

Abstract
When inappropriate (non-physiologic) estrogens affect organisms at critical times of estrogen sensitivity, disruption of normal endocrine functions can result. Non-physiologic estrogen mimetics (environmental, dietary, pharmaceutical) can signal rapidly and potently via the membrane versions of estrogen receptors, as can physiologic estrogens. Both physiologic and non-physiologic estrogens activate multiple signaling pathways, leading to altered cellular functions (eg. peptide release, cell proliferation or death, transport). Xenoestrogens’ mimicry of physiologic estrogens is imperfect. When superimposed, xenoestrogens can alter endogenous estrogens’ signaling and thereby disrupt normal signaling pathways, leading to malfunctions in many tissue types. Though these xenoestrogen actions occur rapidly via nongenomic signaling pathways, they can be sustained with continuing ligand stimulation, combinations of ligands, and signaling that perpetuates downstream, eventually also impinging on genomic regulation by controlling the activation state of transcription factors. Because via these pathways estrogens and xenoestrogens cause nonmonotonic stimulation patterns, they must be carefully tested for activity and toxicity over wide dose ranges. Nongenomic actions of xenoestrogens in combination with each other, and with physiologic estrogens, are still largely unexplored from these mechanistic perspectives.
Keywords: estrogens, nonmonotonic, nongenomic, receptors, xenoestrogens, women’s health
1.0 Introduction1
Estrogens are triple-edged swords. If women have too little of them they can experience problems such as reproductive failure, bone loss, hot flashes, skin changes, and some cardiovascular system vulnerabilities and cognitive declines [1]. Too much of them can result in cancers such as for the breast, uterus, colon, and pituitary [2], or other malfunctions such as blood clots [3] and nausea/eating disorders [4, 5]. Exposure to the wrong estrogens (xenoestrogenic mimetics) could result in endocrine disruption of functions normally mediated by physiologic estrogens [6]. There are many different types of estrogens to consider as candidates for estrogenic or estro-disruptive cellular actions. Since many tissues of males also have estrogen receptors, they will also respond to both physiologic estrogens and xenoestrogens. Some of these actions in both males and females could be of the organizational (nonreversible) types that occur during development [7].
Compounds that have estrogenic effects can act in several ways. Acting through an estrogen receptor (ER) in the cell nucleus, they can directly change the expression of genes via binding to DNA response elements, or binding to other transcription factors that bind to response elements [8]. Acting via an ER at the surface of the cell, they can rapidly initiate cascades of chemical signals (specific ions, lipids, cyclic nucleotides, etc.) which then percolate through a series of kinases and phosphatases to control their eventual targets by adjusting their phosphorylation levels [9, 10]. While these membrane-initiated actions generally happen rapidly, they may take some time to travel to the functional end of the pathways or to build up levels of products that change function. They may also be sustained by repeated reactivation and perpetuation down signaling cascades. Post-translational modifications brought on by nongenomic signaling can have a variety and multiplicity of downstream effects on functional molecules. Of these (and other) possible estrogen-induced mechanisms, only the genomic pathway has yet been extensively examined, and xenoestrogens are very weak via that mechanism. Data are beginning to emerge indicating that xenoestrogens may be much more potent via the non-nuclear (nongenomic, membrane-initiated) mechanisms.
2.0 Different kinds of ERs, their different subcellular distribution, and association with different cellular signaling mechanisms
Historically, genomic (directly transcriptional) responses to steroids acting via their nuclear receptor mediators have been the most studied and thoroughly described with respect to signaling partners, modulators, and biochemical products (RNAs and proteins) [11]. Though very rapid responses to estrogens have been observed for decades [12–14], only recently have separate nongenomic receptor-mediated signaling mechanisms been assigned to them. A variety of ERs (α, β, and GPER) have been linked to nongenomic estrogenic responses, including some ERα splice variants [15, 16]. Though ERs α and β are highly homologous in sequence and structure [17], the GPER (formerly known as GPR30) is of an entirely different receptor class homologous to other seven transmembrane G protein-coupled receptors [18]. Another class of so-called orphan (without clear ligand assignments) receptors, the estrogen receptor-related (ERR) receptors, has so far not been implicated in rapid responses and nongenomic signaling. It is still unclear why such a variety of ERs would be necessary to mediate the effects of estrogens. However, there are quite a few different estrogens (see section 3 below) and this may offer one reason, as we learn more about selectivity of some ligands for certain receptor forms [19]. However, it is interesting that when more than one ER is present in the same tissue or cell type for either genomic or nongenomic responses, ERα tends to be the driver of responses, while ERβ and GPER, when in the presence of ERα, tend to antagonize its responses [20–22].
Among the unique correlations of rapid nongenomic actions with a receptor is one linking recognition of a specific receptor epitope (see Figure 1) to a rapid nongenomic response time-frame. The hinge region epitope for the H151 antibody (Ab) has very interesting properties [23]. When this receptor region is blocked by Ab binding in live unpermeabilized cells (meaning that the Ab can only see the membrane form of receptor), rapid responses to estrogens in those cells are blocked [24]; however, when that same Ab is applied to cells after estrogens are administered, recognition of the epitope by the Ab is blocked for several minutes [25]. In addition, an Ab applied to a very nearby epitope (epitope R3/4; also recognized by Ab ER75 [26]), in the absence of estrogens, triggers the same estrogenic response (prolactin release) as does the E2 ligand [24, 25]. This is an interesting connection between membrane receptor specific subtype (ERα) recognition and a rapid functional response. Perhaps careful testing of the many Abs now available for different ER subtype epitopes can make some parallel connections and uncover some new therapeutic uses for such Abs.
Figure 1. An ERα domain map showing a number of different epitopes with functional properties.

Hinge region epitopes for Abs R4 vs. H151 have special properties with respect to triggering or blocking (respectively) rapid actions, as described in the text. Although the 600 amino acid rat receptor is shown, some of the Abs were raised to the 595 amino acid human receptor (H151, H222, C542, symbols in red) or the 599 amino acid mouse receptor (MC20, rodent Ab symbols in purple), so these amino acid ranges (shown below each Ab symbol) will be approximate for the rat receptor. For reference to functional domain landmarks, AF-1 and AF-2 are the ligand independent and ligand-dependent transcription transactivation domains for nuclear ERα, though the function of these domains in the membrane receptor are likely to be interaction sites for proteins other than partnering transcription co-modulators.
3.0 So many different kinds of estrogens
3.1 Other physiological estrogens
Besides the most often studied estrogen -- cycle-dominant estradiol (E2) -- there are other prominent physiologic estrogens with significant impact at different life stages, such as E1 (estrone, elevated postmenopausally) and E3 (estriol, elevated during pregnancy). There are also many modified physiologic estrogens or metabolites, such as catechol estrogens, methoxy estrogens, sulfated estrogens, etc. [27, 28]. These other physiologic estrogens have long been labeled weak estrogens because they were tested exclusively via the genomic signaling pathway. Now we find that some of them (that have so far been tested) are actually quite potent via the nongenomic signaling pathway [29–32]. Their ability to act potently may relate to actions at particular life stages of women in which these hormones are quite prominent. In pregnancy E3 levels climb steadily until at parturition they are the predominant estrogen available in the circulation; abnormally low amounts of E3 are associated with fetal risk for diseases like Down’s syndrome [33] and eclampsia [34]. In peri- and post-menopause, the levels of E1 rise until they become a dominant hormonal influence [35]. It is at such times that lifelong exposures to some estrogens begin to cause tumors in a variety of estrogen target organs with high receptor numbers (the most sensitive). It is interesting that at this same time, signaling may switch from hormones that are predominantly known for their potent genomic actions (E2) to those that act potently via only the nongenomic pathway (E1 and E3). Is this a protective mechanism at a tumor-prone time? Are the high levels of E3 present at the end of pregnancy also protective -- against eventual estrogen-induced tumor induction in exposed fetuses or pregnant women?
3.2 Environmental estrogens
There are also many different classes of environmental (toxic contaminant) estrogens. Products containing these compounds litter our landfills and leach into our land and water sources (plastics, industrial surfactants, pesticides). Some xenoestrogens such as pesticides (eg. dieldrin, endosulfan) and plastics monomers such as bisphenol A (BPA) have known disease associations [36], Though the mechanisms are not well understood. BPA has become a frequent topic of news reports and regulatory agency debates because of its prevalence in the environment [37], detectable levels in more than 90% of Americans and other populations [38], its actions on a variety of tissues and cell types [39, 40], and at vulnerable developmental stages (eg. developmental factors in asthma, breast cancer, and diabetes in rodent systems [41–43]). These compounds clearly affect functions in sensitive animal models [44], yet their activities could not be explained by genomic cellular mechanisms and the better known nuclear forms of ERs. We and others have recently started approaching this problem by studying the much-neglected atypical or nonclassical estrogenic signaling mechanisms -- that is, the rapid membrane-initiated estrogenic signaling or nongenomic pathway [45]. We also have begun to ask these questions about ordered sets of environmental estrogen compounds that vary incrementally in their structural features, to try to decipher what makes a compound a good nongenomic pathway estrogen [46], as compared to the many studies already determining what makes a good nuclear signaling pathway estrogen.
3.3 Pharmaceutical estrogens
There are also a large number of pharmaceutical estrogens either purified from naturally occurring sources (eg. equine estrogens) or produced synthetically, such as diethylstilbestrol (DES), contraceptive estrogens, antiestrogens (which are often tissue-selective estrogen receptor modulators [SERMs]). Again, how these estrogens mediate their actions is mostly known only for the nuclear transcriptional actions of ERs. Information about their membrane receptor-initiated signaling is nonexistent, or just beginning to be examined.
3.4 Phytoestrogens and their possible role as therapeutics
Many plant estrogens are available both via foods (many of them major components of typical Asian diets) [47] and as dietary supplements marketed by the health food industry. Prominent among these are the isoflavones (present in soy-based foods) and coumesterol (from sprouts and red clover). The prevalence of grape products (especially red grapes, red wine) and nuts in some diets are touted as the reason certain cultures gain health benefits from the actions of the phytoestrogen provided by these foods – resveratrol [48]. Molds that grow on grains can become dietary estrogens when they contaminate food items made from these grains (eg. zearalenone [49]). Much speculation has arisen about the benefits or risks of these dietary components on disease incidences [47] and about the actual ingredient in foods that cause the effects (for instance isoflavones vs. other unidentified compounds that accompany isoflavones in soy-based foods [50]). Another complication in interpreting the beneficial vs. harmful actions of the soy isoflavone genistein is its well-known alternative role as an inhibitor of tyrosine kinases and other enzymes important to cellular signaling mechanisms [51].
Are there any estrogens that may alleviate the effects of estrogen withdrawal at surgical or natural menopause, while not promoting tumors? Phytoestrogens have been suggested as a possible subtype to fulfill this therapeutic role [47]. They have been called very weak estrogens because of their poor performance in transcription assays [52], and were originally thought to act by just replacing more potent estrogens on nuclear receptors. Yet they do have functional effects by themselves [53, 54], both acute (e.g. genistein causing uterine bleeding) and more subtle long-term effects (across populations of women who eat lots of them and have better bone health, and lower cardiovascular and cancer risks). However, the exposure of nursing infants to high concentrations of phytoestrogens in soy-based formulas has been noted as a possibly inappropriate developmental exposure [55]. We recently found that though phytoestrogens do not have the same effects as a powerful tumor-inducing estrogen DES in a rodent model for pituitary cancer, even when present at high (though still dietarily obtainable) concentrations, they do, however, exacerbate increased cell nuclear size and size variability induced by the carcinogen DES, perhaps related to induction of aneuploidy [56, 57]. We will have to examine phytoestrogens and their combinations more carefully in many cancer-causing or –inhibiting tissue scenarios now that we know that they can operate potently via the nongenomic signaling pathway [57, 58], with possible complications of nonmonotonic concentration-dependence (see section 4).
3.5 Selective membrane estrogen receptor modulators (SmERMS)
We and others have studied a variety of estrogens and compared their estrogenicity across many different signaling assays. Though weak in the genomic pathway, many have recently been found to be very potent in the nongenomic signaling pathway (reviewed in [9, 59, 60]), dependent upon the tissue, developmental or reproductive stage, and other regulatory circumstances; so the story of their actions is very complex. By comparing their actions in different tissues we find that they are selective membrane estrogen receptor modulators, which we have dubbed SmERMs. This undoubtedly stems from the availability of different signaling partner molecules in different cell types, as they have been shown to directly interact with these partners in different tissues [61, 62], just as nuclear ERs are well known to have different signaling partners (co-modulators) in different tissues [63].
4.0 Nonmonotonic and oscillating responses and their causes
A curious feature typical of the actions of both physiologic estrogens and xenoestrogens via nongenomic signaling mechanisms is their oscillating time courses and nonmonotonic concentration-responses. What is the basis of these patterns characteristic of these complex responses? Possible mechanisms have been previously reviewed including different receptor levels, subpopulations, oligomerization, compartmentalization, and non-receptor-mediated effects, [9, 64–66]. However, we now know that estrogens activate a myriad of signaling cascades, probably dictated by the complement of ERs engaged, and by the availability of signaling partners in given tissues and cell types [10, 59, 67]. To borrow an analogy from the principles of electrical circuitry, the lesson of global signaling analysis is that cells seem to be hooked up “in parallel” instead of “in series,” as we used to think of signaling pathways. We have examined several pathways that are simultaneously activated by estrogens in pituitary cells. By introducing pathway-specific inhibitors at different time points along the way toward eventual extracellular-regulated kinase (ERK) activation in these cells, we determined that signaling initiated by different estrogens passes down these pathways at different rates [68]. The same is probably true for activation of specific signaling cascades by different concentrations or combinations of estrogens. If one considers the contribution of multiple pathways in a time- and dose-dependent manner, then a composite picture of these results impinging upon the same target [for example, a downstream MAPK (mitogen-activated protein kinase) such as ERK], could sum to an oscillating pattern. See a theoretical example of this in Fig. 2. Here Pathway A ends in ERK activation (its signaling endpoint) early or in response to low concentrations of a triggering estrogenic ligand. Pathway B has the same ERK activation endpoint, but that pathway signal arrives at ERK-phosphorylating MEKs later, or in response to higher concentrations of the estrogenic ligand. But since both pathways are activated by the same ligand-receptor interaction, the endpoint integrates these actions. Therefore, apparent nonmonotonic dose responses and oscillating temporal patterns of activation are generated. Of course, more than two pathways could participate, making a more complex summation with even more potential oscillations, or even sustained activations.
Figure 2. The contribution of multiple pathways to a composite endpoint can result in oscillations.
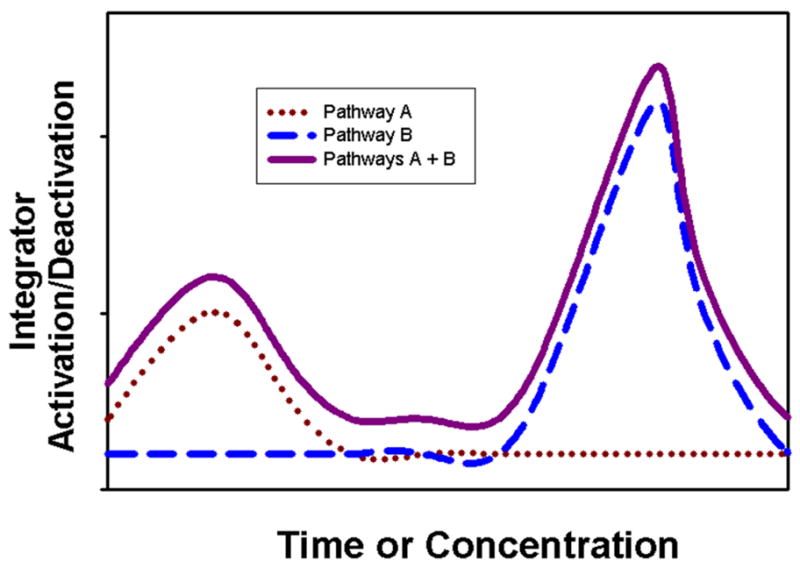
This theoretical diagram of the contributions of Pathway A in conjunction with Pathway B can be applied to both temporal and dose considerations. An example of a common endpoint could be the phosphorylation level of a MAPK. Here Pathway A ends in endpoint activation early, or in response to low concentrations of a triggering estrogenic ligand. Pathway B has the same activation endpoint, but that signaling cascade arrives at the endpoint later, or in response to higher concentrations of the estrogenic ligand. However, since both pathways are activated by the same ligand-receptor interaction, the endpoint integrates these actions, the composite of which oscillates.
In addition, other conditions could also contribute to nonmonotonic responses. Different receptor targets might oppose one another. For instance, phospho-ERK activation by estrogens could be opposed by phosphatase activation by estrogens, and the timing or concentration dependence of these effects could be different. We and others have determined that specific phosphatases are responsible for some of these fluctuating ERK activities induced by estrogens or other activators in cells [69–72]. It has long been observed that hormonal responses decrease again after they reach an asymptote, and this phenomenon is called the hormesis effect [65, 73]. Therefore, the more potent are hormonal effects, the greater their potential for inhibition at higher exposure levels. This could also be brought on by combinations of hormones or their mimetics acting via the same receptor. Hormetic inhibitions are very common in hormonal responses of many kinds, and are thought to be safety mechanisms to prevent overstimulation or conserve cellular resources.
In the end, one of the most important practical questions to answer about the toxicity of these compounds is: at which concentration(s) are they active? Because everyone previously believed xenoestrogens to be weak estrogens (via the nuclear pathway), the large majority of earlier scientific studies about them have been at very high concentrations (μM-mM). Due to the nonmonotonic nature of their responses, many of the tested effects may have missed the concentration ranges in which these compounds are the most effective. Our work and that of others suggests that xenoestrogens can be active down to the pM and fM level in some cellular assays of signaling or function [46, 74, 75]. The ability to see actions at such low concentrations depends upon sensitive, quantitative cellular response assay systems devoid of contaminating estrogens in the control samples. In addition, use of non-transfected cell systems avoids artifacts due to overexpression and heteroexpression of receptors, perhaps with appropriate signaling partners in short supply. Low concentrations of many xenoestrogens are common in the environment, and without this information about low concentration-induced responses, we are incorrectly assuming that xenoestrogens are ineffective and harmless. With such complicated response patterns, these kinds of toxicity/effectiveness testing must be done with wider and more detailed examinations of doses and times. Extrapolation of results from very high doses to predict lowest effective doses is no longer acceptable.
5.0 Types of nongenomic or membrane-initiated functional endpoints and their relationship to the speed of the response
Nongenomic steroid responses are often characterized as rapid responses. However, just because such responses initiate rapidly does not mean they can’t be sustained over long periods of time. In addition, some responses are very proximal to the initial signal, and others further downstream, requiring more time to reach the eventual target. Rapid initiation and response progression speed is used experimentally to rule out a genomic mechanism, but may not describe the entire course or persistence of the response. The following sections supply examples of different types of nongenomic responses.
5.1 Simple activations and deactivations – eg. transporters and enzymes
Some changes elicited by steroids and their mimetics are very rapid and have simple and immediate functional outcomes. One example is changes to the activity of a transporter. We found that estrogens can quickly reverse the direction of the dopamine transporter (DAT), which probably involves a conformational change caused either by direct interaction of the ER with the transporter, or by initiation of signaling cascades that in the end modify the transporter post-translationally [29, 76]. In PC12 cells at concentrations where both E2 and BPA can cause a high efflux via the DAT, they accomplish this in different ways. E2 causes mERα (mediating stimulatory effects) to traffic to the membrane while mERβ (mediating inhibitory effects) leaves the membrane. But when BPA causes high efflux, it does so by causing more DAT to traffic to the membrane. In contrast, BPA at a concentration that inhibits efflux causes all three types of ERs (α, β, and GPER) and the DAT to leave the membrane [74]. DAT modulation usually results in changes to dopamine uptake, but strikingly, both estrogens [29, 77] and amphetamines [78–80] instead cause efflux by reversing the transport direction. Another example of such a direct change to a functional protein is the nongenomic estrogenic regulation of enzymes, including enzymes that create the physiologic estrogenic hormones themselves and their metabolites [81, 82].
5.2 Affecting multiple components of the nongenomic signaling system – eg. secretion
Some functional endpoints may require several steps, and so could involve nongenomic actions on several components. For example, estrogens and xenoestrogens cause pituitary cells to release prolactin stored in secretory vesicles very rapidly. However, after this initial release from vesicles poised at the membrane, other vesicles must travel to and then dock at the membrane [83]. Our studies in this system indicate that while rising Ca++ levels can trigger fusion of the secretory vesicle with the cell surface (release), probably other signaling mechanisms also contribute to the secretory response to estrogens. Other compounds that trigger Ca++ changes very effectively, cannot release prolactin as effectively as estrogens [84]. So in this case several signaling pathways probably need to be engaged, driving multiple actions required to maximally release peptides from cells.
5.3 Complex combinations of genomic and nongenomic actions – eg. cell proliferation and apoptosis
Estrogens and xenoestrogens can also cause cells to proliferate or die. Examples of estrogen-induced proliferative effects occur in the pituitary and the breast [21, 31, 57, 85]; estrogens can cause prostate cancer cells to die [86, 87]. Steroid-induced apoptosis via nongenomic mechanisms is also seen in other tissue types such as glucocorticoid-induced killing of lymphoma cells [88]. These kinds of responses to estrogens are a composite of both rapid (nongenomic) and slow (genomic) mechanisms. Membrane-initiated signaling travels downstream via multiple signaling pathways and is integrated at kinase “nodes” (MAPKs) that sum many incoming signals (including those coming from estrogens, xenoestrogens, and other ligands). MAPKs accomplish this summation by displaying a degree of phosphorylation (activation). Then targets downstream of the MAPK node eventually lead to cell division, cell death, or even retooling (differentiation) depending upon the tissue and types of MAPKs activated or inactivated [59]. Thus they coordinate complicated global cell changes. MAPKs that are thought to selectively activate cell death pathways (JNKs and p38) may take some time to manifest their ultimate functions because of the long series of enzymatic activations, target protein cleavages, resultant protein migrations, and degradations that eventually deconstruct cells.
In addition, major signaling modes can intermingle. Estrogens can either induce or inhibit these pathways, via a complex mixture of enzymatic mechanisms and transcriptional control. Some transcription factors are downstream targets of kinases, and can be rapidly activated by phosphorylation [57]. For example, ERKs activate (phosphorylate) downstream transcription factors such as Elk, ATF2, AP1, and CREB [89–92]. Such activated transcription factors thereby transform the initial nongenomic action into an eventual genomic consequence. Therefore, the terms genomic and nongenomic become somewhat inaccurate in following these actions to their final signaling and functional endpoints.
6.0 Combinations of physiologic estrogens with xenoestrogens
In real life xenoestrogens are rarely present by themselves; in humans and animals environmental or dietary estrogens usually signal on top of a pre-existing level of life stage-dependent physiologic estrogen signaling. Thus it is important to understand how added xenoestrogens affect endogenous physiologic estrogen signaling mechanisms. So far, we have learned that depending upon their concentration, alkylphenol and BPA xenoestrogens can either enhance or inhibit the signaling activities (ERK activation, Ca++) and functions (prolactin release, cell proliferation) elicited by endogenous estrogens in pituitary cells [93, 94] spanning a wide exposure range (fM to μM). We also showed the ability of phytoestrogens to modify actions of physiological estrogens in pituitary [57]; and some xenoestrogens to block efflux via the DAT in PC12 cells [74]. It is unfortunately very difficult to predict disruption toxicities from single point assays, because of the combined nonmonotonic effects of each of the combined estrogens. In general, the lowest xenoestrogen concentrations enhance the activity induced by a physiologic estrogen, and the highest (nM concentrations in our studies) inhibit the actions of physiologic estrogens. But while this is generally true, it is not universally true. That is, sometimes xenoestrogens enhance the hormonal endpoint at every concentration, sometimes they inhibit the endpoint at every concentration, and sometimes they fluctuate back and forth between inhibition and enhancement for compounds with extreme nonmonotonic response curves. This means that each compound will have to be studied very carefully, over a range of concentrations, and in a range of cell types.
7.0 Summary
We cannot extrapolate from well-behaved dose-responses to predict the actions of xenoestrogens, either by themselves, or in disrupting the actions of physiologic estrogens. Xenoestrogens do not follow the expected and simple “dose makes the poison” rules. In fact, the complexity of the signaling mechanisms makes it imperative for researchers to test the whole dose range of exposure to these compounds to decide which produce dangerous (or therapeutic) effects. In addition, these effects can vary in different cell types, tissues, and organs and for different xenoestrogens. Thus we are just beginning to learn how complicated the questions are, and mapping out a strategy for testing each potential xenoestrogen thoroughly, so we can make responsible predictions and set guidelines, especially for exposures during times of particular developmental or tissue vulnerabilities.
Acknowledgments
This work was supported by National Institutes of Health grant ES015292, the UTMB Center for Addiction Research, the UTMB Toxicology Training Grant (T32-ES07254) and the American Institute for Cancer Research. The authors acknowledge the expert skills of Dr. David Konkel, who helped with editing our manuscript.
Footnotes
Abbreviations: Ab (antibody); DAT (dopamine transporter); DES (diethylstilbestrol); E2 (estradiol); E3 (estriol); E1 (estrone); ERK (extracellular-regulated kinase); ERR (estrogen receptor-related receptor); ER (estrogen receptor); GPER (G protein-coupled estrogen receptor); MAPK (mitogen-activated protein kinase); MEK (kinase activators of ERK); mER (membrane estrogen receptor); SERMs (selective estrogen receptor modulators); SmERMs (selective membrane estrogen receptor modulators)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Cheryl S. Watson, Email: cswatson@utmb.edu.
Yow-Juin Jeng, Email: yjeng@utmb.edu.
Jutatip Guptarak, Email: juguptat@utmb.edu.
References
- 1.Wren BG. The benefits of oestrogen following menopause: why hormone replacement therapy should be offered to postmenopausal women. Med J Aust. 2009;190:321–325. doi: 10.5694/j.1326-5377.2009.tb02423.x. [DOI] [PubMed] [Google Scholar]
- 2.Shifren JL, Schiff I. Role of hormone therapy in the management of menopause. Obstet Gynecol. 2010;115:839–855. doi: 10.1097/AOG.0b013e3181d41191. [DOI] [PubMed] [Google Scholar]
- 3.Tchaikovski SN, Rosing J. Mechanisms of estrogen-induced venous thromboembolism. Thromb Res. 2010;126:5–11. doi: 10.1016/j.thromres.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 4.Curran MP, Wagstaff AJ. Spotlight on estradiol and norgestimate as hormone replacement therapy in postmenopausal women. Treat Endocrinol. 2002;1:127–129. doi: 10.2165/00024677-200201020-00006. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson M, Naessen S, Dahlman I, Linden HA, Gustafsson JA, Dahlman-Wright K. Association of estrogen receptor beta gene polymorphisms with bulimic disease in women. Mol Psychiatry. 2004;9:28–34. doi: 10.1038/sj.mp.4001402. [DOI] [PubMed] [Google Scholar]
- 6.Colborn T, vom Saal FS, Soto AM. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ Health Perspect. 1993;101:378–384. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLachlan JA. Environmental signaling: what embryos and evolution teach us about endocrine disrupting chemicals. Endocr Rev. 2001;22:319–341. doi: 10.1210/edrv.22.3.0432. [DOI] [PubMed] [Google Scholar]
- 8.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson CS, Gametchu B. Proteins of multiple classes participate in nongenomic steroid actions. Exp Biol Med. 2003;228:1272–1281. doi: 10.1177/153537020322801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans RM. The nuclear receptor superfamily: a rosetta stone for physiology. Mol Endocrinol. 2005;19:1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- 12.Watson CS, Gametchu B. Membrane-initiated steroid actions and the proteins that mediate them. Proc Soc Exp Biol Med. 1999;220:9–19. doi: 10.1046/j.1525-1373.1999.d01-2.x. [DOI] [PubMed] [Google Scholar]
- 13.Pietras RJ, Szego CM. Cell membrane estrogen receptors resurface. Nature Medicine. 1999;5:1330. doi: 10.1038/70877. [DOI] [PubMed] [Google Scholar]
- 14.Szego CM. Parallels in the modes of action of peptide and steroid hormones: membrane effects and cellular entry. In: Kearns KW, editor. Structure and Function of the Gonadotropins. Plenum Publishing Corp; New York: 1978. pp. 431–472. [Google Scholar]
- 15.Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. 2003;100:4807–4812. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol. 2010;24:709–721. doi: 10.1210/me.2009-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-Å. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocr. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 19.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocr. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 20.Alyea RA, Laurence SE, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Watson CS. The roles of membrane estrogen receptor subtypes in modulating dopamine transporters in PC-12 cells. J Neurochem. 2008;106:1525–1533. doi: 10.1111/j.1471-4159.2008.05491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocr. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 22.Helguero LA, Lindberg K, Gardmo C, Schwend T, Gustafsson JA, Haldosen LA. Different roles of estrogen receptors alpha and beta in the regulation of E-cadherin protein levels in a mouse mammary epithelial cell line. Cancer Res. 2008;68:8695–8704. doi: 10.1158/0008-5472.CAN-08-0788. [DOI] [PubMed] [Google Scholar]
- 23.Knotts TA, Orkiszewski RS, Cook RG, Edwards DP, Weigel NL. Identification of a phosphorylation site in the hinge region of the human progesterone receptor and additional amino-terminal phosphorylation sites. J Biol Chem. 2001;276:8475–8483. doi: 10.1074/jbc.M009805200. [DOI] [PubMed] [Google Scholar]
- 24.Norfleet AM, Clarke C, Gametchu B, Watson CS. Antibodies to the estrogen receptor-α modulate prolactin release from rat pituitary tumor cells through plasma membrane estrogen receptors PMCID:10627290. FASEB J. 2000;14:157–165. doi: 10.1096/fasebj.14.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson CS, Campbell CH, Gametchu B. Membrane estrogen receptors on rat pituitary tumor cells: Immunoidentification and responses to estradiol and xenoestrogens. Experimental Physiology. 1999;84:1013–1022. doi: 10.1111/j.1469-445x.1999.01903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furlow JD, Ahrens H, Mueller GC, Gorski J. Antisera to a synthetic peptide recognize native and denatured rat estrogen receptors. Endocr. 1990;127:1028–1032. doi: 10.1210/endo-127-3-1028. [DOI] [PubMed] [Google Scholar]
- 27.Greenlee H, Chen Y, Kabat GC, Wang Q, Kibriya MG, Gurvich I, Sepkovic DW, Bradlow HL, Senie RT, Santella RM, Ahsan H. Variants in estrogen metabolism and biosynthesis genes and urinary estrogen metabolites in women with a family history of breast cancer. Breast Cancer Res Treat. 2007;102:111–117. doi: 10.1007/s10549-006-9308-7. [DOI] [PubMed] [Google Scholar]
- 28.Riza E, dos SS, De Stavola IB, Bradlow HL, Sepkovic DW, Linos D, Linos A. Urinary estrogen metabolites and mammographic parenchymal patterns in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2001;10:627–634. [PubMed] [Google Scholar]
- 29.Alyea RA, Watson CS. Nongenomic mechanisms of physiological estrogen-mediated dopamine efflux. BMC Neurosci. 2009;10:59. doi: 10.1186/1471-2202-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson CS, Jeng YJ, Kochukov MY. Nongenomic actions of estradiol compared with estrone and estriol in pituitary tumor cell signaling and proliferation. FASEB J. 2008;22:3328–3336. doi: 10.1096/fj.08-107672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarz S, Pohl P. Steroids and opioid receptors. J Steroid Biochem Mol Biol. 1994;48:391–402. doi: 10.1016/0960-0760(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 33.Benn PA. Advances in prenatal screening for Down syndrome: I. general principles and second trimester testing. Clin Chim Acta. 2002;323:1–16. doi: 10.1016/s0009-8981(02)00186-9. [DOI] [PubMed] [Google Scholar]
- 34.Shenhav S, Gemer O, Volodarsky M, Zohav E, Segal S. Midtrimester triple test levels in women with severe preeclampsia and HELLP syndrome. Acta Obstet Gynecol Scand. 2003;82:912–915. doi: 10.1034/j.1600-0412.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- 35.Greenspan FS, Gardner DG. Appendix: Normal Hormone Reference Ranges. In: Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology. 7. Lange Medical Books, McGraw Hill; New York: 2004. pp. 925–926. [Google Scholar]
- 36.McLachlan JA. Environmental signaling: what embryos and evolution teach us about endocrine disrupting chemicals. Endocr Rev. 2001;22:319–341. doi: 10.1210/edrv.22.3.0432. [DOI] [PubMed] [Google Scholar]
- 37.Stahlhut RW, Welshons WV, Swan SH. Bisphenol A data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ Health Perspect. 2009;117:784–789. doi: 10.1289/ehp.0800376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113:391–395. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, Eriksen M, Farabollini F, Guillette LJ, Jr, Hauser R, Heindel JJ, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Keri RA, Knudsen KE, Laufer H, LeBlanc GA, Marcus M, McLachlan JA, Myers JP, Nadal A, Newbold RR, Olea N, Prins GS, Richter CA, Rubin BS, Sonnenschein C, Soto AM, Talsness CE, Vandenbergh JG, Vandenberg LN, Walser-Kuntz DR, Watson CS, Welshons WV, Wetherill Y, Zoeller RT. Chapel Hill bisphenol A expert panel consensus statement: integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod Toxicol. 2007;24:131–138. doi: 10.1016/j.reprotox.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol. 2007;24:178–198. doi: 10.1016/j.reprotox.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Midoro-Horiuti T, Tiwari R, Watson CS, Goldblum RM. Maternal bisphenol a exposure promotes the development of experimental asthma in mouse pups. Environ Health Perspect. 2010;118:273–277. doi: 10.1289/ehp.0901259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, Quesada I, Nadal A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118:1243–1250. doi: 10.1289/ehp.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soto AM, Vandenberg LN, Maffini MV, Sonnenschein C. Does breast cancer start in the womb? Basic Clin Pharmacol Toxicol. 2008;102:125–133. doi: 10.1111/j.1742-7843.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Talsness CE, Andrade AJ, Kuriyama SN, Taylor JA, vom Saal FS. Components of plastic: experimental studies in animals and relevance for human health. Philos Trans R Soc Lond B Biol Sci. 2009;364:2079–2096. doi: 10.1098/rstb.2008.0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watson CS. Signaling themes shared between peptide and steroid hormones at the plasma membrane. Sci STKE. 1999;1999:E1. doi: 10.1126/stke.1999.12.pe1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kochukov MY, Jeng Y-J, Watson CS. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomammotropes. Env Health Perspect. 2009;117:723–730. doi: 10.1289/ehp.0800182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adlercreutz H. Phytoestrogens: epidemiology and a possible role in cancer protection. [Review] Environ Health Perspect 103 Suppl. 1995;7:103–112. doi: 10.1289/ehp.95103s7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 49.Takemura H, Shim JY, Sayama K, Tsubura A, Zhu BT, Shimoi K. Characterization of the estrogenic activities of zearalenone and zeranol in vivo and in vitro. J Steroid Biochem Mol Biol. 2007;103:170–177. doi: 10.1016/j.jsbmb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Lin Y, Meijer GW, Vermeer MA, Trautwein EA. Soy protein enhances the cholesterol-lowering effect of plant sterol esters in cholesterol-fed hamsters. J Nutr. 2004;134:143–148. doi: 10.1093/jn/134.1.143. [DOI] [PubMed] [Google Scholar]
- 51.Kousidou OC, Tzanakakis GN, Karamanos NK. Effects of the natural isoflavonoid genistein on growth, signaling pathways and gene expression of matrix macromolecules by breast cancer cells. Mini Rev Med Chem. 2006;6:331–337. doi: 10.2174/138955706776073420. [DOI] [PubMed] [Google Scholar]
- 52.Gaido KW, Leonard LS, Lovell S, Gould JC, Babai D, Portier CJ, McDonnell DP. Evaluation of chemicals with endocrine modulating activity in a yeast- based steroid hormone receptor gene transcription assay. Toxicol Appl Pharmacol. 1997;143:205–212. doi: 10.1006/taap.1996.8069. [DOI] [PubMed] [Google Scholar]
- 53.Whitten PL, Patisaul HB. Cross-species and interassay comparisons of phytoestrogen action [Review] Environ Health Perspect. 2001;109:5–20. doi: 10.1289/ehp.01109s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker VL, Leitman D, Jaffe RB. Selective estrogen receptor modulators in reproductive medicine and biology. Obstet Gynecol Surv. 2000;55:S21–S47. doi: 10.1097/00006254-200007001-00001. [DOI] [PubMed] [Google Scholar]
- 55.Adlercreutz H, Yamada T, Wahala K, Watanabe S. Maternal and neonatal phytoestrogens in Japanese women during birth. Am J Obstet Gynecol. 1999;180:737–743. doi: 10.1016/s0002-9378(99)70281-4. [DOI] [PubMed] [Google Scholar]
- 56.Pati D, Haddad BR, Haegele A, Thompson H, Kittrell FS, Shepard A, Montagna C, Zhang N, Ge G, Otta SK, McCarthy M, Ullrich RL, Medina D. Hormone-induced chromosomal instability in p53-null mammary epithelium. Cancer Res. 2004;64:5608–5616. doi: 10.1158/0008-5472.CAN-03-0629. [DOI] [PubMed] [Google Scholar]
- 57.Jeng YJ, Watson CS. Proliferative and anti-proliferative effects of dietary levels of phytoestrogens in rat pituitary GH3/B6/F10 cells - the involvement of rapidly activated kinases and caspases. BMC Cancer. 2009;9:334. doi: 10.1186/1471-2407-9-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeng YJ, Kochukov MY, Watson CS. Membrane estrogen receptor-alpha-mediated nongenomic actions of phytoestrogens in GH3/B6/F10 pituitary tumor cells. J Mol Signal. 2009;4:2. doi: 10.1186/1750-2187-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belcheva MM, Coscia CJ. Diversity of G protein-coupled receptor signaling pathways to ERK/MAP kinase. Neurosignals. 2002;11:34–44. doi: 10.1159/000057320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watson CS, Gametchu B, Norfleet AM, Campbell CH, Thomas ML. Rapid nongenomic actions of estrogens [Review] Women and Cancer. 1998;1:21–28. [Google Scholar]
- 61.Boonyaratanakornkit V, Edwards DP. Receptor mechanisms of rapid extranuclear signalling initiated by steroid hormones. Essays Biochem. 2004;40:105–120. doi: 10.1042/bse0400105. [DOI] [PubMed] [Google Scholar]
- 62.Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–353. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 63.Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13:55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- 64.Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weltje L, vom Saal FS, Oehlmann J. Reproductive stimulation by low doses of xenoestrogens contrasts with the view of hormesis as an adaptive response. Hum Exp Toxicol. 2005;24:431–437. doi: 10.1191/0960327105ht551oa. [DOI] [PubMed] [Google Scholar]
- 66.Conolly RB, Lutz WK. Nonmonotonic Dose-Response Relationships: Mechanistic Basis, Kinetic Modeling, and Implications for Risk Assessment. Toxicological Sciences. 2004;77:151–157. doi: 10.1093/toxsci/kfh007. [DOI] [PubMed] [Google Scholar]
- 67.Watson CS, Norfleet AM, Pappas TC, Gametchu B. Rapid actions of estrogens in GH3/B6 pituitiary tumor cells via a plasma membrane version of estrogen receptor-α. Steroids. 1999;64:5–13. doi: 10.1016/s0039-128x(98)00107-x. [DOI] [PubMed] [Google Scholar]
- 68.Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69:181–192. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bermudez O, Marchetti S, Pages G, Gimond C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene. 2008;27:3685–3691. doi: 10.1038/sj.onc.1211040. [DOI] [PubMed] [Google Scholar]
- 70.Yu LG, Packman LC, Weldon M, Hamlett J, Rhodes JM. Protein Phosphatase 2A, a Negative Regulator of the ERK Signaling Pathway, Is Activated by Tyrosine Phosphorylation of Putative HLA Class II-associated Protein I (PHAPI)/pp32 in Response to the Antiproliferative Lectin, Jacalin. Journal of Biological Chemistry. 2004;279:41377–41383. doi: 10.1074/jbc.M400017200. [DOI] [PubMed] [Google Scholar]
- 71.Zivadinovic D, Watson CS. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7:R130–R144. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, Zhang B, Wang M, Carr BI. Cdc25A and ERK interaction: EGFR-independent ERK activation by a protein phosphatase Cdc25A inhibitor, compound 5. J Cell Physiol. 2005;204:437–444. doi: 10.1002/jcp.20297. [DOI] [PubMed] [Google Scholar]
- 73.Calabrese EJ. Getting the dose-response wrong: why hormesis became marginalized and the threshold model accepted. Arch Toxicol. 2009;83:227–247. doi: 10.1007/s00204-009-0411-5. [DOI] [PubMed] [Google Scholar]
- 74.Alyea RA, Watson CS. Differential regulation of dopamine transporter function and location by low concentrations of environmental estrogens and 17beta-estradiol. Environ Health Perspect. 2009;117:778–783. doi: 10.1289/ehp.0800026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Espinosa MJ, Silva E, Granada A, Molina-Molina JM, Fernandez MF, Aguilar-Garduno C, Olea-Serrano F, Kortenkamp A, Olea N. Assessment of the total effective xenoestrogen burden in extracts of human placentas. Biomarkers. 2009;14:271–277. doi: 10.1080/13547500902893744. [DOI] [PubMed] [Google Scholar]
- 76.Foster JD, Cervinski MA, Gorentla BK, Vaughan RA. Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol. 2006:197–214. doi: 10.1007/3-540-29784-7_10. [DOI] [PubMed] [Google Scholar]
- 77.Watson CS, Alyea RA, Hawkins BE, Thomas ML, Cunningham KA, Jakubas AA. Estradiol effects on the dopamine transporter - protein levels, subcellular location, and function. J Mol Signal. 2006;1:5. doi: 10.1186/1750-2187-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becker JB, Rudick CN. Rapid effects of estrogen or progesterone on the amphetamine-induced increase in striatal dopamine are enhanced by estrogen priming: a microdialysis study. Pharmacol Biochem Behav. 1999;64:53–57. doi: 10.1016/s0091-3057(99)00091-x. [DOI] [PubMed] [Google Scholar]
- 79.Becker JB. Estrogen rapidly potentiates amphetamine-induced striatal dopamine release and rotational behavior during microdialysis. Neurosci Lett. 1990;118:169–171. doi: 10.1016/0304-3940(90)90618-j. [DOI] [PubMed] [Google Scholar]
- 80.Binda F, Dipace C, Bowton E, Robertson SD, Lute BJ, Fog JU, Zhang M, Sen N, Colbran RJ, Gnegy ME, Gether U, Javitch JA, Erreger K, Galli A. Syntaxin 1A Interaction with the Dopamine Transporter Promotes Amphetamine-Induced Dopamine Efflux. Mol Pharmacol. 2008;74:1101–1108. doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fan J, Traore K, Li W, Amri H, Huang H, Wu C, Chen H, Zirkin B, Papadopoulos V. Molecular Mechanisms Mediating the Effect of Mono-(2-Ethylhexyl) Phthalate on Hormone-Stimulated Steroidogenesis in MA-10 Mouse Tumor Leydig Cells. Endocr. 2010;151:3348–3362. doi: 10.1210/en.2010-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Catalano S, Barone I, Giordano C, Rizza P, Qi H, Gu G, Malivindi R, Bonofiglio D, Ando S. Rapid estradiol/ERalpha signaling enhances aromatase enzymatic activity in breast cancer cells. Mol Endocrinol. 2009;23:1634–1645. doi: 10.1210/me.2009-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodriguez-Boulan E, Kreitzer G, Musch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005;6:233–247. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- 84.Bulayeva NN, Wozniak A, Lash LL, Watson CS. Mechanisms of membrane estrogen receptor-{alpha}-mediated rapid stimulation of Ca2+ levels and prolactin release in a pituitary cell line. Am J Physiol Endocrinol Metab. 2005;288:E388–E397. doi: 10.1152/ajpendo.00349.2004. [DOI] [PubMed] [Google Scholar]
- 85.Chun TY, Gorski J. High concentrations of bisphenol A induce cell growth and prolactin secretion in an estrogen-responsive pituitary tumor cell line. Toxicol Appl Pharmacol. 2000;162:161–165. doi: 10.1006/taap.1999.8840. [DOI] [PubMed] [Google Scholar]
- 86.Carruba G. Estrogen and prostate cancer: an eclipsed truth in an androgen-dominated scenario. J Cell Biochem. 2007;102:899–911. doi: 10.1002/jcb.21529. [DOI] [PubMed] [Google Scholar]
- 87.Jarred RA, Cancilla B, Prins GS, Thayer KA, Cunha GR, Risbridger GP. Evidence That Estrogens Directly Alter Androgen-Regulated Prostate Development. Endocrinology. 2000;141:3471–3477. doi: 10.1210/endo.141.9.7648. [DOI] [PubMed] [Google Scholar]
- 88.Gametchu B, Watson CS. Correlation of membrane glucocorticoid receptor levels with glucocorticoid-induced apoptotic competence using mutant leukemic and lymphoma cells lines. Journal of Cellular Biochemistry. 2002;87:133–146. doi: 10.1002/jcb.10288. [DOI] [PubMed] [Google Scholar]
- 89.Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 90.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 91.Quesada I, Fuentes E, Viso-Leon MC, Soria B, Ripoll C, Nadal A. Low doses of the endocrine disruptor bisphenol-A and the native hormone 17beta-estradiol rapidly activate transcription factor CREB. FASEB J. 2002;16:1671–1673. doi: 10.1096/fj.02-0313fje. [DOI] [PubMed] [Google Scholar]
- 92.Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–256. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- 93.Jeng YJ, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter ERK phosphorylation profiles in rat pituitary cells. Environ Health Perspect. 2011;119:104–112. doi: 10.1289/ehp.1002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeng YJ, Kochukov M, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter calcium and kinase responses, prolactin release, and membrane estrogen receptor trafficking in rat pituitary cells. Environ Health. 2010;9:61. doi: 10.1186/1476-069X-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]