecenyl succinic anhydride, and 1.0 2,4,6-tri(dimethylaminomethyl)phenol). Thin sections were stained with 2% uranyl acetate and Reynolds lead citrate and examined with a Philips 410LS transmission electron microscope (TEM) operated at 60 kV. Micrographs were systematically and randomly taken in the CA1 dendrite region at magnifications of ×21,000 and ×54,000. The number of active zones (the sites of synaptic vesicle docking and neurotransmitter release) per visual field (approximately 12.5 μm2 at ×21,000) and their average lengths were then analyzed using image processing software.
Statistical analysis
Unpaired two-tailed student’s t test and analysis of variance tests were performed to determine the level of significance. Data are expressed as mean ± standard error (M ± SE). The level of significance was determined at p<0.05
Results
HIVE SCID mice
To investigate the mechanisms underlying HIV-1 neuropathogenesis, we have developed a murine model of fulminate human disease by injecting HIV-1ADA-infected MDM (HIVMDM) into the subcortical brain region (the basal ganglia) of SCID mice. Animals injected with HIV-1 MDM recapitulate HIVE (Persidsky et al. 1996; Anderson et al. 2003). To confirm the validity of HIVE mouse model, histopathological examination of brain tissue was performed. Paraformaldehyde-fixed paraffin-embedded coronal brain sections of MDM-injected (control) and HIV-1 MDM-injected SCID mice were immunolabeled with antibodies to GFAP, CD68, and HIV-1p24 antigen (Dako, Carpinteria, CA, USA). GFAP immunolabeling revealed more prevalent glioastrocytic activation in HIVMDM-injected animals (Fig. 1b) compared to controls (Fig. 1a). The injected HIVMDMs were detected within the injected area by an anti-CD68 antibody (Fig. 1c). Using an antibody for HIV-1p24, we found multinucleated giant cells, a neuropathological hallmark of HIVE in the brains of virus-infected MDM injected animals (Fig. 1d).
Fig. 1.
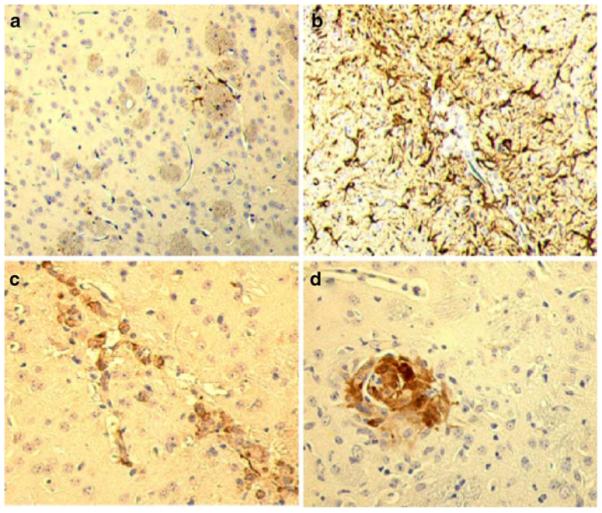
HIVE in SCID mice. HIV-1ADA-infected MDM were stereotactically injected into the basal ganglia of SCID mice. Paraformaldehyde-fixed paraffin-embedded coronal brain sections were immunolabeled with GFAP, human CD68, and HIV-1 p24. In comparison with brain sections from control animals (a), widespread GFAP-positive astrocyte reactions surround the injection site of HIV-1ADA-infected human MDM (b). Human MDM were determined by immunostaining with CD68 and found to be localized within the injection area (c). An example of HIV-1 p24 positive multinucleated giant cells found in a SCID mouse brain 7 days after injection (d). Original magnification ×200
Learning and memory are protected by 4-AP
Cognitive decline is one of the most prominent symptoms seen in AIDS patients with HAD. To determine whether brain injection of HIVMDM impairs animal learning and memory and to evaluate the effects of 4-AP on animal behavior, we performed a radial arm water maze test to detect potential cognitive deficits. In order to evaluate demonstrable cognition, the number of incorrect arm choices (errors) and the time to find the hidden platform (escape latency) were measured 6–8 and 13–15 days after brain injection.
In agreement with previous findings (Anderson et al. 2003, 2004), HIVE SCID mice demonstrated impaired performance in tests assessing spatial learning and memory. Although measurements were not significant at every block, intraday trends 1 week after inoculation indicate consistent learning deficiencies in HIVMDM-injected SCID mice when compared to the control group. For example, in block five on the first day, the average time to find the platform (31.2±6.4 s, n=15) and the average number of errors (4.1±1.0 errors, n=15) were significantly (p<0.05) higher in HIVE animals than the MDM-injected (control) animals (13.7±2.5 s, n=12; 1.3±0.4 errors, n=12), demonstrating an impairment of spatial learning and memory in HIVE animals (Fig. 2a). Furthermore, the average escape latency (7.5±0.8 s, n=12) and average number of errors (0.7±0.3 s, n=12) were significantly lowered (p<0.01) with administration of 4-AP to HIVE mice, suggesting the blockade of voltage-gated K channels can ameliorate HIV-1-associated impairments of animal behavior. Similarly, for all three testing days 1 week after injection, the HIVE mice showed a significantly longer escape latency compared to both the control (uninfected MDM-injected) and 4-AP-treated HIVE mice (Fig. 2c; p<0.01). For example, the average trial time for HIVE mice on day three (17.2±1.8 s, n=60) was significantly slower than those for both the control mice (8.7±0.7 s, n=60, p<0.001) and the 4-AP-treated HIVE mice (7.0±0.6 s, n=60, p<0.001). As expected, an assessment of errors with respect to days mirrored this result (Fig. 2d, p<0.01). To evaluate the possibility that HIVE mice lessened animals’ anxiety in the water or motor skills, we conducted swim speed tests before and after HIV-1 MDM brain injections. As shown in Table 1, no significant difference between HIVMDM-injected groups was observed (n=3 in each group).
Fig. 2.

4-AP ameliorates learning and memory deficits in HIVE mice. Water maze escape latency and error analysis were used to evaluate spatial learning and memory behavior. SCID mice were injected with either HIV-1ADA-infected macrophages (HIVE) or uninfected macrophages (Control), and a subgroup of HIVE mice were subsequently i.p. injected each day with 5 mg/kg 4-AP. A 3-day protocol, consisting of five blocks of three trials each, for a total of 15 trials per day, was performed 1 and 2 weeks after injection. The amount of time required to discover the hidden platform (escape latency) and the number of incorrect arm choices (errors) were recorded for each trial. One week after inoculation, block analysis by escape latency (a) and errors (b) indicates slower learning, inter-day forgetting, and submaximal performance plateaus in HIVE animals, which were not seen with 4-AP administration. Day analysis reveals significant deficits (in both escape latency and errors) in HIVE animals at weeks 1 (c, d) and 2 (e, f) after inoculation, as well as recovered performance in 4-AP treated HIVE mice. The y-axes in a, c, and e are escape latency in seconds, and in b, d, and f are error frequency
Table 1.
Swimming speed (centimeter/second)
| Control | HIV | HIV + 4-AP | |
|---|---|---|---|
| Before injection | 14.71±0.45a | 12.98±0.34 | 13.67±0.48 |
| After injection | 13.26±0.44 | 11.34±0.40 | 11.90±0.36 |
Mean ± SE. Each group has three animals
In order to determine if the neurotoxic effect of HIVE diminished new learning and memory, a second week of water maze testing was performed. On the first day of testing, the MDM and the HIVE groups performed about the same with respect to both escape latency and errors (Fig 2e, f). However, results from the second and third testing days parallel the results of week 1, suggesting persistent HIV-1 related damage (Fig 2e, f; p<0.001). Notably, on each day in the second week of testing, the 4-AP treated group outperformed the HIVE group with significantly faster trials times and fewer errors (Fig 2e, f; p<0.001). For example, the average escape latency for HIVE mice on day 3 of the second week was 17.5±2.6 s (n=75), which was significantly slower than in the uninfected macrophage and 4-AP treated groups (6.7±0.6 s, n=60, p<0.001; 4.6±0.2 s, n=60, p<0.001). Taken together, these behavioral studies revealed deficits in learning (intraday) and memory (intraweek, interweek) in HIVE SCID mice, while demonstrating significantly improved learning and memory scores in response to 4-AP treatment.
4-AP attenuated HIVE LTP inhibitions
In our animal behavior study, we found 4-AP ameliorates HIVMDM-induced impairment of spatial learning and memory in HIVE mice. Next, we sought to determine whether these HIVE-associated changes in animal behavior would correlate with altered synaptic transmission and plasticity and to examine whether Kv channel antagonist 4-AP would have a protective effect. Hippocampal slices were therefore harvested from sacrificed animals at 1 (days 6–8) and 2 weeks (days 13–15) after injection for the study of LTP, a long-lasting enhancement in synaptic strength which is widely believed be the cellular correlate of learning and memory.
As reported previously, high-frequency stimulation of the Schaffer-collateral pathway reliably induced LTP in the CA1 region of the hippocampus in unmanipulated animals (Anderson et al. 2003), with an average LTP magnitude of 205.7±12.9% of basal level (n=5, data not shown) measured 60 min after HFS. Using the identical protocol, HIVE mice exhibited a significant decrease in LTP magnitude compared with animals injected with uninfected MDMs (Fig. 3). The average LTP magnitude measured 60 min after HFS was 153.3±27.9% of the basal level (n=11) in HIVMDM-injected mice, compared with an average LTP magnitude of 206.0±63.4% in MDM-injected mice (n=10). Consistent with our previous findings (Anderson et al. 2003), this statistically significant difference (p<0.05) indicates the injection of HIVMDM inhibits LTP.
Fig. 3.
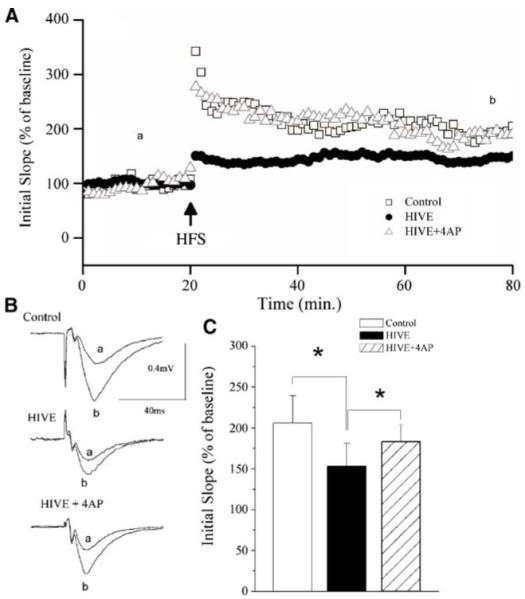
4-AP attenuates HIV-induced inhibition of LTP in the CA1 region of SCID mouse hippocampal slices. SCID mice were injected with HIV-1ADA-infected macrophages or uninfected macrophages, and a subgroup of those injected with HIV-1ADA-infected macrophages were daily administered with 4-AP (5 mg/kg). A The time course and magnitude of LTP in the Schaffer collateral to CA1 synapses recorded from hippocampal slices prepared from SCID mice injected with either HIV-1ADA-infected macrophages (HIVE), uninfected macrophages (control), or HIV-1ADA-infected macrophages and 4-AP (HIVE+4AP). The graph plots the initial slope of the evoked EPSPs recorded from the CA1 dendrite field (stratum radium) in response to constant current stimuli. HFS (500 ms×2) was delivered at the time indicated by an arrow. Each point in the graph is an average of three consecutive sweeps. Note that injection of HIV-1ADA-infected macrophages inhibited LTP (circle) and administration of 4-AP abolished the HIV-1ADA-associated inhibition (triangle). B Individual fEPSPs taken from different time points (a) before and (b) after HFS. C The bar graph showing the average LTP during the last 20 min of each group, demonstrating that HIV-1 inhibits LTP and systemic administration of a Kv channel antagonist, 4-AP, significantly blocked the HIV-1-associated inhibition
Furthermore, this HIVMDM-associated inhibition of LTP could be prevented with systemic administration of 4-AP. The LTP magnitude recorded in HIVE mice with administration of 4-AP was 183.3±20.3% of the basal level (n=6), which was statistically different from the LTP magnitude recorded in HIVE mice without 4-AP administration (p<0.05). This suggests that the K+ channel antagonist 4-AP can ameliorate HIVE associated LTP inhibition. These results show the neurotoxic effects HIVE on electrophysiological properties related to learning and memory and demonstrate that a K+ channel antagonist can restore diminished function attributable to HIV-1ADA-infected MDM.
HIVE and hippocampal synapses
Previous in vitro studies in our laboratory have demonstrated HIV-1-infected MDM culture supernatant inhibits synaptic transmission in the CA1 region of rat hippocampus through presynaptic mechanisms (Xiong et al. 1999). The inhibitory effect of HIVMDM on hippocampal LTP has now been recapitulated in SCID mouse with HIVE. To further investigate the possible nature of HIV/MDM inhibition, electron microscopy was performed in the CA1 region of the hippocampus to assess any evident changes in synaptic morphology and to determine if these changes would be effected by 4-AP treatment.
After completion of the radial arm water maze behavior test, animals were sacrificed and hippocampal brain tissues were prepared using standard procedures for electron microscopy. Micrographs of the CA1 dendrite region were systematically and randomly collected by transmission electron microscopy and evaluated for the number and length of active zones present (Fig. 4). Analysis of these electron micrographs revealed no significant differences between the number of active zones in uninfected MDM-injected (7.4±2.0, n=40 visual fields) and HIVE animals with (6.8±2.1, n=40) or without 4-AP treatment (7.1±2.3, n=40). However, the lengths of the active zones in HIVE animals (214.3±61.3 nm, n=296 synapses) were significantly (p<0.01) reduced compared to those of MDM-injected (control) animals (252.3±67.7 nm, n=272). Furthermore, the HIVMDM-associated reduction in active zone length was prevented by systemic administration of 4-AP. The average active zone length in the 4-AP injected group was 262.5±70.2 (n=284), which was significantly different than the average length observed in untreated HIVMDM-injected animals (p<0.01). These results demonstrate inoculation of SCID mice with HIVMDM damages synaptic structures in the hippocampus, a brain region associated with learning and memory, and that this damage is prevented by systemic administration of the K+ current blocker 4-AP.
Fig. 4.
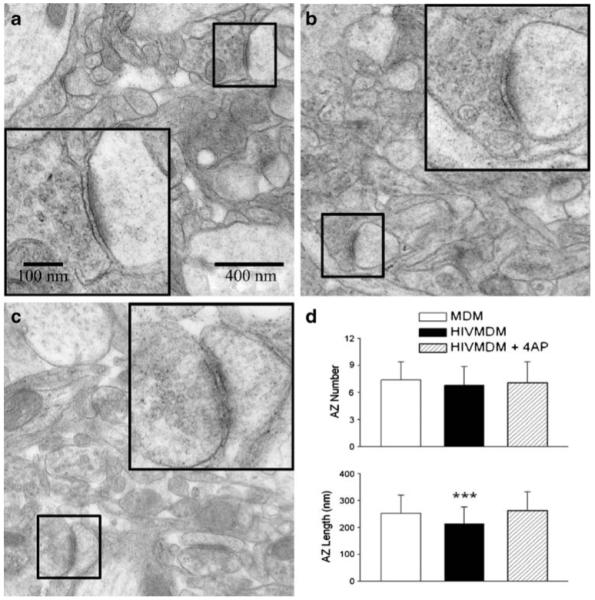
Reduction of active zone length in HIVE mice and its protection by 4-AP. After completion of the water maze, hippocampal slices were fixed and embedded. One hundred-nanometer thin sections were then cut in the neuropil and stained with uranyl acetate and lead citrate. Micrographs were taken using a TEM at magnifications of ×21,000 and ×54,000. The number of active zones per visual field (approximately 12.5 μm2) and their average length were then determined for all groups (three animals in each group). No difference was found in the number of active zones between groups; however, active zone lengths in HIVE animals were found to be significantly shorter than both those in SCID mice injected with uninfected macrophage and HIVE animals treated with 4-AP (d). Representative micrographs taken from mice injected with uninfected macrophage (a), HIV-1ADA-infected macrophage (b), and HIV-1ADA-infected macrophage with daily 4-AP i.p. treatments (c). Examples of active zones for each group are shown at original (small boxes) and high magnification (large inset boxes)
Discussion
Kv channels play a crucial role in the generation of neuronal electrical activity. They repolarize action potentials (APs), set interspike intervals, modulate the resting membrane potential, regulate the AP duration, and stabilize the membrane potential of both excitable and nonexcitable cells. As such, Kv channels are the main regulators of both a neuron’s ability to fire APs and the shape of the AP waveform. As the number of AP or the AP firing patterns encode information (Reike et al. 1997), modulation of Kv channel activity may alter information processing (i.e., learning and memory). Moreover, activations of Kv channels are involved in apoptotic volume decrease due to efflux of K+, Cl−, and H2O and ultimately resulting in apoptosis (Remillard and Yuan 2004). Thus, the dysfunction of Kv channels is believed to be an important link in the mechanisms of memory disturbance. In this study, we demonstrated that systemically administered 4-AP, a non-specific Kv channel blocker, ameliorates cognitive deficits in a murine model of HIVE.
In past studies, an animal model system for HIVE was developed (Persidsky et al. 1996; Persidsky and Gendelman 1997; Anderson et al. 2003, 2004). Animals in this model recapitulate human HIVE following stereotactic injection of HIV-1ADA-infected MDM into the basal ganglia, an area severely affected in HIVE (Glass et al. 1993; Wiley and Achim 1994) and develop behavior abnormalities (Avgeropoulos et al. 1998; Anderson et al. 2003; Sas et al. 2007). In the present study, we validated the inoculation of SCID mice with HIV-1ADA-infected MDM as a model for HAD by demonstrating deficiencies in animal learning and memory and LTP induction in the hippocampus. In order to test whether systemic administration of 4-AP could produce protective effects on HIV-1-associated impairment in spatial learning and memory, we first established a water maze protocol by which we could reliably quantify the cognitive deficits in animals with HIVE. Although measurements were not significant at every block, intraday trends indicate learning deficiencies in SCID mice injected with HIV-infected MDM when compared to those injected with uninfected MDM. Intraweek trends provide further evidence for learning and memory deficiencies in HIVE animals. While learning occurred as early as the second trial in the control group, HIVE animals exhibited substantial deficiencies in learning and memory on all testing dates, with the exception of the first day (day 10) in the second week. On this day, the goal platform position was changed in order to test new learning and memory. As the new task required “forgetting” the old platform position and learning the new one, the control group may have been remembering the previous platform position, while the HIVE group could have both forgotten the old platform position and floundered in efforts to learn the new one. In all cases of HIVE-associated impairment in learning and memory, administration of 4-AP significantly improved animal behavior in water maze tests. These results suggest that Kv channels may be involved in the viral neuropathogenesis and that the blockade of Kv channels can protect neuronal cells from HIV-1-associated injury.
The protective effects of 4-AP administration were also demonstrated in electrophysiological studies of synaptic transmission and neuronal plasticity by HIVE induction. In SCID mice injected with HIV-1-infected MDM, significant impairment of LTP induction was detected in the CA1 region of the hippocampus. Our experiments showed that the daily administration of 4-AP exerts significant protection against this HIV-1-induced impairment. These findings parallel well the results obtained in animal behavior studies, providing further evidence to support a channelopathy hypothesis for HAND as proposed by Gelman and his colleagues (Gelman et al. 2004). It should be noted that as the total duration of the experiment constituted a relatively acute exposure to the suspected neurotoxic products of HIV-1ADA-infected macrophage compared to the chronic condition experienced by HAND patients, these results may represent a less fulminant form of HAND. However, in the context of the gradual onset and varying degrees of HAND, our experiments may nonetheless capture its mechanism.
Having established a role for Kv channel dysfunction in HAND using behavioral and electrophysiological tests, we further sought to associate these results with any observed physical changes in the structures involved in LTP and learning and memory. Golgi staining studies performed in our laboratory (data not shown) revealed reduced dendritic arborization in the hippocampus, which could account for some of the observed deficiencies. To further examine the potential mechanism(s) underlying the observed deficiencies, we performed electron microscopy to analyze synaptic morphology. In particular, we examined the number and length of active zones in the hippocampus, based on our previous results showing HIV-1-infected MDM-conditioned media inhibits LTP via a presynaptic mechanism (Xiong et al. 1999). Given the reduced dendritic arbor in our Golgi stain studies, we expected to observe a decrease in the number of active zones, but instead found similar numbers in all three groups. The lengths of the active zones in the CA1 regions of slices taken from HIVE mice, however, were significantly reduced compared to controls. This reduction was not observed when HIVMDM-injected mice were treated daily with 4-AP. It should be noted that while the length of the active zone in part depends on the shape, orientation, and section depth with respect to the synapse, our large sample size accounts for these natural variations. The significantly shorter active zones in the HIVE group likely indicate a reduction in the typical activity-dependent increases in active zone size, which would normally be expected to accompany the spatial learning task training performed in the weeks prior to sacrifice (Lisman and Harris 1993; Malenka and Bear 2004). As the reduced active zone length reflects a reduction in three-dimensional synaptic surface, it could reasonably be assumed several factors of synaptic transmission would be diminished, thereby compromising memory integrity. Thus, the greater active zone length in the 4-AP treated group gives us morphological evidence in agreement with our functional observations, allowing us to conclude based on our evidence that Kv channels play an important role in viral neuropathogenesis.
As Kv channels are expressed both in neuronal cells and macrophages (Gallin 1991; Mackenzie et al. 2003; Vicente et al. 2003), the systemic administration of 4-AP may provide neuroprotection by blocking Kv channels expressed not only in the host neurons but also the injected macrophages. Studies have shown that increased Kv channel conductance is necessary for macrophage activation and cytokine production (Blunck et al. 2001; Irvine et al. 2007), and upregulated Kv expression is correlated with immune cell activation (Rus et al. 2005). Kv channel blockers have now been used to alleviate the neuropathology and symptoms of experimental autoimmune encephalomyelitis (Beeton et al. 2001) through their ability to reduce immune cell activation (Beeton et al. 2001; Vicente et al. 2003), proliferation (Beeton et al. 2001; Vicente et al. 2003), and production of immune active molecules, such as interleukins and tumor necrosis factor (Haslberger et al. 1992; Maruyama et al. 1994; Beeton et al. 2001; Qiu et al. 2002). Thus, systemic administration of 4-AP may reduce macrophage activation and resultant cytokine production, contributing to the overall protective effect as seen in this study. On the other hand, the macrophage-secreted immune active molecules may affect neuronal Kv channels, resulting to neuronal dysfunction or injury. We have demonstrated that the conditioned media recovered from HIV-1-infected human MDM, which contains soluble immune active molecules, enhanced neuronal outward K+ currents and induced neuronal injury through activation of neuronal Kv channels (Hu et al. 2007). This MDM-conditioned media-induced neuronal injury was blocked by Kv channel antagonist 4-AP or tetraethylammonium, indicating that neuronal Kv channels are involved in HIV-1-associated neuronal injury. Thereby, the neuroprotective effects of systemic administered 4-AP may be a consequence of the blockade of neuronal Kv channels. In either case, the particular involvement of macrophage and neuronal Kv channels in HAD pathology and the possible benefits of 4-AP treatment deserve consideration.
While in our model of HIV-associated dementia 4-AP treatment has proven beneficial, consideration of the possible effect of 4-AP on nonneuronal Kv channels (e.g., cardiac Kv channels) must be taken into account in potential clinical application. During the course of our experiment, one animal receiving daily 4-AP injections died, possibly due to complications related to cardiac potassium channels. In fairness, it should be noted that all other treated animals survived, moving and swimming freely, indistinguishable in their gross motor behavior from animals that were not treated. In addition, the use of 4-AP is not without historical foundation. It originally formed the central ring of tacrine, the first drug prescribed to treat Alzheimer’s type dementia, and has since yielded benefits in the treatment of neuroinflammatory diseases such as multiple sclerosis (Stefoski et al. 1987; Davis et al. 1990; Bever 1994; Bever et al. 1994). These results suggest that Kv channel antagonists may be a potential adjunctive treatment for HAND.
Acknowledgments
The authors thank Mr. Tom Bargar and Dr. Jorge F. Rodriquez-Sierra for their assistance in electron microscopic studies. The authors extend a special thanks to Ms. Julie Ditter and Ms. Robin Taylor for their excellent administrative support. This work was supported by NIH grants R01 2NS041862.
Grant support: NIH NINDS 2R01 NS04862.
Contributor Information
James P. Keblesh, Neurophysiology Laboratory, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Center for Neurovirology and Neurodegenerative Disorders, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA
Huanyu Dou, Center for Neurovirology and Neurodegenerative Disorders, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Howard E. Gendelman, Center for Neurovirology and Neurodegenerative Disorders, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA
Huangui Xiong, Neurophysiology Laboratory, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Center for Neurovirology and Neurodegenerative Disorders, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
References
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F. Neuronal apoptosis in HIV infection in adults. Neuropathol Appl Neurobiol. 1995;21:218–227. doi: 10.1111/j.1365-2990.1995.tb01053.x. doi:10.1111/j.1365-2990.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Anderson ER, Boyle J, Zink WE, Persidsky Y, Gendelman HE, Xiong H. Hippocampal synaptic dysfunction in a murine model of human immunodeficiency virus type 1 encephalitis. Neuroscience. 2003;118:359–369. doi: 10.1016/s0306-4522(02)00925-9. doi:10.1016/S0306-4522(02)00925-9. [DOI] [PubMed] [Google Scholar]
- Anderson ER, Gendelman HE, Xiong H. Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci. 2004;24:7194–7198. doi: 10.1523/JNEUROSCI.1933-04.2004. doi:10.1523/JNEUROSCI.1933-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. doi:10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgeropoulos N, Kelley B, Middaugh L, Arrigo S, Persidsky Y, Gendelman HE, Tyor WR. SCID mice with HIV encephalitis develop behavioral abnormalities. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:13–20. doi: 10.1097/00042560-199805010-00003. [DOI] [PubMed] [Google Scholar]
- Beeton C, Barbaria J, Giraud P, Devaux J, Benoliel AM, Gola M, Sabatier JM, Bernard D, Crest M, Beraud E. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol. 2001;166:936–944. doi: 10.4049/jimmunol.166.2.936. [DOI] [PubMed] [Google Scholar]
- Bever CT., Jr The current status of studies of aminopyridines in patients with multiple sclerosis. Ann Neurol. 1994;36(Suppl):S118–S121. doi: 10.1002/ana.410360728. doi:10.1002/ana.410360728. [DOI] [PubMed] [Google Scholar]
- Bever CT, Jr, Young D, Anderson PA, Krumholz A, Conway K, Leslie J, Eddington N, Plaisance KI, Panitch HS, Dhib-Jalbut S, et al. The effects of 4-aminopyridine in multiple sclerosis patients: results of a randomized, placebo-controlled, doubleblind, concentration-controlled, crossover trial. Neurology. 1994;44:1054–1059. doi: 10.1212/wnl.44.6.1054. [DOI] [PubMed] [Google Scholar]
- Blunck R, Scheel O, Muller M, Brandenburg K, Seitzer U, Seydel U. New insights into endotoxin-induced activation of macrophages: involvement of a K+ channel in transmembrane signaling. J Immunol. 2001;166:1009–1015. doi: 10.4049/jimmunol.166.2.1009. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814–1820. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- Davis FA, Stefoski D, Rush J. Orally administered 4-aminopyridine improves clinical signs in multiple sclerosis. Ann Neurol. 1990;27:186–192. doi: 10.1002/ana.410270215. doi:10.1002/ana.410270215. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Deutsch R, Heaton RK, Marcotte TD, McCutchan JA, Nelson JA, Abramson I, Thal LJ, Atkinson JH, Wallace MR, Grant I, San Diego HIV Neuro-behavioral Research Center Group Neurocognitive impairment is an independent risk factor for death in HIV infection. Arch Neurol. 1997;54:416–424. doi: 10.1001/archneur.1997.00550160054016. [DOI] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E, HNRC Group. HIV Neurobehavioral Research Center Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox L, Alford M, Achim C, Mallory M, Masliah E. Neurodegeneration of somatostatin-immunoreactive neurons in HIV encephalitis. J Neuropathol Exp Neurol. 1997;56:360–368. doi: 10.1097/00005072-199704000-00004. doi:10.1097/00005072-199704000-00004. [DOI] [PubMed] [Google Scholar]
- Gallin EK. Ion channels in leukocytes. Physiol Rev. 1991;71:775–811. doi: 10.1152/physrev.1991.71.3.775. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, James HJ, Sharer LR, Perry SW, Saito Y, Kazee AM, Blumberg BM, Epstein LG. Apoptotic neurons in brains from paediatric patients with HIV-1 encephalitis and progressive encephalopathy. Neuropathol Appl Neurobiol. 1995;21:208–217. doi: 10.1111/j.1365-2990.1995.tb01052.x. doi:10.1111/j.1365-2990.1995.tb01052.x. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Soukup VM, Schuenke KW, Keherly MJ, Holzer C, 3rd, Richey FJ, Lahart CJ. Acquired neuronal channelopathies in HIV-associated dementia. J Neuroimmunol. 2004;157:111–119. doi: 10.1016/j.jneuroim.2004.08.044. doi:10.1016/j.jneuroim.2004.08.044. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Zheng J, Coulter CL, Ghorpade A, Che M, Thylin M, Rubocki R, Persidsky Y, Hahn F, Reinhard J, Jr, Swindells S. Suppression of inflammatory neurotoxins by highly active anti-retroviral therapy in human immunodeficiency virus-associated dementia. J Infect Dis. 1998;178:1000–1007. doi: 10.1086/515693. doi:10.1086/515693. [DOI] [PubMed] [Google Scholar]
- Ghelardini C, Galeotti N, Bartolini A. Influence of potassium channel modulators on cognitive processes in mice. Br J Pharmacol. 1998;123:1079–1084. doi: 10.1038/sj.bjp.0701709. doi:10.1038/sj.bjp.0701709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Storm JF, Reuter D, Fedorov NB, Shao LR, Leicher T, Pongs O, Silva AJ. Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvbeta1.1-deficient mice with impaired learning. Learn Mem. 1998;5:257–273. [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Peters M, Vernon J. Modulation of excitability as a learning and memory mechanism: a molecular genetic perspective. Physiol Behav. 2001;73:803–810. doi: 10.1016/s0031-9384(01)00517-0. doi:10.1016/S0031-9384(01)00517-0. [DOI] [PubMed] [Google Scholar]
- Glass JD, Wesselingh SL, Selnes OA, McArthur JC. Clinical neuropathologic correlation in HIV-associated dementia. Neurology. 1993;43:2230–2237. doi: 10.1212/wnl.43.11.2230. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. doi:10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. doi:10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Haslberger A, Romanin C, Koerber R. Membrane potential modulates release of tumor necrosis factor in lipopolysaccharide-stimulated mouse macrophages. Mol Biol Cell. 1992;3:451–460. doi: 10.1091/mbc.3.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Liu J, Xiong H. Macrophage activates delayed-rectifier type K+ channels in cultured rat hippocampal neurons; The 37th Annual Meeting of the Society for Neuroscience, Nov 3 to 7; San Diego, California. 2007. [Google Scholar]
- Irvine E, Keblesh J, Liu J, Xiong H. Voltage-gated potassium channel modulation of neurotoxic activity in human immunodeficiency virus type-1(HIV-1)-infected macrophages. J Neuroimmune Pharmacol. 2007;2:265–269. doi: 10.1007/s11481-007-9072-4. doi:10.1007/s11481-007-9072-4. [DOI] [PubMed] [Google Scholar]
- Kandanearatchi A, Williams B, Everall IP. Assessing the efficacy of highly active antiretroviral therapy in the brain. Brain Pathol. 2003;13:104–110. doi: 10.1111/j.1750-3639.2003.tb00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. doi:10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Langford TD, Letendre SL, Larrea GJ, Masliah E. Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol. 2003;13:195–210. doi: 10.1111/j.1750-3639.2003.tb00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Harris KM. Quantal analysis and synaptic anatomy—integrating two views of hippocampal plasticity. Trends Neurosci. 1993;16:141–147. doi: 10.1016/0166-2236(93)90122-3. doi:10.1016/0166-2236(93)90122-3. [DOI] [PubMed] [Google Scholar]
- Mackenzie AB, Chirakkal H, North RA. Kv1.3 potassium channels in human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2003;285:L862–L868. doi: 10.1152/ajplung.00095.2003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. doi:10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maruyama N, Kakuta Y, Yamauchi K, Ohkawara Y, Aizawa T, Ohrui T, Nara M, Oshiro T, Ohno I, Tamura G, et al. Quinine inhibits production of tumor necrosis factor-alpha from human alveolar macrophages. Am J Respir Cell Mol Biol. 1994;10:514–520. doi: 10.1165/ajrcmb.10.5.8179913. [DOI] [PubMed] [Google Scholar]
- Masliah E, Ge N, Achim C, Hansen L, Wiley C. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51:585–593. doi: 10.1097/00005072-199211000-00003. doi:10.1097/00005072-199211000-00003. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I, HNRC Group. The HIV Neurobehavioral Research Center Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. doi:10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. doi:10.1080/713831484. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. doi:10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- McCutchan JA, Wu JW, Robertson K, Koletar SL, Ellis RJ, Cohn S, Taylor M, Woods S, Heaton R, Currier J, Williams PL. HIV suppression by HAART preserves cognitive function in advanced, immune-reconstituted AIDS patients. AIDS. 2007;21:1109–1117. doi: 10.1097/QAD.0b013e3280ef6acd. doi:10.1097/QAD.0b013e3280ef6acd. [DOI] [PubMed] [Google Scholar]
- Messier C, Mourre C, Bontempi B, Sif J, Lazdunski M, Destrade C. Effect of apamin, a toxin that inhibits Ca(2+)-dependent K+ channels, on learning and memory processes. Brain Res. 1991;551:322–326. doi: 10.1016/0006-8993(91)90950-z. doi:10.1016/0006-8993(91)90950-Z. [DOI] [PubMed] [Google Scholar]
- Nath A. Pathobiology of human immunodeficiency virus dementia. Semin Neurol. 1999;19:113–127. doi: 10.1055/s-2008-1040830. doi:10.1055/s-2008-1040830. [DOI] [PubMed] [Google Scholar]
- Neuenburg JK, Brodt HR, Herndier BG, Bickel M, Bacchetti P, Price RW, Grant RM, Schlote W. HIV-related neuropathology, 1985 to 1999: rising prevalence of HIV encephalopathy in the era of highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2002;31:171–177. doi: 10.1097/00126334-200210010-00007. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Braaten AJ, Hall CD, Robertson KR. Better quality of life with neuropsychological improvement on HAART. Health Qual Life Outcomes. 2006;4:11. doi: 10.1186/1477-7525-4-11. doi:10.1186/1477-7525-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Development of laboratory and animal model systems for HIV-1 encephalitis and its associated dementia. J Leukoc Biol. 1997;62:100–106. doi: 10.1002/jlb.62.1.100. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet H, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello S, Gendelman H. Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol. 1996;149:1027–1053. [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Roberts B. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 1995;146:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- Qiu MR, Campbell TJ, Breit SN. A potassium ion channel is involved in cytokine production by activated human macrophages. Clin Exp Immunol. 2002;130:67–74. doi: 10.1046/j.1365-2249.2002.01965.x. doi:10.1046/j.1365-2249.2002.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reike F, de Warland R, Ruyter van Steveninck R, Bialek W. Spikes: exploring the neural code. MIT; Cambridge, MA: 1997. [Google Scholar]
- Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol. 2004;286:L49–L67. doi: 10.1152/ajplung.00041.2003. doi:10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- Rus H, Pardo CA, Hu L, Darrah E, Cudrici C, Niculescu T, Niculescu F, Mullen KM, Allie R, Guo L, Wulff H, Beeton C, Judge SI, Kerr DA, Knaus HG, Chandy KG, Calabresi PA. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A. 2005;102:11094–11099. doi: 10.1073/pnas.0501770102. doi:10.1073/pnas.0501770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, Becker JT, Cohen B, McArthur JC. HIV-associated neurologic disease incidence changes: multicenter AIDS Cohort Study, 1990–1998. Neurology. 2001;56:257–260. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. doi:10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- Sacktor N, Nakasujja N, Skolasky R, Robertson K, Wong M, Musisi S, Ronald A, Katabira E. Antiretroviral therapy improves cognitive impairment in HIV+ individuals in sub-Saharan Africa. Neurology. 2006;67:311–314. doi: 10.1212/01.wnl.0000225183.74521.72. doi:10.1212/01.wnl.0000225183.74521.72. [DOI] [PubMed] [Google Scholar]
- Sas AR, Bimonte-Nelson HA, Tyor WR. Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of interferon-alpha in the brain. AIDS. 2007;21:2151–2159. doi: 10.1097/QAD.0b013e3282f08c2f. doi:10.1097/QAD.0b013e3282f08c2f. [DOI] [PubMed] [Google Scholar]
- Solntseva EI, Bukanova Iu V, Skrebitskii VG. Memory and potassium channels. Usp Fiziol Nauk. 2003;34:16–25. [PubMed] [Google Scholar]
- Stefoski D, Davis FA, Faut M, Schauf CL. 4-Aminopyridine improves clinical signs in multiple sclerosis. Ann Neurol. 1987;21:71–77. doi: 10.1002/ana.410210113. doi:10.1002/ana.410210113. [DOI] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Galgani S, Narciso P, Ferri F, Sebastiani G, D’Amato C, Affricano C, Pigorini F, Pau FM, De Felici A, Benedetto A. Positive and sustained effects of highly active antiretroviral therapy on HIV-1-associated neurocognitive impairment. AIDS. 1999;13:1889–1897. doi: 10.1097/00002030-199910010-00011. doi:10.1097/00002030-199910010-00011. [DOI] [PubMed] [Google Scholar]
- Vicente R, Escalada A, Coma M, Fuster G, Sanchez-Tillo E, Lopez-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278:46307–46320. doi: 10.1074/jbc.M304388200. doi:10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Achim C. Human immunodeficiency virus encephalitis is the pathological correlate of dementia in acquired immunodeficiency syndrome. Ann Neurol. 1994;36:673–676. doi: 10.1002/ana.410360422. doi:10.1002/ana.410360422 published erratum appears in Ann Neurol 1995 Jan;37(1):140. [DOI] [PubMed] [Google Scholar]
- Xiong H, Zheng J, Thylin M, Gendelman HE. Unraveling the mechanisms of neurotoxicity in HIV type 1-associated dementia: inhibition of neuronal synaptic transmission by macrophage secretory products. AIDS Res Hum Retroviruses. 1999;15:57–63. doi: 10.1089/088922299311718. doi:10.1089/088922299311718. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. doi:10.1016/S0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]