

Abstract
Motivation: Biomedical ontologies have proved to be valuable tools for data analysis and data interoperability. Protein–ligand interactions are key players in drug discovery and development; however, existing public ontologies that describe the knowledge space of biomolecular interactions do not cover all aspects relevant to pharmaceutical modelling and simulation.
Results: The protein–ligand interaction ontology (PLIO) was developed around three main concepts, namely target, ligand and interaction, and was enriched by adding synonyms, useful annotations and references. The quality of the ontology was assessed based on structural, functional and usability features. Validation of the lexicalized ontology by means of natural language processing (NLP)-based methods showed a satisfactory performance (F-score = 81%). Through integration into our information retrieval environment we can demonstrate that PLIO supports lexical search in PubMed abstracts. The usefulness of PLIO is demonstrated by two use-case scenarios and it is shown that PLIO is able to capture both confirmatory and new knowledge from simulation and empirical studies.
Availability: The PLIO ontology is made freely available to the public at http://www.scai.fraunhofer.de/bioinformatics/downloads.html.
Contact: martin.hofmann-apitius@scai.fraunhofer.de
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Biological and medical ontologies are formal representations of biomedical knowledge. They have been proved to be very useful for the communication of biomedical information through controlled vocabularies, definitions and proper metadata annotation (Bodenreider et al., 2005; Rubin et al., 2008). Numerous examples have demonstrated their value for data-mining and knowledge-discovery approaches. Ontologies have been used for automated reasoning (Héja et al., 2008), for large-scale annotation of entire genomes (Ashburner et al., 2000; Thomas et al., 2003), for data mining in microarray data (Whetzel et al., 2006), for prediction of biomolecular interactions (Yoshikawa et al., 2004) and for semantic and ontological search in unstructured information sources such as scientific text (Doms and Schroeder, 2005; Spasic et al., 2005; http://www.nlm.nih.gov/mesh/meshhome.html).
The biological domain has developed a large portfolio of widely accepted and widely used ontologies (http://www.ebi.ac.uk/ontology-lookup/) including gene ontology (Ashburner et al., 2000), the sequence ontology (Eilbeck et al., 2005) and the microarray gene expression database ontology (Whetzel et al., 2006). In parallel, the medical sector has generated its own portfolio with, e.g. the foundational model of anatomy (Rosse et al., 2003), SNOMED (Systematized Nomenclature of Medicine; Spackman et al., 1997) and ICD (International Statistical Classification of Diseases and Related Health Problems; http://www.who.int/classifications/icd/en/). However, relevant knowledge in the pharmaceutical sector has not yet been addressed by the public scientific community. Some proprietary efforts to organize the knowledge relevant for pharma industry exist. To our state of knowledge, the BioWisdom pharma ontology (Broekstra et al., 2004; http://www.biowisdom.com/2010/04/metawise/) is the only ontological resource representing a substantial part of the pharma world. This proprietary ontology comprises substantial evidence (extracted from literature) and incorporates a broad spectrum of public sources. In fact, a good part of the BioWisdom ontology underlying their ontology framework is taken from public ontologies. However, these public ontologies are organized in a way that new options for ontology alignment and reasoning are created.
Motivated by the public–private research project ‘Neuroallianz’, a nationally funded project on joint academic–industrial drug discovery and development in the area of dementia (http://www.bmbf.de/en/10540.php), we have started to develop an ontology representing knowledge about protein–ligand interactions. With this article, we present ‘protein–ligand interaction ontology’ (PLIO). PLIO is representing knowledge about the interaction of proteins and ligands (including drugs) and has a different scope and conceptual resolution than the molecular interaction ontology (Cote et al., 2006). An important feature of PLIO is that it links directly from an ontology framework describing protein–ligand interactions to the mathematical formulas relevant for the computation of some of the entities represented in the ontology. To our knowledge, this is the first example for an ontology, which directly links from a knowledge representation to the mathematical building blocks that describe the leaves of the ontology in mathematical terms. It is noteworthy that although we have adopted the top-level formal ontology structure during the construction of PLIO, our attempt was concentrated on keeping the concept definitions close to expressions in natural language. Thus, the hierarchical structure of the ontology can serve as a robust navigation tree for terminology integration and text-mining applications.
2 METHODS
PLIO was constructed according to the ontology building life cycle (Gómez-Pérez et al., 2004). To be compliant with the construction of formal ontologies, we followed the principle criteria of the top-level ontologies using the basic formal ontology upper level concepts (Grenon et al., 2004). The Protégé OWL editor was used for building the ontology in Ontology Web Language (OWL) format (http://protege.stanford.edu/~overview/).
Scope and domain coverage of the PLIO was defined by answering to a set of competency questions (cf. Supplementary Table S1). These questions capture different levels of complexity that the ontology must represent and are used to identify the key concepts and relationships between them.
2.1 Knowledge acquisition and conceptualization
Concepts were extracted from the UMLS web browser (Unified Medical Language System; http://www.nlm.nih.gov/research/umls/), International Union of Pure and Applied Chemistry (IUPAC) gold book (http://goldbook.iupac.org/), International Union of Pharmocology Committee on Receptor Nomenclature and Drug classification (Neubig et al., 2003), glossary of terms used in medicinal chemistry (IUPAC recommendations; Wermuth et al., 1998), web search and the protein–protein interaction ontology (http://bioportal.bioontology.org/visualize/39508). With the help of competency questions, a set of main concepts was identified and corresponding relationships between the parent and children classes were established. Each entity includes a specific description including name, synonym(s), reference(s) and—when appropriate—mathematical formula(s) as well as links to relevant web services which might be available for the computation of values for the entity.
2.2 Terminology analysis and concept enrichment
Transformation of the ontology OWL format into a dictionary file was achieved using a Java program. The program extracts the concept names and the corresponding synonyms from the ontology OWL structure and assigns unique identifiers to each concept. This dictionary was incorporated into ProMiner, our named entity recognition software (Fluck et al., 2007). In a subsequent step, the major super-class concepts were used as keywords for search in PubMed and from the result list of each concept search, several abstracts were chosen randomly. After compiling all abstracts, a corpus of 500 PubMed abstracts with informative contents about protein–ligand interaction was formed and randomly divided into a training set (250 abstracts), which was used for extracting the terminology manually and building the dictionary, and an annotation set for developing the gold standard (250 abstracts). From the latter, a test set of 100 abstracts was selected. In order to create the reference gold standard, suitable annotation guidelines were developed so that the annotator is guided to keep the breadth and depth of the ontology in mind and to consider not only the super-class concepts but also their corresponding sub-class concepts as well as their synonyms for annotation; for example, the term ‘hydrogen bond’ (a synonym of hydrogen bonding) is annotated under the class ‘interaction type’ as it is the subclass of the class ‘non-bonded interaction’ which is itself the subclass of the class ‘intermolecular interaction’ which is itself the subclass of the class ‘interaction type’ The following classes were chosen for manual annotation: ligand-binding site, interaction simulation, interaction detection, interaction type, ligand activity, ligand-binding site property, ligand complex, and thermodynamics of protein–ligand interactions. These classes cover the scope of the PLIO and represent its main concepts.
Using these annotation guidelines, both training and test sets were manually annotated by means of the Knowtator tool (http://knowtator.sourceforge.net/). For enrichment purposes (optimizing the dictionary), the training set was analysed for false-negative entities, which—after individual expert evaluation—were added to the PLIO terminology. The test set served as the gold standard set as well, because the evaluation process requires the performance comparison between the automatically and manually annotated text from the same set.
2.3 Evaluation
PLIO was assessed for its structural and functional features using the NeON Toolkit (http://www.neon-toolkit.org/) and AgreementMaker (Cruz et al., 2009).
To evaluate the quality of the ontology in terms of measuring the boundaries of the knowledge domain it captures, precision, recall and F-score values were calculated. These values were computed based on the longest string match found between automatically annotated words by ProMiner, and the (human) gold standard annotation for each abstract in the selected corpus. The following formulas were used for the computation of recall, precision and F-score values (Morgan et al., 2008):
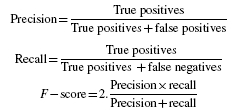 |
where true positives are the number of entities that were found by ProMiner and that matched the annotation in the gold standard; false positives are the number of entities that were (automatically) annotated by ProMiner but could not be matched to annotations in the (expert annotated) gold standard and false negatives are the number of entities that were not found by ProMiner when compared with the manual gold standard annotation.
2.4 Visualization of concepts through the text
To visualize the named entities embedded in the ontology, PLIO was integrated into our SCAIView software (Friedrich et al., 2008). SCAIView is a visualization interface for ProMiner annotations and displays named entities by markup of the text (e.g. PubMed abstracts). The key feature of SCAIView is the possibility to perform ontological search in biomedical text using concept hierarchies and synonyms associated with each concept in PLIO. For use of PLIO in SCAIView, the hierarchical organization of the ontology was preserved by transforming the ontology OWL file into XML format.
3 RESULTS
3.1 PLIO structure and contents
PLIO captures a wide range of key concepts specific to the knowledge domain of protein–ligand interaction including forces that govern the protein–ligand complex formation (e.g. van der Waals and electrostatics), interaction descriptors (e.g. pharmacophore and interaction fingerprint), interaction detection methods (e.g. nuclear magnetic resonance and X-ray), methods by which protein–ligand interactions can be simulated and predicted (e.g. molecular dynamics and docking), classification of ligand activities (e.g. biological activity and binding activity), classification of ligand modes of action (e.g. agonist and inhibitor), classification of binding sites (e.g. allosteric site and orthosteric site), and structure–activity relationships (e.g. QSAR and COMFA).
The high-level semantic framework of PLIO comprises three basic classes (i.e. protein, ligand and interaction) describing rather different types of entities (Fig. 1).
Fig. 1.

Top-level classes of PLIO. Root classes and the type of their relations are depicted.
The ‘protein biological unit’ concept reflects the complex protein structure. Sub-classes to this concept describe topological areas in the protein where a ligand can bind, i.e. ligand-binding site. This concept captures different categories of ligand-binding sites found in literature, as well as chemical and geometrical properties of the ligand-binding site. It was included in PLIO because small molecules (ligands; drugs) can bind to and specifically interact with ligand-binding sites so that a certain biological response or a chain of biological events is triggered as a result.
Normally, the outcome of an interaction between a ligand and its biological target(s) is determined by the potential of the ligand to induce a certain type of activity upon binding to its target (e.g. biological activity, binding activity and intrinsic activity). Therefore, the ‘activity’ concept was included in PLIO to capture specific features of the ligand to induce such activities.
The ‘biological activity’ concept encompasses structure–activity relationships (Fig. 2) but it is also important to characterise a ligand in terms of its ability to generate and reproduce a response, and for this reason ‘intrinsic activity’ and ‘efficacy’ were included. The concept ‘binding activity’ characterises those features that are important for ligand binding to a certain protein (e.g. affinity, specificity, selectivity and cooperative binding).
Fig. 2.

The concept ‘binding activity’ was included in PLIO to characterise the features that are important for ligand activity against a certain biological target.
The concept ‘interaction’ reflects the main features and different interaction types occurring between protein and ligand upon binding, and techniques by which protein–ligand interactions can be detected and simulated. Prediction of an interaction is represented by the ‘interaction detection’ and the ‘interaction simulation’ concepts respectively. Additionally, the interaction concept captures different chemoinformatic descriptors such as ‘pharmacophore’ and ‘structural interaction fingerprint’′, as well as interaction descriptors (electrostatic, quantum chemical, thermodynamics, geometrical, constitutional and topological descriptors).
For the sake of ontology completeness, concepts reflecting physical interactions have been annotated with their corresponding formulas and web services in addition to the common annotation fields (reference, synonym list and definition). The purpose is to hyperlink appropriate ontology leaves to online mathematical equations and/or web services that compute the corresponding functions. Figure 3 illustrates the ontology concept ‘Ki’ (equilibrium dissociation constant) annotated with its mathematical foundation (formulas field) and relevant software/web service (software field) through which users can access the link and calculate Ki online. The entire ontology consists of 375 entities interlinked by 10 semantic relation types (Table 1).
Fig. 3.

The ontology concept ‘Ki’ (equilibrium dissociation constant) annotated with its mathematical formula and web services.
Table 1.
Semantic relation types used in the PLIO ontology
| Relationship | Ontology example |
|---|---|
| binds_to | Ligand ‘binds_to’ ligand-binding site |
| can_be_formulated_as | Structure–activity relationship ‘can_be_formulated_as’ quantitative structure activity relationships |
| Defines | Activity landscape ‘defines’ biological activity |
| described_by | Thermodynamics of protein–ligand interactions ‘decsribed_by’ Gibbs free energy of binding |
| has_a | Activity landscape ‘has_a’ selectivity cliff |
| interacts_with | Ligand ‘interacts_with’ protein |
| is_a | Ligand ‘is_a’ conformer |
| is_proportional_to | Intrinsic activity ‘is proportional_to’ efficacy |
| located_in | Protein function site ‘located_in’ protein domain |
| part_of | Protein domain is ‘part_of’ protein fragment |
In case that a sub-class could be related to more than one super-class, multiple-inheritance connections were introduced; for example, if both ligand-binding site and ligand have aromatic rings, a π -stacking interaction is possible between them. As physicochemical properties characterise both protein-binding site and the corresponding ligand, multiple inheritances were established between these two concepts by means of the following relationships:
ligand ‘has a’ physicochemical properties;
ligand ‘has a’ surface property;
ligand ‘has a’ volume property and
ligand ‘has a’ electrostatic potential.
3.2 PLIO evaluation
Assessment of the quality of PLIO was based on both structural and functional criteria. Table 2 summarises the structural features of the ontology.
Table 2.
Structural characterization of the PLIO ontology
| Features | Diameter | Depth | No. of concepts | No. of leaves |
|---|---|---|---|---|
| Classes | 375 | 13 | 371 | 271 |
| Properties | 13 | 0 | 12 | 12 |
Analysis of PLIO using the XD analysis tool (eXtereme Design annotation tools; http://neon-toolkit.org/wiki/XDTools) verified that each entity is related at least to one other entity through some ontology axiom (i.e. no isolated entity), each entity is the instance of something (i.e. no missing type), and there is no intersection of classes in the domain or range of properties. We further evaluated the boundaries of the knowledge domain addressed by our ontology through aligning it with the two closest related ontologies, small-molecule ontology (SMO; Choi et al., 2010) and drug interaction ontology (DIO; Yoshikawa et al., 2004). Using a parametric string matcher algorithm, it could be shown that PLIO covers topics not previously captured by existing ontologies. At the same time, the low percentages of overlap between PLIO and these ontologies implies that PLIO has still maintained its coherence to the neighbouring knowledge domains (Table 3).
Table 3.
Ontology matching between PLIO and two related ontologies, namely SMO and DIO
| Reference ontology | Alignment algorithm | Threshold (%) | Global class match (%) | Local class matches (above threshold) (%) |
|---|---|---|---|---|
| SMO | Parametric String Matcher | 60 | 5.6 | Chem_physical property: 63.7 Interaction: 87.8 Transport: 93.0 Protein: 100 Modulation: 95.6 |
| DIO | Parametric String Matcher | 75 | 30.9 | Competitive inhibition: 82.9 Inhibition: 79.0 Enzymes: 97.0 Proteins: 91.8 |
Thresholds have been manually optimized for obtaining the highest overlap between source and target concepts through the increase of recall.
In comparison, SMO does not capture the features responsible for molecular recognition events and DIO ignores intra- and inter-molecular forces that govern the interactions between molecules.
To set the scope of the PLIO ontology, three competency questions were sketched. Answering the competency questions requires sufficient ontological coverage to capture the concepts of the domain (Supplementary Table S1).
After enrichment analysis of the training set (see Section 2), 81 concepts were enriched with synonyms and 25 new concepts were added to the PLIO.
The terminology behind PLIO supports 1321 synonyms (on average 3.5 synonyms per concept). Evaluation of the terminology showed a satisfactory performance on an independent test corpus of 100 Medline abstracts (Table 4).
Table 4.
Results of the ontology evaluation using NLP-based approach
| Descriptions of assessment | Precision | Recall | F-score |
|---|---|---|---|
| Independent test set of 100 abstracts | 0.94 | 0.72 | 0.8154 |
3.3 Usability profile
PLIO provides users with 1051 entity annotation axioms for all instances and classes. The coverage of relevant information in the ontology has been increased by adding 75 formula annotations and several software hyperlinks. Through integration of PLIO in SCAIView (Friedrich et al., 2008), we could make the ontology easily navigable as a tree and, at the same time, visualize the markup of PLIO concepts tagged in PubMed abstracts.
3.4 Use cases
There are numerous publications that either report on findings generated by simulation of protein–ligand interactions (e.g. docking and molecular dynamics simulations), or report on empirical experiments testing protein–ligand interactions in binding assays and other biochemical tests. PLIO—when used for semantic annotation of PubMed abstracts—can be employed to systematically screen the published knowledge and, for example, compare simulation-based and empirical knowledge on protein–ligand interactions. In the following use-case scenarios, we demonstrate that PLIO can be used to find confirmatory statements (i.e. protein–ligand interaction simulation experiments confirm empirical findings), or statements that indicate that, for example, through simulation new insights can be gained beyond the state of knowledge resulting from empirical experiments (and vice versa).
3.4.1 HIV-1 protease flaps
The human immunodeficiency virus type 1 aspartic protease (HIV1 protease) is a protein produced by HIV virus and the major drug target for acquired immune deficiency syndrome (AIDS) therapy. For development of potent inhibitors against HIV virus, the mechanism of protein–ligand binding should be understood using experimental and simulation approaches. In this application scenario, the main focus is HIV-1 protease flaps. HIV protease flaps are flexible and provide an access to the active site when they are in open conformation (Scott and Schiffer, 2000). Flaps are responsible for substrate penetration inside HIV1 protease and product release from the active site (Nicholson et al., 1995). PLIO ontological search was used to find relevant knowledge from both simulation and biochemical studies. The results were manually classified into confirmatory and information gain statements extracted from PubMed abstracts using SCAIView ontological search (Table 5). Table 5 shows that document retrieval based on PLIO terms can successfully discriminate between simulation and experimental knowledge related to HIV-1 protease flaps. In the case of HIV-1 protease flaps, the simulation knowledge is in agreement with the experimental (biochemical) knowledge represented in a different set of PubMed abstracts. Accordingly, the simulation results suggest that the flaps can be in ensemble of conformations between ‘semi-open’, ‘open’ and ‘curled’ conformation. The experimental results strongly support this hypothesis. The information gain column represents additional information related to the HIV-1 flap region gained from the results of ontological search. This information can be used for improved characterization and understanding of HIV-1 protease flaps and designing successful therapeutic inhibitors.
Table 5.
The knowledge statements from both simulation and experimental results related to HIV-1 protease flaps captured with the help of PLIO in PubMed abstracts and grouped into confirmatory and information gain statements
| Confirmatory statement | Information gain |
|---|---|
| Simulation: | Simulation: |
| (i) Flap conformations: semi-open, open and curled conformations (PMID: 16188477) | (i) HIV-1 showed that the monomer displayed considerable flexibility in the interfacial portions of the flap, the N- and C-termini, and, to a lesser extent, the active site (PMID: 8460108) |
| (ii) Flaps may exist in the ensemble of conformations between closed and opened (PMID: 17786489) | (ii) The highly flexible tips of the flaps, with the sequence Gly–Gly–Ile–Gly–Gly, are seen curling back into the protein and thereby burying many hydrophobic residues (PMID: 11188690) |
| Experiment: | Experiment: |
| (i) Each conformer of flaps population in apo HIV-1 protease described as ‘tucked/curled’, ‘closed’, ‘semi-open’ and ‘wide-open’ (PMID: 19788299) | (i) In flap region is observed a small increase in the amplitude of internal motion on the sub-nanosecond timescale and several residues in the flap region are mobile on the conformational exchange timescale, millisecond to microseconds (PMID: 12824484) |
| (ii) The tips of flaps in the unligated protease dimer interact with each other in solution (PMID: 17894346) |
3.4.2 Adenosine receptor antagonists
Adenosine receptors belong to the class of G-protein-coupled receptors, known as GPCRs. In this application scenario, affinity of N(6)-cyclopentlyladenosine (CPA), N(6)-cyclohexyl adenosine (CHA) and 2-chloro-N6-cyclopentyladenosine (CCPA) ligands against adenosine receptors is investigated. The goal is to compare the experimental and simulation knowledge related to the affinities of these ligands (Table 6) using PLIO ontological search in SCAIView. The results indicate that the high affinity of CPA and CCPA ligands against adenosine receptor 1 is confirmed by comparing the simulation knowledge from one PubMed abstract with the biochemical experimental knowledge from another abstract. The information gain column lists additional information regarding the interaction of CPA, CHA and CCPA ligands. It contains information related to interaction preferences of these ligands with their target-binding sites and electrostatic similarity features to other ligands. Obviously, the simulation-based findings add information that goes beyond what was found using experimental, biochemical tests.
Table 6.
Relevant statements from both, simulation and experimental results related to adenosine receptor inhibitors captured by PLIO in PubMed abstracts are listed
| Confirmatory statement | Information gain |
|---|---|
| Experiment: (i) Selective adenosine receptor agonists: CPA, CHA, CCPA, 2′Me-CCPA, NECA, IB-MECA. | Experiment: (i) Enhancers are able to increase the non-bonded interactions of the binding site with agonists as CHA, CPA, MeCPA and MeCCPA. (PMID: 12144931) |
| 2′-Me-CCPA was confirmed to be the most selective, high affinity agonist at human A(1) receptor with a Ki value of 3.3 nM and 2903- and 341-fold selective versus human A(2A) and A(3) receptors, respectively. (PMID: 15743197) | Simulation: (ii) Common binding site was found for CPA, CCPA, and NECA agonists. (PMID: 15174168) |
| Simulation: CPA CCPA Docking studies explained the lower affinity of N(6)-3-(R)-tetrahydrofur anyl-substituted compounds at bovine A(1)AR compared to that of N(6)-cyclopentyl analogues, showing that the oxygen of the tetrahydrofuranyl ring establishes unfavourable electrostatic interactions with the CO oxygen of Asn254. (PMID: 17933541) | Simulation: (iii) CPA and DPCPX show greater electrostatic similarity when the aromatic rings are superimposed according to the flipped model, in which the xanthine ring is rotated around its horizontal axis. (PMID: 7751869) |
| Simulation: (i) The binding cavity of A(1)AR is smaller than of the A(2a)AR. For this reason less bulky ligands like CPA are able to give close interactions with the A(1)AR. (PMID: 16427161) | Simulation: (iv) In the docking exploration, it was found that 2′-Me-CCPA was able to form a number of interactions with several polar residues in the transmembrane helices TM-3, TM-6, and TM-7 of bA(1)AR which were not preserved in the molecular dynamics simulation of 3′-Me-CCPA/bA(1)AR complex. (PMID: 15743197) |
| Experiment: (ii) The most active compound was found to be 3′-Me-CPA which displayed a K(i) value of 0.35 microM at A(1) receptor and a selectivity for A(1) versus A(2A) and A(3) receptors higher than 28-fold. (PMID: 15743197) |
The key statements in these publications were classified into confirmatory and information gain type of statements.
4 DISCUSSION
Currently, the existing ontologies (e.g. SMO, MIO and DIO) describe molecular interactions at the complex macro-scale level as a biological event but they do not address the fundamental physics behind an interaction occurring between a ligand and its target. Besides differences in scope between PLIO and the ontologies mentioned above, the main difference lies in the characterization of interactions between target and ligand. PLIO is a focused ontology in terms of representing protein–ligand interactions at the micro-scale level (e.g. electrostatic interaction, van der Waals interaction and covalent bonding), explicitly representing the major known features involved in protein–ligand interactions from different points of view such as biophysics, chemoinformatics, molecular modelling, and experimental methodology. For example, the interaction concept in DIO is represented as the event when ligand (an effecter) interacts with its biological target (objects) and triggers certain output (biological event). This biological event representation of interaction, which is present in both DIO and SMO, is another way to look at the physical interactions between molecules; however, this kind of representation ignores intra- and inter-molecular forces that govern the interactions.
In the design of PLIO, special attention has been paid to the usability profile of the ontology which addresses to what extent the set of annotations and metadata of the ontology contributes to its usability and application by end users. For this reason, PLIO is distinct from other ontologies in providing extra mathematical and web hyperlink annotations so that quantitative concepts and parameters can be directly calculated online. When integrated into our information retrieval system, PLIO leveraged the efficiency of semantic information retrieval and knowledge representation by providing the possibility to perform ontological search in two directions: in depth using concepts hierarchy and in breadth utilizing synonyms associated with each concept. It not only detected the established knowledge but also allowed for gain of information which otherwise could not be explicitly detected. This approach enables users to exploit the added value of gained information for generation of novel hypotheses.
5 CONCLUSIONS
In combination with text-mining technologies, PLIO and its lexicon create a powerful ontology-driven search engine which is able to answer complex questions in the area of protein–ligand interaction. It facilitates knowledge and information retrieval that helps scientists to find diverse information on a certain drug target or a protein–ligand complex from simulation and experimental knowledge scattered throughout the literature. This work represents a first attempt to develop an open, public PLIO and we do not claim that PLIO covers the entire knowledge in that domain. Thus, like other ontologies, PLIO needs continuous improvement and is proposed to the scientific community as an ontology that is open for further contribution.
5.1 Outlook
The value of an ontology representing knowledge relevant for the pharmaceutical drug discovery and development process has been demonstrated by the commercial success of BioWisdom and the uptake of their ontology framework in major pharmaceutical companies. Their ontology, however, remains proprietary and, thus, will not be widely and openly shared with the scientific community. The lack of an open ontology in the pharmaceutical area has prompted us to start to work on this topic in small, well-defined areas of knowledge where we feel competent to contribute to the yet-to-be-generated, large, public pharma ontology. Therefore, we consider PLIO as an ‘ontology draft’ which forms only a small section of the proposed public pharma ontology. We intend to make this and any following building block of the proposed pharma ontology freely available to the public and we invite the scientific community to help improving this ontology.
ACKNOWLEDGEMENTS
The authors wish to thank Harsha Gurulingappa, Theo Mevissen and Juliane Fluck for their support in text mining.
Funding: B-IT (Bonn-Aachen International Center for Information Technology, University of Bonn) foundation (to O.I., E.Y. and A.W.).
Conflict of Interest: none declared.
REFERENCES
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenreider O, et al. Biomedical ontologies. Pac. Symp. Biocomput. 2005;10:76–78. doi: 10.1142/9789812704856_0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekstra J, et al. Proceedings of the WWW'04 workshop on Application Design, Development and Implementation Issues in the Semantic Web. New York: 2004. The drug ontology project for Elsevier. [Google Scholar]
- Choi J, et al. A semantic web ontology for small molecules and their biological targets. J. Chem. Inf. Model. 2010;50:732–741. doi: 10.1021/ci900461j. [DOI] [PubMed] [Google Scholar]
- Cote RG, et al. The ontology lookup service, a lightweight cross-platform tool for controlled vocabulary queries. BMC Bioinformatics. 2006;7:97. doi: 10.1186/1471-2105-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz IF, et al. International Conference on Very Large Databases. Lyon, France: 2009. AgreementMaker: efficient matching for large real-world schemas and ontologies; pp. 1586–1589. [Google Scholar]
- Doms A, et al. Nucleic Acids Res. 2005. GoPubMed: exploring PubMed with the gene ontology; p. 33. W783–W786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilbeck K, et al. The sequence ontology: a tool for unification of genome annotations. Genome Biol. 2005;6:R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluck J, et al. Second BioCreAtIvE Challenge Workshop: Critical Assessment of Information Extraction in Molecular Biology. 2007. ProMiner: recognition of human gene and protein names using regularly updated dictionaries. Fundación CNIO Carlos III, Madrid, Spain, pp. 149–151. [Google Scholar]
- Friedrich CM, et al. @neuLink: a service-oriented application for biomedical knowledge discovery. In: Solomonides T, et al., editors. HealthGrid 2008, Proceedings of HealthGrid 2008. Amsterdam: IOS Press; 2008. pp. 165–172. [PubMed] [Google Scholar]
- Gómez-Pérez A, et al. Ontological Engineering. Springer, Berlin: 2004. [Google Scholar]
- Grenon P, et al. Biodynamic ontology: applying BFO in the biomedical domain. In: Pisanelli DM, editor. Ontologies in Medicine. Amsterdam: IOS Press; 2004. pp. 20–38. [PubMed] [Google Scholar]
- Héja G, et al. Ontological analysis of SNOMED CT. BMC Med. Inform. Decis. 2008;8(Suppl. 1):S8. doi: 10.1186/1472-6947-8-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AA, et al. Overview of BioCreative II gene normalization. Genome Biol. 2008;9(Suppl. 2):S3. doi: 10.1186/gb-2008-9-s2-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR, et al. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 2003;55:597–606. doi: 10.1124/pr.55.4.4. [DOI] [PubMed] [Google Scholar]
- Nicholson LK, et al. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 1995;2:274–280. doi: 10.1038/nsb0495-274. [DOI] [PubMed] [Google Scholar]
- Rosse C, et al. A reference ontology for bioinformatics: the Foundational Model of Anatomy. J Biomed. Inform. 2003;36:478–500. doi: 10.1016/j.jbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Rubin DL, et al. Biomedical ontologies: a functional perspective. Brief Bioinformatics. 2008;9:75–90. doi: 10.1093/bib/bbm059. [DOI] [PubMed] [Google Scholar]
- Scott WR, Schiffer CA. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure. 2000;8:1259–1265. doi: 10.1016/s0969-2126(00)00537-2. [DOI] [PubMed] [Google Scholar]
- Spackman KA, et al. Proceedings of AMIA Annual Fall Symposium. Nashville, USA: 1997. SNOMED RT: a reference terminology for health care; pp. 640–644. [PMC free article] [PubMed] [Google Scholar]
- Spasic I, et al. Text mining and ontologies in biomedicine: making sense of raw text. Brief Bioinformatics. 2005;6:239–251. doi: 10.1093/bib/6.3.239. [DOI] [PubMed] [Google Scholar]
- Thomas PD, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermuth CG, et al. Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998) Pure Appl. Chem. 1998;70:1129–1143. [Google Scholar]
- Whetzel PL, et al. The MGED ontology: a resource for semantics-based description of microarray experiments. Bioinformatics. 2006;22:866–873. doi: 10.1093/bioinformatics/btl005. [DOI] [PubMed] [Google Scholar]
- Yoshikawa S, et al. Drug interaction ontology (DIO) for inferences of possible drug–drug interactions. Stud. Health Technol. Inform. 2004;107:454–458. [PubMed] [Google Scholar]