

Abstract
Objective
To elucidate the non-redundant roles of JNK1 and JNK2 in antigen-induced arthritis (AIA).
Methods
Mice that were genetically disrupted in Jnk1 or Jnk2 were primed with methylated BSA (mBSA) in complete Freund’s adjuvant and then given an intraarticular challenge with mBSA in the knee on day 21. Bone marrow chimeras were generated and similarly treated. Joints were harvested and prepared for histological assessment. T cell responses were verified by cytokine and proliferation responses, and relative immunoglobulin responses were measured by ELISA. Cytokine mRNA expression levels were measured by qPCR. Thioglycollate and zymosan A elicited macrophage recruitment was tested in vivo and migration was tested in vitro. The peptide inhibitor D-JNKi was injected daily starting four days after intraarticular mBSA injection in wild type (WT) mice and inflammation was histologically scored.
Results
JNK1-deficient, but not JNK2-deficient mice, had a reduction in inflammatory infiltration and joint damage. This effect was primarily restricted to hematopoetic cells, but B and T cell responses were preserved in mBSA-injected mice. JNK1-deficient macrophages produced cytokines and chemokines comparably to WT counterparts. However, macrophage migration was impaired in vivo and in vitro. Targeting JNK with the peptide inhibitor D-JNKi dramatically reduced inflammation and joint destruction in WT mice.
Conclusions
AIA is dependent on JNK1, but not JNK2. JNK1 is a promising molecular target for reducing autoimmune inflammation as its inhibition impairs macrophage migration.
INTRODUCTION
RA is an autoimmune disease characterized by excessive autoantibody production, aggressive synoviocyte behavior, and abundant cytokine production (1). In addition to synovial inflammation, the aggressive tissue front called pannus invades and destroys local articular structures. The introduction of biological therapy for RA and other inflammatory diseases represents a major therapeutic advance. Biological agents such as tumor necrosis factor-α (TNF) blockers disrupt interactions of effector molecules with their receptors, but many patients have an inadequate response (2). However, orally bioavailable small molecules that target key intracellular signaling molecules might regulate both cytokine production and cytokine action, affecting more than one function and thus, be more effective (3).
Mitogen-activated protein kinases (MAPK) are expressed and activated in the RA synovium (4) and have been the focus of drug development efforts due to their prominent role in the regulation of cytokines, chemokines, degradative enzymes, migration, programmed cell death, and cell proliferation (5–7). Jun N-terminal kinases (JNK), which belong to the MAPK family, are highly activated in isolated RA fibroblast-like synoviocytes (FLS) and in the synovium itself (8, 9). They play a major role in cytokine production and extracellular matrix regulation through production of matrix metalloproteinases (MMP). However, the functions of JNK extend beyond MMP and cytokine expression and also involve cell proliferation, apoptosis (10), angiogenesis (11), and migration (12).
The JNKs are encoded by three separate loci, Jnk1-3, of which Jnk1 and Jnk2 are ubiquitously expressed, whereas Jnk3 is mainly expressed in heart, testis, and brain (13). Separate roles of JNK1 and JNK2 in the pathogenesis of arthritis are yet to be fully defined as in some models they appear to have compensatory and redundant functions. JNK1 is not essential for inflammatory arthritis in TNF-transgenic mice (14), suggesting that signaling through JNK2 might compensate for the deficiency in JNK1 in that model. In addition JNK2-deficient mice exhibited only a modest decrease in cartilage damage in a model of passive collagen-induced arthritis (15). On the other hand, a JNK inhibitor, SP600125, was mildly anti-inflammatory in rat adjuvant-induced arthritis, and also provided striking protection against bone and cartilage destruction (9). However, off-target effects of SP600125 could have potentially contributed to some of these effects (16, 17).
In this report, we show that JNK1, but not JNK2, is essential for pathogenesis of antigen-induced arthritis (AIA). JNK1 deficiency attenuates arthritis induction and joint destruction through multiple mechanisms including reduced synovial inflammatory infiltration, inflammatory cytokine production and MMP expression. Of particular importance, the ability of macrophages to migrate was impaired, even in the presence of potent chemokine stimulation. These results indicate that JNK1 plays a key role in the pathogenesis of arthritis and could serve as a new therapeutic target for RA.
METHODS
Mice
Jnk1−/− and Jnk2−/− mice were previously described (18, 19). Mice used in these experiments were 8–12 weeks age females. C57Bl/6 and CD45.1 congenic mice were purchased from the Jackson Laboratories (Bar Harbor, ME). The mice were bred and maintained under standard conditions in the University of California, San Diego animal facility that is accredited by the American Association for Accreditation of Laboratory Animal Care. All animal protocols received prior approval by the institutional review board.
Reagents
Lipopolysaccharide (LPS; Escherichia coli 0111:B4) and zymosan A were purchased from Sigma (St. Louis, MO). Zymosan A was suspended in PBS, boiled for 1 h, rinsed with PBS three times, and resuspended in PBS at 1 mg/ml. Thioglycollate (TG) was from Difco Laboratories. MCP-1 (monocyte chemotactic protein-1) and TNF were purchased from R&D systems (Minneapolis, MN), and IL-1β was purchased from PeproTech, Inc. (Rocky Hill, NJ).
Antigen-induced arthritis (AIA) induction
Experimental AIA was induced by a subcutaneous injection of 100 μg of mBSA emulsified in 100μl of complete Freund’s adjuvant (CFA) in the flank, and one week later by an intradermal injection of 100 μg of mBSA/CFA in the tailbase. Two weeks after these injections, arthritis was induced by intraarticular (i.a.) injection of 60 μg of mBSA in 10 μl of saline into the right knee joint. The left knee was injected with PBS to serve as a control. Disease was assessed 10 days post-intraarticular injection by histological analysis as described below.
Histology
Joints were fixed in 10% formalin, decalcified in 10% EDTA for 2–3 weeks, trimmed, and embedded. Sections were prepared from the tissue blocks and stained with hematoxylin and eosin (H&E) or Safranin O-fast green to determine proteoglycan content. A semiquantitative scoring system was used to assess synovial inflammation, extra-articular inflammation, erosion and proteoglycan loss (0–5 scale) as previously described (20). Histological analyses were performed in a blinded manner.
Immunohistochemistry
Sections from decalcified fixed tissues were incubated overnight at 4°C with rat anti-F4/80 antibody (MCA497R) from AbD Serotec (Raleigh, NC) at a 1:200 dilution. Antigen retrieval was with citrate buffer (DAKO S1700) at 96°C for 20 minutes.
Bone marrow chimeras
Adult mice were lethally irradiated with 11 Gy. Bone marrow cells harvested from femurs and tibias of donors were washed in serum-free medium and counted. Recipients were injected with 107 cells in 200 μl of serum-free RPMI intravenously. Chimerism was verified by flow cytometry for the appropriate CD45 allele after eight weeks.
Real-time quantitative (q) PCR
Joints were dissected to remove extra-articular tissue, and snap frozen in liquid nitrogen. The specimens were pulverized and total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) and reverse-transcribed with random hexamers and Superscript II Kit (Invitrogen). qPCR was performed with SYBR Green PCR Master Mix Kit (Applied Biosystems). The relative amounts of transcripts were compared to those of 18S rRNA and normalized to untreated samples by the ΔΔCt method. Primer sequences are available upon request.
T cell proliferation assay
Mouse spleen cells were isolated and washed in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 10 mM Hepes, 1 mM sodium pyruvate, 50 mM 2-mercaptoethanol, 1% L-glutamine, and 100 units/ml of penicillin/streptomycin. Erythrocytes were lysed and after washing, cells were counted and 2×105 cells were placed in each well of a sterile, U-bottomed microculture plate in medium with 12.5 or 25 μg/ml of mBSA. Supernatants were harvested for cytokines after 48h or cultures were maintained at 37°C for 4 days for proliferation assays. Sixteen hrs before harvesting, 1 μCi of 3H-thymidine was added in 25 μl of RPMI. Cultures were harvested with a cell harvester and 3H-thymidine incorporation was determined.
Determination of serum antibodies
Methylated BSA-specific antibodies of various isotypes (IgG, IgG1, IgG2a, IgG2b, IgG3) were measured in sera by ELISA. Antigen was coated on microtiter plates at a concentration of 1 μg/ml. Antibody titers were assessed by 2-fold serial dilutions of sera, followed by detection of bound mouse Ig with a 1:500 dilution of peroxidase-conjugated rabbit anti-mouse Ig. O-phenylenediamine was used as substrate for the peroxidase reactions.
Flow cytometry
Antibodies used by flow cytometry included CD45.2-FITC, CD45.1-PE, F4/80-FITC and Gr-1-PE (BD Biosciences, San Jose, CA). Antibodies against integrin-α2-PE, integrin-α4-PE, integrin-β1-PE and integrin-β2-PE were purchased from Sigma (St Louis, MO)
Cytokine and chemokine quantification
MCP-1, IFNγ, IL-4, IL-10 and IL-17 amounts were measured by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN), following manufacturer’s protocol.
Zymosan A and thioglycollate-induced peritonitis
Peritonitis was induced by intraperitoneal (i.p.) injection with 2 ml of 3% sterile TG medium or 1 ml with 400ug zymosan A. Mice were sacrificed after 6 hrs or after 3 days and peritoneal cells were removed by lavage with 5 ml of PBS. Lavage fluids were separated by centrifugation and supernatants were used for chemokine analysis. Cell yield was obtained by counting and the relative percentages of neutrophils (measured in mice sacrificed after 6 hrs of TG or zymosan A injection) or macrophages (measured in mice killed after 3 days of TG or zymosan A injection) were determined by flow cytometry after staining with anti-F4/80 and anti-Gr-1 antibodies.
In vitro migration
TG-induced macrophages were harvested from Jnk1−/− and WT mice, and 5×105 cells per well were dispersed into 96 well plates with central stoppers (Oris™, Platypus Technologies, LLC, Madison, WI) and allowed to settle overnight. The stoppers were removed except for the baseline control wells. The cells were treated with MCP-1 (R&D Systems, Minneapolis, MN), and varying serum concentrations for 4 hrs. The cells were then stained with calcein AM (Invitrogen) and the fluorescence (485/535) of the cells that migrated to the center of the well was measured through a mask with small aperatures aligned with the center of the wells using a fluorimeter (Wallac 1420, PerkinElmer, Waltham, MA).
Immunoblot
Peritoneal macrophages were disrupted in lysis buffer (PhosphoSafe™, Novagen, Gibbstown, NJ) containing a protease inhibitor cocktail. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. Blots were probed with antibodies against phospho-JNK (Cell Signaling Technology, Danvers, MA), JNK1 and JNK1/2 (BD, Pharmingen), CCR5 (Novus Biologicals, Littelton, CO) and actin (Sigma, St Louis, MO). Horseradish peroxidase-conjugated anti-IgG (Santa Cruz Biotechnology Inc, Santa Cruz, CA) was used as secondary antibody. Membranes were developed using a chemiluminescence system (ECL detection reagent: Amersham Life Science, Aylesbury, UK).
Fibroblast like synoviocytes (FLS) and bone marrow-derived macrophages (BMDM)
Briefly, synovial tissue was collected, minced, and incubated with 0.5 mg/ml collagenase VIII (Sigma, St. Louis, MO) in serum-free Dulbecco’s modified Eagle’s medium (DMEM; Gibco) for 2 hrs at 37°C, washed extensively, and cultured in DMEM supplemented with 10% fetal calf serum, penicillin, streptomycin, and L-glutamine in an humidified 5% CO2 atmosphere. FLS from passages 3–9 were used. To generate BMDM, bone marrow cells were cultured in DMEM (Invitrogen) with 10% FBS and 20% L929 supernatant containing macrophage-stimulating factor for 6 days and were replated for the assays as indicated.
D-JNKi peptide treatment
D-JNKi, whose sequence is DQSRPVQPFLNLTTPRKPR-PP-RRRQRRKKRG and a TAT control peptide PP-RRRQRRKKRG were synthesized by the Protein Chemistry Department at the Research Institute of Molecular Pathology and were kindly provided by Dr. E. Wagner. Specific inhibition of JNK activation by D-JNKi was shown previously (21). D-JNKi or TAT peptides diluted in PBS were injected i.p. at 20 μg/g body weight daily, starting on day 4 after mBSA i.a. injection and until day 10.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Mann Whitney U test was used for pair-wise comparisons. ANOVA was used for multiple group comparisons with Bonferonni post hoc comparisons for multiple pair-wise comparisons. All statistical analyses were performed using PRISM version 4.0b (GraphPad Software, San Diego, California). Results were considered significant for p<0.05.
RESULTS
JNK1 is required for antigen-induced arthritis
To examine the role of JNK1 and JNK2 in a model of chronic inflammatory arthritis, C57Bl/6 mice deficient for either JNK isoform were compared to WT mice for incidence and severity of AIA. Histopathological analysis showed a reduction in inflammatory cell infiltration, joint destruction and cartilage damage in Jnk1−/− mice (Fig. 1A and B). JNK1-deficient mice had significantly lower scores for all histological indices compared to WT and JNK2-deficient mice (p<0.05 by ANOVA with Bonferroni post hoc comparison, n=14–16/group).
Figure 1. JNK1 is critical for effector function in AIA.

A) Representative H&E and Safranin O stained sections of knee joints on day 10 of AIA induction in WT and JNK-deficient mice (n=14–16/group). Original magnification 200x. B) Sections above were scored for inflammatory infiltration, bone erosion and cartilage damage. Jnk1−/− mice had significantly lower scores than WT and Jnk2−/− mice. Results are expressed as means ± SEM.* p< 0.05 vs. WT and Jnk2−/− mice. C) RNA samples from whole joints (6 mice/group) were analyzed by qPCR in triplicate for expression of the indicated genes. mRNA amounts were normalized to 18S rRNA and fold induction is relative to PBS-injected joints of the same genotype. Induction of cytokine and MMP3 expression was less in Jnk1−/− mice. Results are expressed as means ± SEM.* p< 0.05 vs. WT and Jnk2−/− mice.
Effect of JNK1 deficiency on cytokines, MMP and adaptive immunity
To evaluate the influence of JNK deficiency on expression of selected participants in inflammatory arthritis, we determined relative expression of IL-1β, TNF, IL-6 and MMP3 mRNAs in paws from these mice by qPCR on day 10 after i.a, mBSA injection. Amounts of mRNA for all four mediators were statistically decreased in Jnk1−/− mice, but not in Jnk2−/− mice, compared to WT controls (Fig. 1C).
AIA requires the generation of mBSA-specific CD4+ T cells and antibodies. Although Jnk1−/−CD4+ T cells showed defects in T cell activation or differentiation in vitro (18, 22, 23), it also was shown that the absence of JNK1 in T cells did not alter their ability to mount a pathogenic autoimmune response to myelin in vivo (24). Consistent with the latter results, Fig. 2A shows that quantitatively similar splenocyte proliferative responses against mBSA were found in cells derived from Jnk1−/−, Jnk2−/− and WT mice. In addition there were no differences in splenocyte IFNγ, IL-4, IL-10 or IL-17 release after in vitro restimulation between the strains (Fig. 2B). Humoral immunity, tested by the relative levels of mBSA-specific antibodies in sera, was also comparable in all strains (Fig. 2C).
Figure 2. Adaptive immunity is preserved in JNK1 deficiency.

A) Cellular immunity was determined by measuring 3H-thymidine incorporation (cpm) after in vitro restimulation of splenocytes from arthritic mice with mBSA. (B) Splenocytes were restimulated in vitro with mBSA and supernatants harvested after 48 hrs. IL-10, IL-17, IFNγ, and IL-4 were assayed by ELISA. (C) Humoral immunity was determined by measuring amounts of anti-mBSA antibody isotypes by ELISA. Results are expressed as means ± SEM for 6 mice/group.
Arthritis is partially dependent on hematopoetic cells and radioresistant cells
To evaluate whether joint inflammation was associated with bone marrow-derived elements other than T or B cells, or with connective tissue, bone marrow chimeras were generated by irradiating WT and Jnk1−/− recipients, and reconstituting them with Jnk1−/− and WT donor bone marrow. After 8 weeks, bone marrow engraftment was confirmed by flow cytometry and AIA was then induced. Development of arthritis was reduced in mice with Jnk1−/− bone marrow-derivedcells (Fig. 3A). As the Jnk1−/− recipients of Jnk1−/− bone marrow had less inflammation than the WT recipients, a contribution from the radioresistant cells could not be excluded. Concordantly, mice harbouring JNK1-deficient bone marrow trended toward lower levels of IL-1β and IL-6 mRNA expression (Fig. 3B).
Figure 3. Arthritis depends primarily on JNK1 in bone marrow-derived cells.
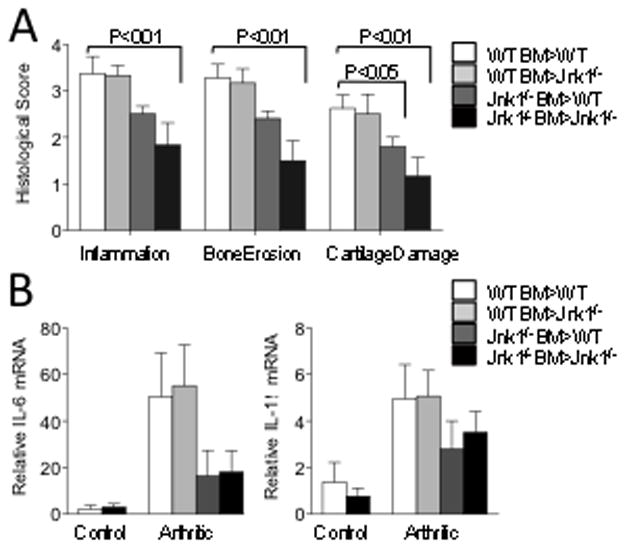
A) WT and Jnk1−/− mice (n=8/group) were irradiated and reconstituted with Jnk1−/− or WT bone marrow as indicated. AIA was induced 8 weeks after bone marrow reconstitution and mice were analyzed 10 days later. One knee from each mouse was prepared for histologic scoring with H&E and safranin O staining. Shown are average inflammation, erosion, and cartilage damage scores ± SEM. B) Whole joint RNA was extracted and amounts of mRNAs were analyzed by qPCR. Cytokine mRNA expression decreased in mice reconstituted with Jnk1−/− bone marrow. Results are expressed as means ± SEM for 5 mice/group.
JNK1 regulates macrophage migration
In this model, JNK1 deficient mice were able to mount an adaptive immune response to mBSA. (Fig 2). However, JNK deficiency in the bone marrow derived cells reduced inflammation in the chimeras. In the innate immune compartment, JNK1 is markedly down-regulated during neutrophil differentiation and maturation (25). Hence we examined macrophages as a likely candidate amongst bone marrow derived cells. To further analyze the role of JNK1 in macrophages, BMDM were stimulated by TNF and IL-1β. We did not detect any differences in chemokine or cytokine gene expression between JNK1-deficient and WT BMDM stimulation with either TNF or IL-1β (Supplementary Fig. 1).
Although Jnk1−/− macrophages retained the ability to express cytokines and chemokines, we noticed a marked reduction in F4/80 positive cells in the affected joints of Jnk1−/− but not Jnk2−/−mice (Fig. 4A), suggesting an impairment of inflammatory cell migration. Several prior reports indicated that JNK was critical to cellular migration (12, 26). To further study the effect of JNK1 specifically on inflammatory cell migration, we used the TG and zymosan peritonitis models. Neutrophil recruitment was assessed by injecting TG or zymosan into the peritoneum of WT, Jnk1−/− and Jnk2−/− mice. After 6 hrs the peritoneal cavities were lavaged and neutrophils were quantified. Neutrophil recruitment was reduced in the Jnk2−/− mice, but not the Jnk1−/− mice (Fig. 4B). The Jnk2−/− neutrophils are functionally deficient in both isoforms of JNK as mature neutrophils express minimal detectable JNK1 (25). However, Jnk1−/− mice had a significantly reduced influx of macrophages, assessed 3 days after injecting TG or zymosan into the peritoneum suggesting that there is little redundancy in JNK1 and JNK2 in macrophage migration (Fig. 4B). TG elicited JNK1-deficient peritoneal macrophages also did not migrate well when stimulated in vitro by non-specific factors in FCS relative to WT macrophages (Fig. 4C). Migration impairment was not due to a decrease in surface integrin expression (Supplementary Fig. 2).
Fig. 4. JNK regulates macrophage migration.

A) Knees were harvested on day 10 after mBSA injection and immunostained with F4/80. Original magnification 200x. Shown are representative stains for 5 samples/group. Fewer F4/80+ cells were present in joints of arthritic Jnk1−/− mice. WT, Jnk1−/− and Jnk2−/− mice (n=4–6/group) were i.p. injected with TG or zymosan A. B) The peritoneum was lavaged after 6 hrs and neutrophil influx was determined by cell count and flow cytometry (Gr-1+F4/80−). Alternatively the peritoneum was lavaged after 72 hrs and macrophage influx was determined as above (F4/80+Gr-1−). Shown are means ± SEM. C) TG- elicited peritoneal macrophages were dispersed into 96 well plates with stoppers that obscured the center of the wells overnight. Most of the stoppers were removed and fresh medium with 10% FCS was added. After 4 hrs the cells were fluorescently stained and the central fluorescence measured through a perforated mask provided in the kit. Shown are representative wells and the relative fluorescence measured from the center of the wells. Jnk1−/− macrophages migrated significantly less than WT macrophages. Results are expressed as means ± SEM. Representative of four independent experiments.
As FCS is not a defined stimulant, we also evaluated migration in response to MCP-1, which is a major chemoattractant for monocytes that acts by binding to a specific cell-surface receptor, CC-chemokine receptor-2 (CCR2). We confirmed a previous report that CCR2 ligation with MCP-1 results in JNK phosphorylation (27) (Fig. 5A). JNK1-deficient macrophages were also compromised in their general migratory response to this stimulus, although their directional response was not directly tested (Fig. 5B). We excluded the possibility that the secretion of MCP-1 from JNK1-deficient fibroblasts or macrophages was impaired; there were no differences in MCP-1 amounts in the peritoneal lavage fluid in vivo after TG injection (Fig. 5C). We also could not detect differences in secretion of MCP-1 from peritoneal macrophages and fibroblasts in vitro after stimulation with LPS, IL-1β or TNF (Fig. 5D). In addition there were no differences in CCR2 expression between WT and JNK1-deficient macrophages (Supplementary Fig. 3).
Figure 5. JNK1-deficient macrophages migrate poorly after MCP-1 stimulation.
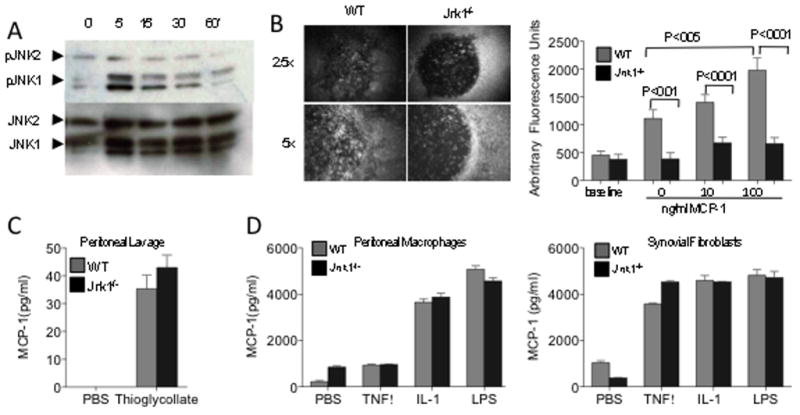
A) Peritoneal macrophages were stimulated in vitro with 100 ng/ml MCP-1 for the indicated times. Cells were lysed and analyzed by immunoblotting for presence of phospho-JNK1/2. B) TG-elicited peritoneal macrophages were dispersed into 96 well plates with stoppers overnight. Most of the stoppers were removed and graded amounts of MCP-1 were added to the medium. Migration into the central clearing was assessed by fluorescently staining the cells after 4 hrs. Baseline fluorescence was measured by removing the stoppers from control wells at 4 hrs. Results are expressed as means ± SEM. Shown are representative examples of macrophage migration into the central area, and the relative fluorescence of wells compared to wells where the stopper was removed at the end of the experiment. Jnk1−/− macrophages migrated significantly less than WT macrophages. Representative of four independent experiments. C) MCP-1 amounts in lavage fluids were assayed by ELISA. Shown are means ± SEM for 5 mice/group. D) MCP-1 amounts in peritoneal macrophages (5×105 cells/ml) and fibroblasts (plated at 1×105 cells/ml) supernatants, 24 hrs after stimulation with IL-1β (1 ng/ml), TNF (100 ng/ml) and LPS (50 ng/ml),. MCP-1 was measured in triplicate by ELISA.
Effect of a permeable peptide JNK inhibitor in the AIA model
To test the therapeutic potential of targeting JNK, we used a specific JNK inhibitor that interferes with both JNK1 and JNK2 signaling to treat AIA. The inhibitor is a short D-amino acid peptide (D-JNKi) that has been modified to be cell permeable and has been used to reduce insulin resistance (28), TNF-induced liver failure (28), and hypoxia-induced retinopathy (11). This peptide inhibitor is a more specific JNK inhibitor than the previously reported small molecular weight inhibitor SP600125 (21). D-JNKi and a control peptide (TAT peptide) were injected i.p. (20μg/g/day) daily starting on day 4 after injecting mBSA in the knee joint. D-JNKi treatment successfully interfered with joint inflammation and destruction (Fig. 6A). Although the cellular infiltration was reduced, this inhibitor also reduced the cytokine production from in vitro stimulated BMDM (Fig. 6B and C). The D-JNKi treatment did not significantly alter T cell proliferation or antigen specific cytokine secretion (Supplementary Fig. 4). These results suggest that the peptide inhibitor has a greater effect than inhibiting JNK1, and inhibiting both JNK1 and JNK2 is impacting both innate immune cell migration and cytokine production.
Figure 6. D-JNKi treatment successfully abrogated joint inflammation and destruction.
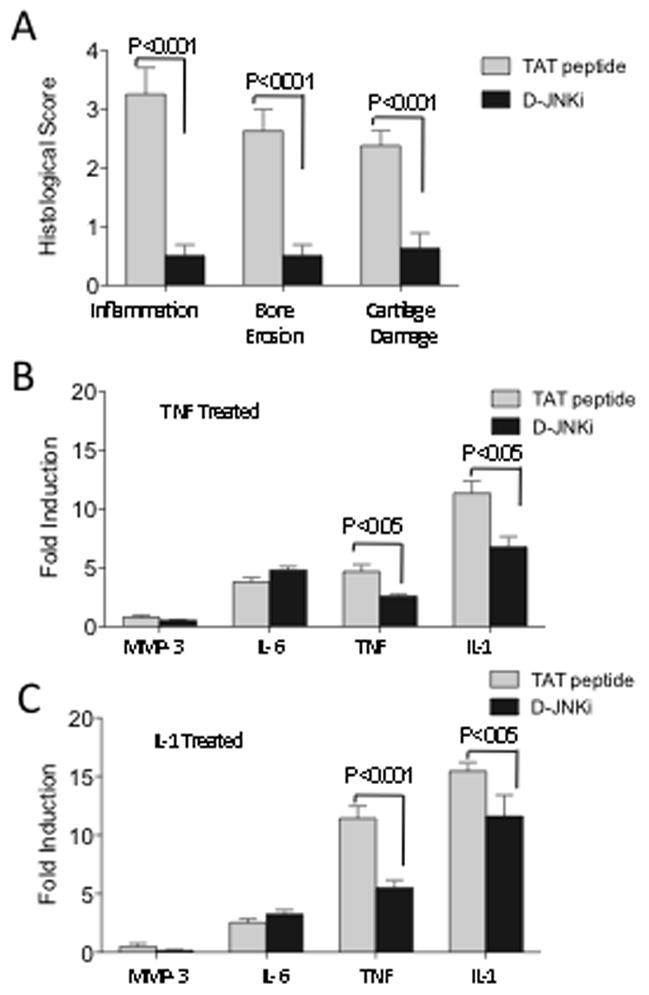
A) AIA was induced in WT mice. D-JNKi or TAT peptides diluted in PBS were injected i.p. at 20 μg/g body weight daily starting on day 4 after mBSA or saline i.a. injection until day 10. Both knees from each mouse were prepared for histological scoring by H&E and safranin O staining. Shown are the average inflammation, erosion and cartilage damage scores ± SEM for 8 mice/group. Sections from D-JNKi treated mice had significantly lower scores (Mann Whitney U test). BMDM were pretreated with 10μM TAT peptide or D-JNKi for 1 hr and then stimulated with B) TNF (10 ng/ml) or C) IL-1β (2 ng/ml) for 4hrs. The cells were lysed, RNA extracted and analyzed by qPCR for expression of the indicated amplicons. The fold induction relative to unstimulated BMDM are shown. Data are pooled from two experiments and significance assessed by Mann Whitney U test.
DISCUSSION
In chronic arthritis, inflammation is perpetuated by continued recruitment of inflammatory cells to the synovium. There is an initial breech of tolerance to joint-associated self-antigens that might precede the onset of disease by years (29). The acute presentation of joint swelling may involve additional factors, including innate cellular subsets. Histological analysis of synovial samples from RA patients and the profile of inflammatory cytokines in the synovial fluid suggest that macrophages play a key role (30, 31). Although biological therapies have targeted cytokines that are produced by macrophages or their receptors, including IL-1β, TNF and IL-6, they are not effective in all patients (2). Another strategy that could successfully intercede with the pathogenesis of inflammatory arthritis would be the use of small molecule inhibitors that disrupt the continued recruitment of inflammatory cells to the synovium and thereby limit the amounts of destructive enzymes and proinflammatory cytokines in the local joint environment.
We sought to examine the efficacy of inhibiting the two major JNK isoforms, JNK1 and JNK2. Using a T cell- and macrophage-dependent arthritis model we examined the impact of JNK1 or JNK2 ablation on pathogenesis. Importantly, JNK1 and JNK2 were not redundant. Where the disruption of Jnk1 resulted in a reduction in inflammatory infiltration into the synovium and a consequent reduction in bone erosion (Fig. 1A) no effects were seen with Jnk2 ablation. The role of JNK in T cell priming remains controversial (18, 19, 22, 23). JNK1 deficiency might skew the Th2 phenotype of CD4+ T cells (23) and diminish their proliferative capacity (19). However, it was also shown that alterations in T cell priming were not intrinsic to the T cells and instead were dependent on macrophage behavior (24). In our study we evaluated the lymphocytes for their antigen specific responses to mBSA and found that specific Jnk1−/− splenocytes proliferated to the same extent as WT counterparts. Similarly we did not detect a difference in the antibody profiles of Jnk1−/− or Jnk2−/− mice. As mature neutrophils have little if any expression of JNK1 we investigated macrophages as the cellular subset primary affected by JNK1 deficiency (25).
Previous reports described the contribution of macrophages to mBSA-induced arthritis (32, 33). Clodronate-mediated ablation of macrophages reduced inflammation and joint destruction in rodents (32). In the AIA model, destruction of bone is due to the release of IL-1, and is not FcγR-dependent (34–36), suggesting that macrophages and not auto-antibodies might play a key role in the chronic phase of disease. Indeed, specific targeting of macrophages severely limited the overall inflammatory effector response (32, 33). Although Jnk1−/− macrophages retain the ability to express cytokines and chemokines, we found a substantial limitation in macrophage migration and recruitment to the synovium in JNK1-deficient mice. Migration of different cell types such as endothelial cells (37), neutrophils (26) or keratinocytes (12) has been associated with JNK1/2 activation. The labile adhesions required for rapid cell migration in response to a variety of stimuli were attributed to the JNK-mediated phosphorylation of paxillin (12). In our study, macrophage migration following different stimuli was significantly impaired in the absence of JNK1. However, neutrophil migration was unaffected as evidenced by their rapid influx to the peritoneum of JNK1-deficient mice following TG and zymosan A injection. This result is not surprising as mature murine neutrophils predominantly express JNK2 and not JNK1 (25).
The inhibition of macrophage migration by targeting MCP-1 with a biological or small molecule CCR2 inhibitor is not effective in RA (38, 39). The synovium contains other monocyte or macrophage chemoattractants (40, 41). Other receptors or chemokines permit the recruitment of macrophages and a strategy for targeting the converging intrinsic mechanisms of macrophage migration might be more effective. An approach that targets selected protein kinases with small molecules might be effective in multiple pathogenic steps. Here we utilized a cell permeable peptide that reduces the activities of both JNK1 and JNK2. This peptide was effective when initiated after the onset of disease as a treatment and reduced the inflammation and joint destruction in this model. The efficacy of the D-JNKi peptide might not be solely attributable to JNK1 inhibition, as this peptide reduces the activity of both JNK1 and JNK2 and there might have been additional off target effects (21).
The inhibition of both JNK isoforms would likely have pluripotent effects, not only on hematopoietic cells but also on FLS, and endothelial cells. This inhibitor might not only limit the migration of macrophages, but also can reduce inflammatory cytokine and MMP production (9, 15), and angiogenesis (11). This notion is supported by a recent report that targeted deletion of both JNK1 and JNK2 in hematopoietic cells markedly reduced TNF production in a hepatitis model (42). Most of JNK functions are redundant between JNK1 and JNK2, as suggested in TNF-transgenic mice, in which JNK1 was not essential for inflammation (14). A dual inhibitor would more potently reduce inflammation as seen with the D-JNKi treated mice compared to the mice genetically disrupted in a single JNK isoform (Figure 1B and 6A). In summary, the JNK1 deficiency or blockade limited the migration of macrophages and reduced joint inflammation.
Supplementary Material
After 4 hrs stimulation with TNF (10 ng/ml) or IL-1β (2 ng/ml) BMDM were lysed, RNA extracted and analyzed by qPCR for expression of the indicated mRNAs. Data are representative of three experiments.
Peritoneal macrophages were harvested and stained with F4/80 and the indicated integrin antibodies, and analyzed by flow cytometry. Data are representative of three mice per genotype.
BMDM protein lysates were separated by SDS-PAGE, blotted and examined for presence of the indicated proteins.
WT mice were immunized with mBSA and boosted on day 7. On day 21, knees were injected with mBSA. Four days after the knee was injected mice were treated with either TAT peptide (n=4) or D-JNKi peptide (n=5) daily i.p. On day 10, mice were sacrificed and the splenocytes were harvested and stimulated in vitro with mBSA (25μg/ml). (A) After 48 hours the supernatants were harvested and assayed for the indicated cytokine by ELISA. (B) Splenocytes from mice primed above with mBSA were removed and restimulated in culture with mBSA (25μg/ml) in the presence of D-JNKi or TAT peptide (10μM), and the supernatants were tested for the indicated cytokines. In parallel cultures, cells from mice treated in vivo with D-JNKi or TAT peptide (C) or splenocytes treated in vitro with D-JNKi or TAT peptide (10μM) (D) were pulsed with 3H-thymidine after 48 hours and harvested 16 hours later for incorporation. Shown are the means ± SEM. There were no significant differences between the groups.
Acknowledgments
This work was supported by grants from the Arthritis Foundation, the Spanish Society of Rheumatology and the National Institutes of Health (AR47825 and AI043477).
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.van Vollenhoven RF. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatol. 2009;5(10):531–41. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- 3.Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69 (Suppl 1):i77–82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner CW, Bitzan P, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43(11):2501–12. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 6.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 7.Sweeney SE, Firestein GS. Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann Rheum Dis. 2006;65(Suppl 3):iii83–8. doi: 10.1136/ard.2006.058388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291(1):124–30. [PubMed] [Google Scholar]
- 9.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108(1):73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57(4–5):283–95. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 11.Guma M, Rius J, Duong-Polk KX, Haddad GG, Lindsey JD, Karin M. Genetic and pharmacological inhibition of JNK ameliorates hypoxia-induced retinopathy through interference with VEGF expression. Proc Natl Acad Sci U S A. 2009;106(21):8760–5. doi: 10.1073/pnas.0902659106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424(6945):219–23. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- 13.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 14.Koller M, Hayer S, Redlich K, Ricci R, David JP, Steiner G, et al. JNK1 is not essential for TNF-mediated joint disease. Arthritis Res Ther. 2005;7(1):R166–73. doi: 10.1186/ar1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002;46(3):818–23. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 16.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanemura S, Momose H, Shimizu N, Kitagawa D, Seo J, Yamasaki T, et al. Blockage by SP600125 of Fcepsilon receptor-induced degranulation and cytokine gene expression in mast cells is mediated through inhibition of phosphatidylinositol 3-kinase signalling pathway. J Biochem. 2009;145(3):345–54. doi: 10.1093/jb/mvn172. [DOI] [PubMed] [Google Scholar]
- 18.Sabapathy K, Hu Y, Kallunki T, Schreiber M, David JP, Jochum W, et al. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr Biol. 1999;9(3):116–25. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- 19.Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J Exp Med. 2001;193(3):317–28. doi: 10.1084/jem.193.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60(12):3642–50. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9(9):1180–6. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 22.Dong C, Yang DD, Tournier C, Whitmarsh AJ, Xu J, Davis RJ, et al. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405(6782):91–4. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- 23.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282(5396):2092–5. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 24.Tran EH, Azuma YT, Chen M, Weston C, Davis RJ, Flavell RA. Inactivation of JNK1 enhances innate IL-10 production and dampens autoimmune inflammation in the brain. Proc Natl Acad Sci U S A. 2006;103(36):13451–6. doi: 10.1073/pnas.0601155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sukhumavasi W, Egan CE, Denkers EY. Mouse neutrophils require JNK2 MAPK for Toxoplasma gondii-induced IL-12p40 and CCL2/MCP-1 release. J Immunol. 2007;179(6):3570–7. doi: 10.4049/jimmunol.179.6.3570. [DOI] [PubMed] [Google Scholar]
- 26.Yeh MC, Mukaro V, Hii CS, Ferrante A. Regulation of neutrophil-mediated killing of Staphylococcus aureus and chemotaxis by c-jun NH2 terminal kinase. J Leukoc Biol. 2010;87(5):925–32. doi: 10.1189/jlb.0609399. [DOI] [PubMed] [Google Scholar]
- 27.Sodhi A, Biswas SK. Monocyte chemoattractant protein-1-induced activation of p42/44 MAPK and c-Jun in murine peritoneal macrophages: a potential pathway for macrophage activation. J Interferon Cytokine Res. 2002;22(5):517–26. doi: 10.1089/10799900252981990. [DOI] [PubMed] [Google Scholar]
- 28.Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124(3):601–13. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50(2):380–6. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 30.Szekanecz Z, Koch AE. Macrophages and their products in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19(3):289–95. doi: 10.1097/BOR.0b013e32805e87ae. [DOI] [PubMed] [Google Scholar]
- 31.Brennan F, Beech J. Update on cytokines in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19(3):296–301. doi: 10.1097/BOR.0b013e32805e87f1. [DOI] [PubMed] [Google Scholar]
- 32.Richards PJ, Williams AS, Goodfellow RM, Williams BD. Liposomal clodronate eliminates synovial macrophages, reduces inflammation and ameliorates joint destruction in antigen-induced arthritis. Rheumatology (Oxford) 1999;38(9):818–25. doi: 10.1093/rheumatology/38.9.818. [DOI] [PubMed] [Google Scholar]
- 33.Lawlor KE, Wong PK, Campbell IK, van Rooijen N, Wicks IP. Acute CD4+ T lymphocyte-dependent interleukin-1-driven arthritis selectively requires interleukin-2 and interleukin-4, joint macrophages, granulocyte-macrophage colony-stimulating factor, interleukin-6, and leukemia inhibitory factor. Arthritis Rheum. 2005;52(12):3749–54. doi: 10.1002/art.21495. [DOI] [PubMed] [Google Scholar]
- 34.van de Loo AA, Arntz OJ, Bakker AC, van Lent PL, Jacobs MJ, van den Berg WB. Role of interleukin 1 in antigen-induced exacerbations of murine arthritis. Am J Pathol. 1995;146(1):239–49. [PMC free article] [PubMed] [Google Scholar]
- 35.van Lent PL, Nabbe K, Blom AB, Holthuysen AE, Sloetjes A, van de Putte LB, et al. Role of activatory Fc gamma RI and Fc gamma RIII and inhibitory Fc gamma RII in inflammation and cartilage destruction during experimental antigen-induced arthritis. Am J Pathol. 2001;159(6):2309–20. doi: 10.1016/s0002-9440(10)63081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Lent PL, van Vuuren AJ, Blom AB, Holthuysen AE, van de Putte LB, van de Winkel JG, et al. Role of Fc receptor gamma chain in inflammation and cartilage damage during experimental antigen-induced arthritis. Arthritis Rheum. 2000;43(4):740–52. doi: 10.1002/1529-0131(200004)43:4<740::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 37.Volin MV, Huynh N, Klosowska K, Reyes RD, Woods JM. Fractalkine-induced endothelial cell migration requires MAP kinase signaling. Pathobiology. 2010;77(1):7–16. doi: 10.1159/000272949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haringman JJ, Gerlag DM, Smeets TJ, Baeten D, van den Bosch F, Bresnihan B, et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2387–92. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 39.Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008;58(7):1931–9. doi: 10.1002/art.23591. [DOI] [PubMed] [Google Scholar]
- 40.Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. 2008;275(18):4448–55. doi: 10.1111/j.1742-4658.2008.06580.x. [DOI] [PubMed] [Google Scholar]
- 41.Murphy G, Caplice N, Molloy M. Fractalkine in rheumatoid arthritis: a review to date. Rheumatology (Oxford) 2008;47(10):1446–51. doi: 10.1093/rheumatology/ken197. [DOI] [PubMed] [Google Scholar]
- 42.Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell. 2009;136(2):249–60. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
After 4 hrs stimulation with TNF (10 ng/ml) or IL-1β (2 ng/ml) BMDM were lysed, RNA extracted and analyzed by qPCR for expression of the indicated mRNAs. Data are representative of three experiments.
Peritoneal macrophages were harvested and stained with F4/80 and the indicated integrin antibodies, and analyzed by flow cytometry. Data are representative of three mice per genotype.
BMDM protein lysates were separated by SDS-PAGE, blotted and examined for presence of the indicated proteins.
WT mice were immunized with mBSA and boosted on day 7. On day 21, knees were injected with mBSA. Four days after the knee was injected mice were treated with either TAT peptide (n=4) or D-JNKi peptide (n=5) daily i.p. On day 10, mice were sacrificed and the splenocytes were harvested and stimulated in vitro with mBSA (25μg/ml). (A) After 48 hours the supernatants were harvested and assayed for the indicated cytokine by ELISA. (B) Splenocytes from mice primed above with mBSA were removed and restimulated in culture with mBSA (25μg/ml) in the presence of D-JNKi or TAT peptide (10μM), and the supernatants were tested for the indicated cytokines. In parallel cultures, cells from mice treated in vivo with D-JNKi or TAT peptide (C) or splenocytes treated in vitro with D-JNKi or TAT peptide (10μM) (D) were pulsed with 3H-thymidine after 48 hours and harvested 16 hours later for incorporation. Shown are the means ± SEM. There were no significant differences between the groups.