

Abstract
Fibrogenesis is a pathological wound repair process that fails to cease, even when the initial insult has been removed. Fibroblasts are principal mediators of fibrosis, and fibroblasts from fibrotic tissues fail to return to their quiescent stage, including when cultured in vitro. In a search for underlying molecular mechanisms, we hypothesized that this perpetuation of fibrogenesis is caused by epigenetic modifications. We demonstrate here that hypermethylation of RASAL1, encoding an inhibitor of the Ras oncoprotein, is associated with the perpetuation of fibroblast activation and fibrogenesis in the kidney. RASAL1 hypermethylation is mediated by the methyltransferase Dnmt1 in renal fibrogenesis, and kidney fibrosis is ameliorated in Dnmt1+/− heterozygous mice. These studies demonstrate that epigenetic modifications may provide a molecular basis for perpetuated fibroblast activation and fibrogenesis in the kidney.
Renal fibrosis is a pathologic scarring process1. A hallmark of fibrosis, as opposed to physiological wound healing, is that the active scarring does not cease once the initial insult has been contained and instead becomes a continuous process1,2. What perpetuates scarring in the setting of fibrosis is not yet known3. Activated fibroblasts are commonly considered main mediators of renal fibrosis3. The term ‘activated’ refers to the observation that fibroblasts associated with physiological repair or fibrosis show an increased proliferative activity, increased production of extracellular matrix constituents and expression of α-smooth muscle actin (α-SMA)3,4. In the setting of fibrosis, such activation is so robust that primary fibroblasts isolated from fibrotic kidneys maintain their activated state even when cultured in vitro5,6. It has been hypothesized that such maintenance of the activated stage reflects the failure of the fibroblasts in the fibrotic kidney to return to their resting state as they do in physiological wound healing1,3. This phenomenon is observed not only in fibrotic kidney fibroblasts but also in activated fibroblasts isolated from any given fibrotic organ4,7–9. Here we hypothesized that epigenetic modifications are a molecular cause for fibrotic fibroblast activation and fibrosis.
Results
Identification of RASAL1 hypermethylation in fibrotic fibroblasts
To test our hypothesis that an epigenetic modification, in particular hypermethylation, is involved in renal fibrogenesis, we treated CD1 mice that had been challenged with folic acid to induce fibrosis with the demethylating agent 5′-azacytidine10,11. Progressive fibrosis and kidney failure was significantly inhibited in the mice that received 5′-azacytidine from day 3 to day 28 after folic acid injection (Fig. 1a–c). Treatment with 5′-azacytidine significantly ameliorated accumulation of activated fibroblasts, as it decreased accumulation of fibroblast-specific protein-1 (FSP-1)-positive fibroblasts (Fig. 1d,e), α-SMA+ myofibroblasts (Fig. 1d,f) and type I collagen in the folic acid–challenged kidneys (Fig. 1d,g). We next assessed the impact of 5′-azacytidine on fibrotic human kidney fibroblasts in vitro. In cell culture experiments, exposure to 5′-azacytidine normalized proliferative activity, type I collagen expression and α-SMA expression of the fibrotic fibroblasts (Supplementary Fig. 1). These observations prompted us to study whether hypermethylation of specific genes in fibroblasts could be involved in both perpetuation of fibroblast activation and renal fibrogenesis.
Figure 1.
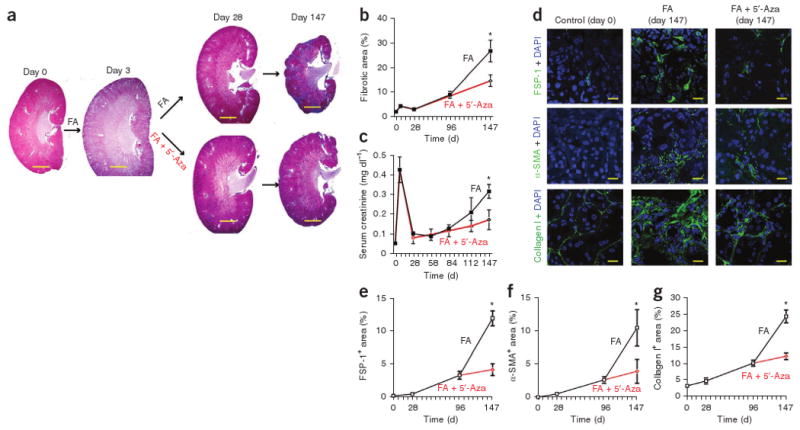
5′-azacytidine ameliorates experimental renal fibrosis. (a) Representative photomicrographs of Masson's trichrome–stained kidney sections from mice that received folic acid (FA) (control) or folic acid and 5′-azacytidine (5′-Aza). Mice were analyzed at 0 d, 3 d, 28 d or 147 d after folic acid injection. Scale bar, 200 μm. (b) Interstitial fibrotic area in Masson's trichrome–stained sections. The graph summarizes average values at the indicated time points of each group. (c) Average serum creatinine concentrations at the indicated time points. (d) Representative confocal photomicrographs of immunofluorescence labeling for FSP-1 (top), α-SMA (middle) or type I collagen (bottom) on kidney sections of control CD1 mice (left column), kidneys of mice after folic acid injection (middle column) and from kidneys of mice that had received both folic acid and 5′-azacytidine (right column). Scale bar, 20 μm. (e–g) Relative stained area in frozen sections that were labeled with antibodies to FSP-1 (e), α-SMA (f) or type I collagen (g). Data are presented as means ± s.e.m. *P < 0.05.
Methylation of DNA occurs on cytosine residues that precede a guanosine in the DNA sequence (the CpG dinucleotide)12. Abnormal methylation of CpG clusters (CpG islands) in the promoter region (referred to as ‘hypermethylation’) often results in silencing of the gene13. In addition to the functional implications of gene inactivation in tumor development, these aberrant hypermethylation patterns also represent excellent genomic targets for diagnostic approaches13–17.
To identify candidate genes that are hypermethylated in fibrotic fibroblasts, we compared primary human fibroblasts from fibrotic kidneys with fibroblasts from nonfibrotic kidneys with a genome-wide methylation screen (sample characteristics are specified in Supplementary Table 1). This screen revealed 12 genes that were methylated in all seven tested fibrotic fibroblast samples and nonmethylated in all tested nonfibrotic fibroblast samples (Supplementary Table 2).
Among the identified genes, RASAL1 stood out as a candidate to facilitate fibroblast activation and progression of renal fibrosis. RASAL1 was one of three genes (out of the 12) that had similar CpG islands in both humans and mice (enabling us to study its role in mice) and that consistently showed decreased expression in fibrotic mouse kidneys (Supplementary Table 2).
Ras proteins in general are signal switch molecules that regulate cell fate by coupling receptor activation to downstream effector pathways that control diverse cellular responses, including proliferation18,19. RASAL1 is a member of the RAS-GAP family, which catalyzes Ras inactivation by binding to GTP-Ras and catalyzing hydrolysis to GDP-Ras20. In cancer cells, growth factor–independent Ras hyperactivity causes stimulation of autonomous cell proliferation21,22. Such Ras hyperactivity is most often caused by gain of function mutations of Ras genes21,22 but can also be caused by loss of Ras-GAPs, including RASAL1 (refs. 23–25). We hypothesized that epigenetic silencing of RASAL1 could have a mechanistic role in fibrotic fibroblast activation. We confirmed by methylation-specific PCR (MSP) and bisulfite genomic sequencing (BGS) that RASAL1 was hypermethylated in all tested fibrotic fibroblasts as compared to nonfibrotic fibroblasts (Fig. 2a,b and Supplementary Table 1). In vitro methylation significantly inhibited RASAL1 promoter activity (Fig. 2c), and, in fibrotic fibroblasts, RASAL1 methylation was associated with substantial repression of RASAL1 expression (Fig. 2d).
Figure 2.

RASAL1 hypermethylation in fibrotic kidney fibroblasts. (a) Electrophoresis of PCR products using primers for methylated RASAL1 (top) and unmethylated RASAL1 (bottom). Primary fibroblasts TK188, TK261, TK270, TK274, TK110 and TK257 were isolated from fibrotic human kidneys; TK124, TK173 and TK210 were derived from nonfibrotic human kidneys. (b) BGS of the indicated cells. Each box is representative of the indicated cell culture; each row of dots in the boxes is representative of the RASAL1 CpG island; each dot is representative of a single CpG. Empty dots indicate unmethylated CpGs; black dots indicate methylated CpGs. Each row represents a single sequenced clone (five for each cell line). We methylated DNA with the bacterial methyltransferase SssI as positive control. (c) Relative activity of pGL4-luciferase plasmid containing unmethylated or methylated RASAL1 promoter. (d) Average RASAL1 expression (by qRT-PCR) of each fibroblast culture (normalized to TK173 nonfibrotic fibroblasts set arbitrarily to 1). AU, arbitrary units. (e) Representative photomicrographs of Masson's trichrome–stained kidneys of control CD1 mice and of CD1 mice 5 months after folic acid injection. Scale bars, 20 μm. (f) Average serum creatinine concentrations of control CD1 mice and of mice with renal fibrosis. (g) Relative Rasal1 expression (qRT-PCR) in kidneys of control CD1 mice and of mice that had received folic acid. Expression in control mice was set to an arbitrary value of 1. (h) Rasal1 methylation, as analyzed by MeDIP. The top picture shows a virtual gel of Rasal1 PCR products of captured (methylated) DNA; the bottom picture shows Rasal1 PCR products of input DNA (to control for equal loading in immunoprecipitation). (i) Immunofluorescence double-labeling with antibodies to Rasal1 (red) and FSP-1 (green). The arrow highlights an FSP-1+Rasal1+ fibroblast. Scale bars, 20 μm. (j) The average number of FSP-1+Rasal1+ cells among all FSP-1+ fibroblasts per group (top) and the number of FSP-1+ fibroblasts per visual field in each group (bottom). Data are presented as means ± s.e.m. ***P < 0.001.
Renal fibrosis is associated with Rasal1 methylation in mice
To gain insights into the role of Rasal1 hypermethylation on the progression of renal fibrosis, we next investigated Rasal1 methylation and Rasal1 expression in fibrotic and nonfibrotic mouse kidneys. Rasal1 expression was decreased in fibrotic kidneys of mice which had been challenged with folic acid (Fig. 2e–g). Using a strategy that combines immunoprecipitation of methylated DNA followed by analysis of the captured methylated DNA by Rasal1-specific PCR (MeDIP)26, we analyzed Rasal1 methylation in total DNA samples from nonfibrotic control kidneys and from fibrotic kidneys. Rasal1 was hypermethylated in all fibrotic kidneys but not in control kidneys (Fig. 2h), confirming that Rasal1 methylation correlates with decreased Rasal1 expression. We next performed immunofluorescence double-labeling experiments with antibodies to Rasal1 and FSP-1 to assess Rasal1 expression specifically by fibroblasts in the kidney (Fig. 2i,j). Whereas ∼76% of FSP-1+ fibroblasts in control kidneys were also positive for Rasal1, the percentage of FSP-1+Rasal1+ fibroblasts among all FSP-1+ fibroblasts was significantly lower in fibrotic kidneys (∼12%), suggesting that fibroblast accumulation in renal fibrogenesis is associated with Rasal1 silencing (Fig. 2i,j). To rule out the possibility that Rasal1 hypermethylation is only relevant in the folic acid–induced nephropathy model, we also assessed Rasal1 methylation in the model of nephrotoxic serum nephritis in C57BL/6 mice27,28. We established that fibrosis in this second model was similarly associated with Rasal1 methylation and decreased Rasal1 expression (Supplementary Fig. 2).
Given that fibrogenesis upon kidney injury correlates with Rasal1 hypermethylation, we next elucidated whether absence of fibrosis upon kidney injury also correlates with absence of Rasal1 methylation. First, we analyzed Rasal1 methylation in the mouse model of ischemia-reperfusion injury, a model of physiological repair similar to wound healing29,30. Our rationale for using this model was that in this model an acute kidney injury is induced (by brief clamping of the renal pedicle), is spontaneously regenerated and, unlike the folic acid nephropathy and nephrotoxic serum nephritis models, does not lead to substantial long-term fibrotic lesions (Fig. 3a,b). MeDIP analysis revealed that Rasal1 was not hypermethylated upon ischemia-reperfusion injury (Fig. 3c). Additionally, we performed secondary analysis on kidney samples with folic acid nephropathy and 5′-azacytidine treatment. Whereas Rasal1 was hypermethylated in fibrotic kidneys of mice that had been challenged with folic acid, Rasal1 was not hypermethylated in the kidneys with ameliorated fibrosis of mice who had received 5′-azacytidine in addition to folic acid (Fig. 3d).
Figure 3.
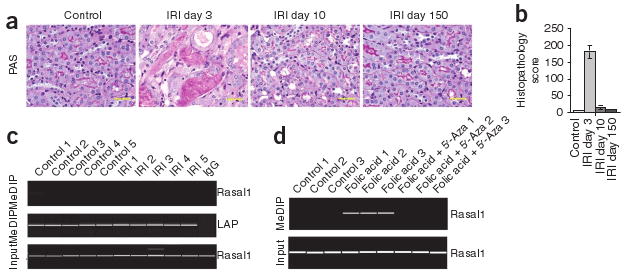
Absence of Rasal1 hypermethylation in kidneys that do not become fibrotic upon injury. (a) Representative photomicrographs of periodic acid–Schiff–stained kidney before and 3, 10 and 150 d after initiation of ischemia-reperfusion injury (IRI). Scale bars, 20 μm. (b) Average histopathological degree of tubular injury over time. (c) Rasal1 methylation upon ischemia-reperfusion injury, as analyzed by MeDIP. Top, PCR analysis of immunoprecipitated methylated DNA with primers specific to Rasal1. Middle, leucine aminopeptidase (LAP) positive control. Bottom, input DNA control. (d) Rasal1 methylation, as analyzed in control kidneys, in fibrotic kidneys of mice that had been challenged with folic acid and in kidneys of mice that had received folic acid and had been also treated with 5′-azacytidine. The picture shows a virtual gel of methylated DNA immunoprecipitation analysis. Data are presented as means ± s.e.m.
Hyperactive Ras contributes to fibroblast activation and fibrosis
We next explored the role of Rasal1 in fibroblast activation in vitro. Rasal1 was hypermethylated and Rasal1 expression was lower in fibrotic mouse kidney fibroblasts compared to nonfibrotic fibroblasts (Fig. 4a,b), as we had observed in human renal fibroblasts. Rasal1 knockdown lowered Rasal1 expression in nonfibrotic fibroblasts to the level observed in fibrotic fibroblasts (Fig. 4b), correlating inversely with Ras activity (Fig. 4c). Such Rasal1 knockdown phenocopied the intrinsic proliferative activity in serum-free medium that we observed in fibrotic fibroblasts (Fig. 4d), as well as in the increased type I collagen expression (Fig. 4e) and α-SMA expression of fibrotic fibroblasts (Fig. 4f). We obtained similar results when we analyzed the role of RASAL1 in the activation of human fibroblasts (Supplementary Fig. 3). In summary, our data suggest that RASAL1 methylation causes increased intrinsic proliferative activity, type I collagen expression and α-SMA expression of fibrotic fibroblasts via Ras hyperactivity (Supplementary Fig. 4).
Figure 4.
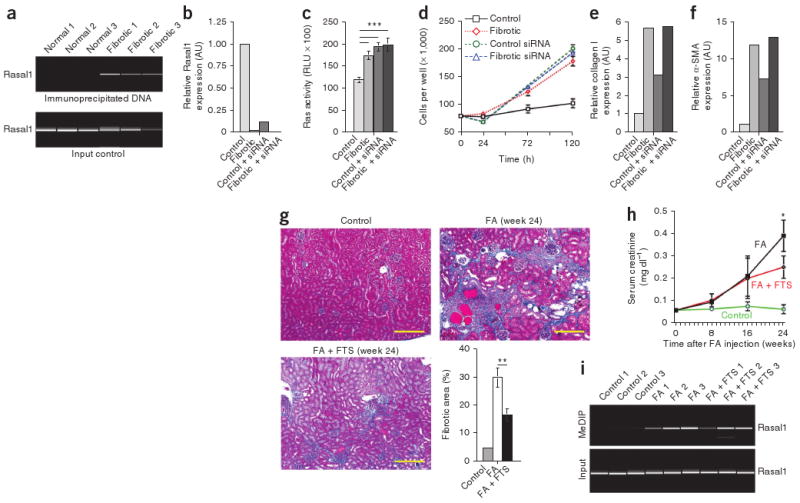
Rasal1 silencing causes renal fibroblast activation via Ras hyperactivity. (a) Methylation of Rasal1 in primary fibroblasts isolated from fibrotic and nonfibrotic mouse kidneys, as analyzed by MeDIP. Top, sample analysis; bottom, input DNA control reaction. (b) Relative Rasal1 expression in control fibroblasts, fibrotic fibroblasts, control fibroblasts transfected with Rasal1-specific siRNA and fibrotic fibroblasts transfected with Rasal1-specific siRNA. (c) Ras activity of nonfibrotic control fibroblasts, nonfibrotic control fibroblasts transfected with Rasal1-specific siRNA, fibrotic fibroblasts and fibrotic fibroblasts transfected with Rasal1-specific siRNA after 3 d of culture in serum-free medium. (d) Average cell counts for nonfibrotic control fibroblasts, nonfibrotic control fibroblasts transfected with Rasal1-specific siRNA, fibrotic fibroblasts and fibrotic fibroblasts transfected with Rasal1-specific siRNA at the indicated time points. (e,f) Relative type I collagen and α-SMA expression, respectively, in each group listed. (g) Representative photomicrographs of Masson's trichrome–stained kidneys of healthy CD1 mice (control), mice who had received a single injection of folic acid (24 weeks after folic acid injection) and mice who had received treatment with the Ras inhibitor FTS in addition to receiving folic acid. Scale bars, 100 μm. The bar graph summarizes the relative fibrotic area in each group. (h) Average serum creatinine concentrations at the indicated time points in the indicated groups. (i) Kidneys of three representative mice in each group were analyzed by MeDIP; the top picture shows amplified methylated Rasal1 DNA, and the bottom picture shows the control input DNA. For b, e and f, control values were arbitrarily set to 1. Data are presented as means ± s.e.m. * P < 0.05; **P < 0.01; ***P < 0.001.
We next explored whether Ras hyperactivity also links Rasal1 silencing to renal fibrogenesis in vivo. We induced fibrosis in CD1 mice with folic acid and gave them either DMSO (vehicle control) or the pan-Ras inhibitor S-trans, transfarnesylthiosalicyclic acid (FTS)18 from day 90 (when fibrotic lesions are already established in this model) to day 150, when we killed the mice. Kidneys of FTS-treated mice showed significantly less fibrosis as compared to the untreated mice (Fig. 4g). FTS had a protective effect on kidney function (Fig. 4h), despite similar Rasal1 methylation in both groups (Fig. 4i). Similarly, FTS administration substantially ameliorated fibrosis and preserved excretory renal function in mice that had been challenged with nephrotoxic serum (Supplementary Fig. 5)31.
Dnmt1 is involved in Rasal1 hypermethylation in fibrogenesis
We next aimed to gain insights into the mechanisms that underlie hypermethylation in renal fibrogenesis. A crucial step in DNA methylation involves DNA methyltransferases (Dnmts) that catalyze methylation of CpG dinucleotides in genomic DNA32,33. There are three known biologically active Dnmts: Dnmt1, Dnmt3a and Dnmt3b (no function has yet been found for Dnmt2)32,33. To gain insights into the possible involvement of Dnmts in renal fibrogenesis, we performed quantitative real-time PCR (qRT-PCR) on total RNA samples isolated from control mouse kidneys and from kidneys that were challenged with folic acid. In the folic acid–challenged kidneys, expression of Dnmt1, but not of Dnmt3a and Dnmt3b, was increased (Fig. 5a). Immunofluorescence double-labeling revealed more nuclear Dnmt1 labeling in kidneys of mice that had received folic acid as compared to control kidneys (Fig. 5b). Whereas most FSP-1+ fibroblasts had Dnmt1+ nuclei, Dnmt1 staining was not restricted to fibroblasts (Fig. 5b). Given these findings, we hypothesized that Dnmt1 has a role in hypermethylation of RASAL1 and in renal fibrogenesis. We next performed chromatin immunoprecipitation (ChIP) assays to establish a link between increased Dnmt1 expression and Rasal1 methylation. ChIP assay showed that Dnmt1 was bound to Rasal1 DNA in folic acid–challenged kidneys, but not in control kidneys (Fig. 5c), suggesting that Dnmt1 is involved in methylation of Rasal1.
Figure 5.
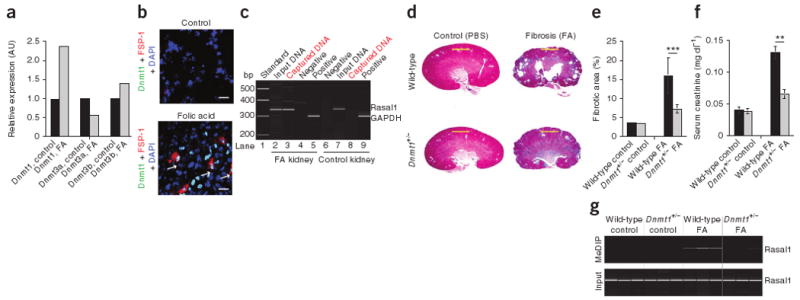
Dnmts in the mouse model of folic acid–induced nephropathy. (a) Relative expression of Dnmt1, DNmt3a and Dnmt3b in nonfibrotic and fibrotic mouse kidneys. Expression in FA-treated kidneys was normalized to kidneys from uninjected control mice, with each control value set arbitrarily to 1. (b) Representative photomicrographs of immunofluorescence labeling with antibodies specific to Dnmt1 (green) and FSP-1 (red). The arrows highlight nuclear DNMT1 staining in FSP-1+ fibroblasts in fibrotic kidneys (bottom). Scale bars, 20 μm. (c) Dnmt1 protein–Rasal1 promoter complexes were captured with Dnmt1-specific antibodies, detected by Rasal1 PCR and analyzed by electrophoresis. (d) Representative photomicrographs of Masson's trichrome–stained kidneys from wild-type control mice (top left), wild-type mice that had received folic acid (top right), Dnmt1+/− control mice (bottom left) and Dnmt1+/− mice that had received folic acid (bottom right). Scale bars, 200 μm. (e) Average fibrotic area in each of the indicated groups. (f) Average serum creatinine concentrations in each of the indicated groups. (g) MeDIP analysis for Rasal1 (top) and the input control DNA (bottom) of DNA isolated from kidneys of each indicated group. Data are presented as means ± s.e.m. **P < 0.01, ***P < 0.001.
To gain further insights into the contribution of Dnmt1 to Rasal1 methylation and progression of renal fibrogenesis, we challenged Dnmt1+/− heterozygous mice (in which Dnmt1 expression is decreased by 70% (ref. 34)) and wild-type control mice with folic acid (all on a C57BL/6 background). Renal fibrosis was reduced in Dnmt1+/− mice as compared to wild-type mice (Fig. 5d–f and Supplementary Fig. 6), correlating with ameliorated Rasal1 methylation (Fig. 5g). Both fibrosis and Rasal1 methylation were similarly reduced in Dnmt1+/− mice upon challenge with nephrotoxic serum (Supplementary Fig. 7).
Two distinct mechanisms of transcriptional silencing
To gain additional insights into Rasal1 expression and Rasal1 methylation, we exposed primary mouse kidney fibroblasts to transforming growth factor-β1 (TGF-β1), which is known to induce fibroblast activation in vitro35–37. TGF-β1 caused decreased Rasal1 mRNA expression within 8 h (Fig. 6a), whereas Rasal1 methylation could be detected after 5 d of TGF-β1 incubation but not yet after 24 h (Fig. 6b). This suggested that TGF-β1 silences Rasal1 expression via a dual mechanism—first via direct (and potentially reversible) transcriptional repression and then via methylation (potentially irreversible). To investigate this hypothesis, we next evaluated the return of Rasal1 expression upon TGF-β1 removal after TGF-β1 incubation for 24 h (when Rasal1 expression is suppressed but the Rasal1 promoter is not yet methylated) and after 5 d (when the Rasal1 promoter is methylated). After 24-h TGF-β1 incubation, Rasal1 expression returned almost to baseline levels when the TGF-β1 was removed, whereas Rasal1 expression remained suppressed when TGF-β1 was removed after a 5-d TGF-β1 incubation (Fig. 6a). Thus, short-term exposure to TGF-β1 caused reversible suppression of Rasal1 without methylation, whereas long-term exposure caused methylation of the Rasal1 promoter, causing its irreversible transcriptional suppression, as is observed in fibrotic fibroblasts (Supplementary Fig. 8). These findings suggest that RASAL1 suppression is a constituent of physiological (and reversible) TGF-β1–induced fibroblast activation and that methylation of the RASAL1 promoter is a mechanism to perpetuate silencing of this common pathway, resulting in sustained fibroblast activation, which is typical of fibrogenesis. In this regard, we found that the transient fibroblast activation, which occurs during physiological repair after ischemia-reperfusion injury in the kidney (which involves short-term TGF-β1 increase), was associated with transient suppression of Rasal1 expression without Rasal1 methylation (Supplementary Fig. 9)38–40.
Figure 6.
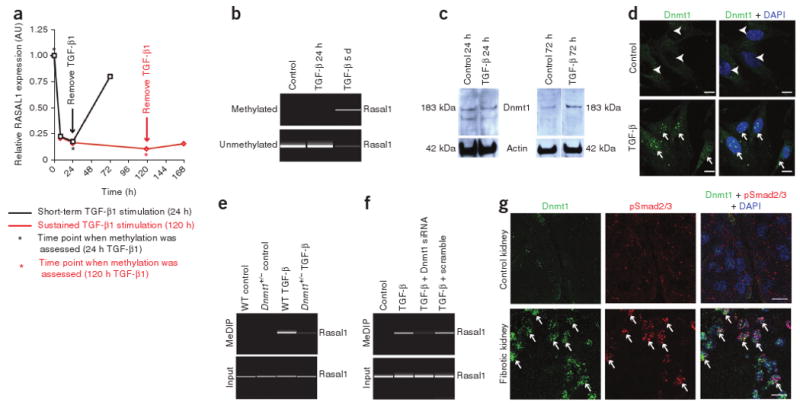
TGF-β1–induced methylation of Rasal1 in kidney fibroblasts is mediated by Dnmt1. (a) Relative Rasal1 expression over time. (b) Rasal1 methylation in primary mouse kidney fibroblasts in response to TGF-β1 exposure for 24 h and 5 d, as indicated by the asterisks in panel a. We analyzed methylation by methylation-specific PCR. Top, a virtual gel of the PCR with primers specific for methylated Rasal1; bottom, the corresponding result when primers specific for nonmethylated Rasal1. (c) Immunoblot with antibodies specific for Dnmt1. (d) Representative photomicrographs of fibroblasts labeled with antibodies to Dnmt1 (green). Fibroblasts were maintained in serum-free medium (control) or exposed to TGF-β1 for 72 h. Arrowheads highlight Dnmt1− nuclei in control fibroblasts, and arrows point to Dnmt1+ nuclei in TGF-β1–treated fibroblasts. (e) Rasal1 methylation in response to exposure to TGF-β1 (for 5 d) in primary fibroblasts isolated from kidneys of wild-type (WT) C57BL/6 mice and from littermate Dnmt1+/− mice. Top, MeDIP sample analysis; bottom, input DNA control reaction. (f) Rasal1 methylation in control fibroblasts, fibroblasts transfected with Dnmt1-specific siRNA, TGF-β1–stimulated fibroblasts and fibroblasts transfected with Dnmt1-specific siRNA and stimulated with TGF-β1 for 5 d. (g) Double-immunofluorescence labeling on control mouse kidneys (top) and on kidneys that were made fibrotic by a single injection of folic acid (bottom) using antibodies to Dnmt1 (green) and phosphorylated Smad2 and Smad3 (pSmad2/3, red). Arrows highlight nuclear pSmad2/3 colocalized with nuclear Dnmt1. Scale bars, 200 μm.
Dnmt1 mediates Rasal1 methylation in kidney fibroblasts
After establishing that TGF-β1 can induce Rasal1 methylation within 5 d, we next used this system to elucidate a possible link between TGF-β1, Dnmt1 activation and Rasal1 methylation. Comparison of serum-starved and TGF-β1–stimulated primary mouse kidney fibroblasts revealed that TGF-β1 induced Dnmt1 expression and caused nuclear translocation of Dnmt1 after 72 h, whereas we could not yet detect increased Dnmt1 expression after 24 h (Fig. 6c,d). To study the role of Dnmt1 in TGF-β1–induced Rasal1 methylation, we isolated primary fibroblasts from kidneys of Dnmt1+/− heterozygous mice and from littermate control mice and exposed them to TGF-β1. Whereas exposure to TGF-β1 caused Rasal1 methylation in wild-type fibroblasts, Rasal1 methylation was ameliorated in TGF-β1– stimulated Dnmt1+/− fibroblasts (Fig. 6e). Dnmt1 knockdown with small interfering RNA similarly ameliorated Rasal1 methylation in response to TGF-β1 (Fig. 6f), suggesting that Rasal1 hypermethylation in response to the profibrotic growth factor TGF-β1 is dependent on Dnmt1.
To correlate the cell culture studies with progression of kidney fibrosis, we next explored whether active TGF-β1 signaling in injured kidneys is associated with increased Dnmt1 expression. We performed immunofluorescence double-labeling of nonfibrotic kidneys from control mice and kidneys of mice that had been challenged with folic acid using antibodies to phosphorylated Smad2 and Smad3, which transmit signals from TGF-β receptors to the nucleus, and antibodies to Dnmt1. Whereas in control kidneys we did not detect phosphorylated Smad2 and Smad3 or Dnmt1, nuclear Dnmt1 immunolabeling in the fibrotic kidneys correlated with nuclear phosphorylated Smad2 and Smad3 staining (indicative of active TGF-β signaling in these cells) (Fig. 6g), suggesting that TGF-β induces Dnmt1 in the injured kidney similar to its effect in vitro.
Discussion
Activated fibroblasts are considered principal mediators of fibrogenesis. Unlike in physiological wound repair, where fibroblast activation is spontaneously reversible, the fibroblast activation associated with fibrogenesis is perpetuated, and fibroblasts maintain their activated phenotype even when cultured in vitro. Here we found that the demethylating agent 5′-azacytidine normalizes the phenotype of fibrotic fibroblasts in vitro and ameliorates experimental kidney fibrosis in mice. Even though we are aware that the effect of 5′-azacytidine is not limited to fibroblasts, this led us to hypothesize that hypermethylation of specific genes in fibroblasts is pivotal in perpetuating fibrogenesis. To gain insights into possible genes that are affected by hypermethylation in fibrotic fibroblasts, we performed a genome-wide methylation screen and identified 12 genes that are selectively hypermethylated in primary human fibrotic fibroblasts. Among these genes, we focused on RASAL1, an inhibitor of Ras. Our studies highlight commonalities between cancer progression and perpetuated fibrogenesis41. The capacity of fibrotic fibroblasts to proliferate in serum-free media until they reach senescence resembles the autonomous growth of cancer cells. Because, to our knowledge, mutated oncogenes cannot yet be linked to fibrogenesis, it is possible that commonalities between cancer and fibrosis are based on common epigenetic modifications. We provide evidence that epigenetic RASAL1 silencing causes fibroblast activation and fibrogenesis, similar to cancer cells that are enabled to proliferate in a growth factor–independent manner upon RASAL1 silencing24,25. Furthermore, it remains to be determined whether the activated phenotype observed in fibrotic fibroblasts and in cancer-associated fibroblasts share common underlying mechanisms. Our studies further highlight differences between fibrogenesis and physiological repair. Fibroblast activation is associated with transcriptional RASAL1 repression in both the settings of physiological kidney repair and pathological fibrogenesis. However, whereas reversible fibroblast activation (typical of physiological repair) is associated with reversible RASAL1 suppression without its hypermethylation, sustained fibroblast activation typical of fibrotic kidneys is associated with irreversible RASAL1 expression due to hypermethylation of the RASAL1 promoter. This suggests that hypermethylation perpetuates fibroblast activation and ultimately fibrogenesis by imprinting pathways of fibroblast activation that are engaged during the transient fibroblast activation typical of physiological repair. We further reveal that hypermethylation associated with fibrogenesis is mediated by the methyltransferase Dnmt1 and that pathological hypermethylation by Dnmt1 can be induced by long-term exposure to the profibrotic growth factor TGF-β1. Additional studies are required to gain further insights into the mechanisms that cause Dnmt1 to hypermethylate specific genes.
Although our studies demonstrated that the RASAL1 hypermethylation correlates strongly with fibrogenesis and absence of RASAL1 methylation correlates with physiological repair upon kidney injury, future studies manipulating RASAL1 in vivo are needed to elucidate whether RASAL1 silencing alone is sufficient to induce fibrogenesis in the kidney or whether it modulates the rate at which induced fibrosis progresses.
Our studies emphasize Ras hyperactivity as a potential therapeutic target for kidney fibrosis, confirming previous studies that suggested a prominent role of Ras signaling in renal fibrogenesis31. Previous studies have shown that increased Ras signaling within the fibrotic microenvironment is due to autocrine and paracrine growth factor stimulation. Our results show a new mechanism of epigenetic RASAL1 silencing causing increased intrinsic Ras-GTPase activity in fibroblasts. These results suggest that the antifibrotic capacity of Ras inhibitors should be further explored.
To our knowledge, this is the first report of hypermethylation as a mechanism of perpetuating fibroblast activation and fibrogenesis. In summary, hypermethylation of specific genes contributes to fibrogenesis. Future studies are needed to elucidate the utility of methylated genes as diagnostic markers to predict fibrosis and the possible therapeutic efficacy of methylation inhibitors in progression of fibrogenesis.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemedicine/.
Supplementary Material
Acknowledgments
This study was partially funded by research grants R01DK081576 (M.Z.), R03DK081687 (M.Z.), R01DK30932 (D.J.S.), R01DK55001 (R.K.), R01CA125550 (R.K.), Mentored Clinical Scientist Development Awards 1K08 CA129204 (E.M.Z.) and K08 DK074558 (M.Z.) from the National Institutes of Health, the American Society of Nephrology Carl W. Gottschalk Scholar Grant (M.Z.), American Heart Association Scientist Development Grant SDG0735602T, Deutsche Forschungsgemeinschaft-Stipendium BE4211/1-1 (W.B.), Champalimaud Metastasis Research Program (R.K.) and research funds from the Beth Israel Deaconess Medical Center for the Division of Matrix Biology. We thank A. Lau and T. Zhong Hu for technical assistance. FSP-1–specific antibodies were a gift from E. Neilson (Vanderbilt University).
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Author Contributions: W.B. performed and designed experiments, analyzed data and edited the manuscript. S.M. performed experiments and analyzed data. E.M.Z. advised, performed experiments, analyzed data and edited the manuscript. G.A.M. and C.A.M. characterized and provided human fibroblasts. H.K. generated Rasal1 antibodies. D.J.S. provided nephrotoxic serum and edited the manuscript. R.K. advised and edited the manuscript. M.Z. designed, performed and supervised experiments, analyzed data and wrote the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Eddy AA. Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol. 1996;7:2495–2508. doi: 10.1681/ASN.V7122495. [DOI] [PubMed] [Google Scholar]
- 2.Kuncio GS, Neilson EG, Haverty T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991;39:550–556. doi: 10.1038/ki.1991.63. [DOI] [PubMed] [Google Scholar]
- 3.Strutz F, Zeisberg M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol. 2006;17:2992–2998. doi: 10.1681/ASN.2006050420. [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 5.Müller GA, Rodemann HP. Characterization of human renal fibroblasts in health and disease: I. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am J Kidney Dis. 1991;17:680–683. doi: 10.1016/s0272-6386(12)80351-9. [DOI] [PubMed] [Google Scholar]
- 6.Rodemann HP, Müller GA. Characterization of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am J Kidney Dis. 1991;17:684–686. doi: 10.1016/s0272-6386(12)80352-0. [DOI] [PubMed] [Google Scholar]
- 7.Weber KT, Brilla CG. Factors associated with reactive and reparative fibrosis of the myocardium. Basic Res Cardiol. 1992;87 1:291–301. doi: 10.1007/978-3-642-72474-9_25. [DOI] [PubMed] [Google Scholar]
- 8.Hinz B, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lafyatis R. Targeting fibrosis in systemic sclerosis. Endocr Metab Immune Disord Drug Targets. 2006;6:395–400. doi: 10.2174/187153006779025766. [DOI] [PubMed] [Google Scholar]
- 10.Lin J, et al. Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat Med. 2005;11:387–393. doi: 10.1038/nm1217. [DOI] [PubMed] [Google Scholar]
- 11.Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: patho-physiology and therapeutic implications. Ann Intern Med. 2001;134:573–586. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- 12.Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 13.Feinberg AP, Vogelstein B. Alterations in DNA methylation in human colon neoplasia. Semin Surg Oncol. 1987;3:149–151. doi: 10.1002/ssu.2980030304. [DOI] [PubMed] [Google Scholar]
- 14.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 15.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353–2364. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]
- 17.Gius D, et al. The epigenome as a molecular marker and target. Cancer. 2005;104:1789–1793. doi: 10.1002/cncr.21395. [DOI] [PubMed] [Google Scholar]
- 18.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 19.Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563–R574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Walker SA, et al. Identification of a Ras GTPase-activating protein regulated by receptor-mediated Ca2+ oscillations. EMBO J. 2004;23:1749–1760. doi: 10.1038/sj.emboj.7600197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 22.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 23.Arun D, Gutmann DH. Recent advances in neurofibromatosis type 1. Curr Opin Neurol. 2004;17:101–105. doi: 10.1097/00019052-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Kolfschoten IG, et al. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell. 2005;121:849–858. doi: 10.1016/j.cell.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Jin H, et al. Epigenetic silencing of a Ca2+-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc Natl Acad Sci USA. 2007;104:12353–12358. doi: 10.1073/pnas.0700153104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–1077. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd CM, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeisberg M, et al. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 29.Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest. 1994;93:2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliver JA, Maarouf O, Cheema FH, Martens TP, Al-Awqati Q. The renal papilla is a niche for adult kidney stem cells. J Clin Invest. 2004;114:795–804. doi: 10.1172/JCI20921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendry BM, Sharpe CC. Targeting Ras genes in kidney disease. Nephron. 2003;93:e129–e133. doi: 10.1159/000070236. [DOI] [PubMed] [Google Scholar]
- 32.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 33.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 34.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 35.Bottinger EP, Bitzer M. TGF-β signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 36.Border WA, Noble NA. Targeting TGF-β for treatment of disease. Nat Med. 1995;1:1000–1001. doi: 10.1038/nm1095-1000. [DOI] [PubMed] [Google Scholar]
- 37.Strutz F, et al. TGF-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2) Kidney Int. 2001;59:579–592. doi: 10.1046/j.1523-1755.2001.059002579.x. [DOI] [PubMed] [Google Scholar]
- 38.Basile DP, et al. Identification of persistently altered gene expression in the kidney after functional recovery from ischemic acute renal failure. Am J Physiol Renal Physiol. 2005;288:F953–F963. doi: 10.1152/ajprenal.00329.2004. [DOI] [PubMed] [Google Scholar]
- 39.Forbes JM, Leaker B, Hewitson TD, Becker GJ, Jones CL. Macrophage and myofibroblast involvement in ischemic acute renal failure is attenuated by endothelin receptor antagonists. Kidney Int. 1999;55:198–208. doi: 10.1046/j.1523-1755.1999.00253.x. [DOI] [PubMed] [Google Scholar]
- 40.Villanueva S, Cespedes C, Vio CP. Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am J Physiol Regul Integr Comp Physiol. 2006;290:R861–R870. doi: 10.1152/ajpregu.00384.2005. [DOI] [PubMed] [Google Scholar]
- 41.Ding M, et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12:1081–1087. doi: 10.1038/nm1460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.