

Abstract
A new series of triazolotriazines variously substituted at the C5 and N7 (5–25) positions was synthesized and fully characterized at the four adenosine receptor (AR) subtypes. In particular, arylacetyl or arylcarbamoyl moieties were introduced at the N7 position, which enhanced affinity at the hA2B and hA3 ARs, respectively, when utilized on the pyrazolo-triazolopyrimidine nucleus as we reported in the past. In general, compounds with a free amino group at the 7 position (5, 6), showed good affinity at the rat (r) A2A AR (range 18.3–96.5 nM), while the introduction of a phenylcarbamoyl moiety at the N7 position (12, 19, 24) slightly increased the affinity at the hA3 AR (range 311–633 nM) with respect to the unsubstituted derivatives. The binding profiles of the synthesized analogues seemed to correlate with the substitutions at the C5 and N7 positions. At the hA2B AR, derivative 5, which contained a free amino group at the 7 position, was the most potent (EC50 3.42 μM) and could represent a starting point for searching new non-xanthine hA2B AR antagonists. Molecular models of the rA2A and hA3 ARs were constructed by homology to the recently reported crystallographic structure of the hA2A AR. A preliminary receptor-driven structure–activity relationship (SAR) based on the analysis of antagonist docking has been provided.
Keywords: G Protein-coupled receptors, Adenosine receptor antagonists, Triazolo-triazine, Ligand–receptor modeling studies
1. Introduction
Activation or blockade of adenosine receptors (ARs) is responsible for a broad range of effects in various organ systems suggesting that regulation of ARs has substantial therapeutic potential. Recent the focus has been increased on the cardioprotective1,2 and neuro-protective3,4 effects associated with AR activation during periods of cardiac and cerebral ischemia, respectively. It has also been proposed that antagonists of distinct AR subtypes may be used in the treatment of asthma5,6 or certain neurological diseases such as Parkinson’s disease.6,7 A complete summary of the physiological roles of ARs and their potential as clinical targets in a variety of disease states have been recently reported.6–11
All ARs are members of the superfamily of G protein-coupled receptors (GPCRs), with four subtypes currently recognized, the A1AR, A2AAR, A2BAR, and A3AR,12 which exert their physiological role by activation or inhibition of several second messenger systems. In particular, the modulation of adenylate cyclase activity could be considered to be the principal signal mediated by these receptors.1,2
In the last 20 years intense medicinal chemical efforts led to the synthesis of a variety of AR agonists and antagonists for the pharmacological characterization of this family of G protein-coupled receptors.13 With respect to antagonists, several classes of heterocyclic derivatives have been reported as AR antagonists with high levels of both affinity and selectivity. In particular, in recent years we investigated in depth the nucleus of triazolo-pyrazolopyrimidine as a basis for designing ARs antagonists. Modulating the substitution at the C5, N7 and N8 positions (Chart 1) led to potent and selective A2A and A3 AR antagonists (compounds 1 and 2).14–21
Chart 1.

Pyrazolo-triazolopyrimidine derivatives as A2A and A3 AR antagonists.
Nevertheless, this class of compounds, like other tricyclic structures, was subject to poor water solubility and more importantly complicated synthetic routes. In consideration of these problems, medicinal chemists recently focused their attention on the synthesis of more simplified heterocyclic derivatives, in particular bicyclic systems such as adenine,22 triazolopyrazine23–25 and triazolotriazine.26–28 One of the most appealing bicyclic cores is represented by the triazolotriazine nucleus, which led in the past to the discovery of ZM241385 (3) that could be considered as one of the most potent A2A AR antagonists yet reported.29,30 In addition, 3 also binds with high affinity at the human (h) A2B AR (28 nM), and its tritiated form is suitable for use as a radioligand in binding studies at this receptor subtype.31 Very recently a large number of triazolotriazine derivatives bearing different substitution at the 5-position have been reported,26 and compound 4 proved to be potent and selective for the A2A in comparison to the A1 AR. Nevertheless, the lack of binding data at the A2B and A3 ARs prevents a comparison with other fully characterized derivatives (Chart 2).26
Chart 2.

Triazolotriazine derivatives as AR antagonists.
Taking into account these experimental observations, we decided to better investigate the potential of modifying this nucleus at the C5 and N7 positions (5–25). In particular, arylacetyl or arylcarbamoyl moieties at the N7 position on the pyrazolo-triazolopyrimidine nucleus enhanced affinity at the A2B and A3 ARs, respectively (Chart 3).17,21
Chart 3.

Molecular simplification approach applied for the design of the novel triazolotriazine analogs.
All the compounds have been fully characterized at the four ARs with the aim of better understanding the structure–activity relationship (SAR) profile of this class of compounds and optimizing the substitution in order to modulate both AR affinity and selectivity. Moreover, a preliminary receptor-driven SAR based on the recently published crystallographic structure of the hA2A AR has been provided.
2. Results and discussion
2.1. Chemistry
The compounds with the free amino group bearing a phenoxy (5) or methylthio (6) residue at the C5 position were prepared as previously described by Caulkett et al.,32 while introduction of a dimethylamino group at the C5 position (7) was performed by heating compound 5 at 120 °C in a sealed tube with an ethanolic solution (33%) of dimethylamine.
Derivatives substituted at the N7 position (8–25) were prepared by treating compounds 5–7 with the appropriate acylchloride (in presence of triethylamine) or arylisocyanate in dioxane at reflux (Scheme 1).
Scheme 1.

Reagents: (i) EtOH, dimethylamine, 120 °C sealed tube, 4 h; (ii) R–N=C=O, dioxane, Et3N, reflux, 12 h; (iii) R–COCl, dioxane, reflux, 12 h.
2.2. Biological activity
The receptor binding affinities of the synthesized compounds (5–25) are reported in Table 1. Binding was conducted at the following ARs: rat A1 (from rat cerebral cortex membranes),33 rat A2A (from rat striatal membranes),34 and hA3 (from HEK-293 cells expressing the hA3 AR).35 [3H]N6-Phenylisopropyladenosine ([3H]R-PIA), (A1)33 [3H]2-[4-[(2-carboxyethyl)-phenyl]ethylamino]-5′-N-ethylc-arbamoyladenosine, ([3H]CGS21680) (A2A)24 and [125I]N6-(4-amino-3-iodobenzyl)-5′-N-methylcarbamoyladenosine ([125I]I-AB-MECA) (A3)26 have been used as radioligands in binding assays. Instead, for evaluating potency at the hA2B AR, due to the lack of a readily available radioligand, the activity of antagonists was determined in adenylyl cyclase experiments in CHO (Chinese hamster ovary) cells expressing the hA2B AR.37,38
Table 1.
Structures and binding profile of synthesized compounds 5–25
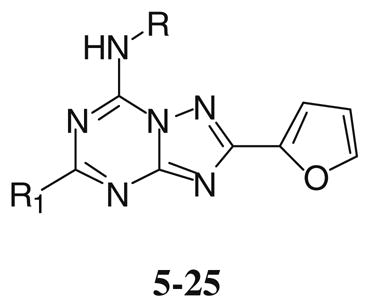 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | R1 | R | rA1 (Ki, nM) % displ. at 10μMa | rA2A (Ki, nM)b | hA2B EC50 (nM)c | hA3 (Ki, nM) or % displ. at 10μMd | rA1/rA2A | rA1/hA3 | rA2A/hA3 |
| 5 | OPh | H | 2,720 ± 680 | 18.3 ± 3.4 | 3,420 ± 200 | 489 ± 63 | 148 | 0.04 | 5.5 |
| 6 | SCH3 | H | 1730 ± 360 | 96.5 ± 36.1 | 14,900 ± 1,700 | 2580 ± 780 | 18 | 0.7 | 0.04 |
| 7 | N(CH3)2 | H | 31,400 ± 8000 | 3800 ± 1230 | 33,700 ± 1,800 | 7270 ± 2230 | 8.3 | 4.3 | 0.5 |
| 8 | SCH3 | COCHPh2 | 557 ± 205 | > 10,000 | > 100,000 | 170 ± 45 | < 0.05 | 3.2 | 59 |
| 9 | SCH3 | COCH2Ph | 1420 ± 260 | 429 ± 38 | > 100,000 | 4200 ± 950 | 3.3 | 0.33 | 0.1 |
| 10 | SCH3 | COCH2-Ph-4OCH3 | 5000 ± 1170 | 1570 ± 660 | 42,400 ± 9,900 | 2100 ± 400 | 3.2 | 2.4 | 0.7 |
| 11 | SCH3 | CO(CH2)4CH3 | 1520 ± 170 | 427 ± 96 | > 100,000 | 24 ± 3% | 3.5 | < 0.04 | < 0.15 |
| 12 | SCH3 | CONHPh | 10,000 ± 2900 | 1660 ± 1120 | 17,100 ± 2,700 | 414 ± 29 | 6 | 24 | 4 |
| 13 | SCH3 | CONH-Ph-4CH3 | 3440 ± 690 | 742 ± 194 | 16,800 ± 3,100 | 2200 ± 54 | 4.6 | 1.5 | 0.3 |
| Compd | R1 | R | rA1 (Ki, nM) | rA2A (Ki, nM) | hA2B EC50 (nM) | hA3 (Ki, nM) or% displ. at 10μM | rA1/rA2A | rA1/hA3 | rA2A/hA3 |
| 14 | OPh | COCHPh2 | <10% inhib | 1880 ± 310 | > 100,000 | 4400 ± 1150 | > 5.3 | > 2.2 | 0.4 |
| 15 | OPh | COCH2Ph | 7260 ± 2820 | 136 ± 48 | > 100,000 | 1020 ± 173 | 53 | 7 | 0.13 |
| 16 | OPh | COCH2-Ph-4-OCH3 | 12 ± 2% inhib | 892 ± 252 | > 100,000 | 5200 ± 1300 | > 11 | > 2 | 0.17 |
| 17 | OPh | CO(CH2)4CH3 | 2900 ± 710 | 189 ± 26 | > 100,000 | 2200 ± 470 | 15 | 1.3 | 0.08 |
| 18 | OPh | CONHPh | 17 ± 5% inhib | 38.9 ± 3.5 | 8,870 ± 1,680 | 633 ± 37 | > 257 | > 16 | 0.06 |
| 19 | OPh | CONH-Ph-4CH3 | 35 ± 5% inhib | 214 ± 72 | 20,000 ± 3,600 | 750 ± 130 | > 46 | > 13 | 0.3 |
| 20 | N(CH3)2 | COCHPh2 | 951 ± 88 | 4090 ± 1360 | > 100,000 | 473 ± 85 | 0.2 | 2 | 8.6 |
| 21 | N(CH3)2 | COCH2Ph | 6080 ± 1040 | 6700 ± 3200 | > 100,000 | 1050 ± 466 | 0.9 | 5.8 | 6.4 |
| 22 | N(CH3)2 | COCH2-Ph-4-OCH3 | 8740 ± 590 | 10,300 ± 1,300 | > 100,000 | 2700 ± 345 | 0.8 | 3.2 | 3.8 |
| 23 | N(CH3)2 | CO(CH2)4CH3 | 1570 ± 380 | > 10,000 | > 100,000 | 2200 ± 370 | < 0.16 | 0.7 | > 4.5 |
| 24 | N(CH3)2 | CONHPh | 3150 ± 550 | 580 ± 100 | > 100,000 | 311 ± 122 | 5.4 | 10 | 1.8 |
| 25 | N(CH3)2 | CONH-Ph-4CH3 | 2720 ± 830 | 2720 ± 2090 | > 100,000 | 9600 ± 1200 | 1 | 0.3 | 0.3 |
Displacement of specific [3H]R-PIA binding (A1) in rat brain membranes.
Displacement of specific [3H]CGS 21680 binding (A2A) in rat striatal membranes.
Measurement of adenylyl cyclase activity in CHO cells stably transfected with human recombinant A2B AR, expressed as EC50 (μM).
Displacement of specific [125I]-I-AB-MECA binding at hA3 receptors expressed in CHO cells. Data are expressed as Ki ± SEM in nM or as % of displacement at 10lM(n = 3–6).
All of the synthesized compounds showed affinities at the four ARs in the high nanomolar or micromolar range without significant levels of selectivity. The analysis of the data reported in Table 1, clearly indicates that in general, with the exception of compound 7, compounds with a free amino group at the 7 position (5 and 6) showed good affinity at the A2A AR (range 18.3–96.5 nM) with quite significant levels of selectivity versus the other AR subtypes.
A significantly different selectivity profile could be noted when the amino group at the 7 positionwas substituted with an acyl group, and most importantly the affinities at the four AR subtypes seemed to be very sensitive to the substitution at both the 5 and 7 positions.
In particular, when a phenylacetyl moiety was introduced at the N7 position (9, 15, and 21) the binding profiles of the derivatives were quite different with respect to the N7 unsubstituted derivatives (5–7), and it was also significantly modified by the substitution at the 5 position. In fact, the presence of a methylthio (9) or phenoxy (15) group at the 5 position enhanced affinity at the A2A AR (range 136–429 nM), while the potency at the other receptor subtypes was poor (range 1–7 μM). In contrast, the presence of a dimethylamino group at the 5 position (21) reduced affinity (1–7 μM) at all the ARs. When the phenylacetyl moiety at the N7 was substituted at the para position with a methoxy group (10, 16, and 22), a general decrease of affinity versus all the ARs were observed in comparison to the unsubstituted derivatives (9, 15, and 21). If a bulky arylacetyl moiety (such as diphenylacetyl-8, 14, and 20) was introduced at the N7 position, a completely different biological profile was evident. In fact, the presence of a bulky substituent at the N7, position combined with a methylthio (8) or dimethylamino (20) group at the 5 position, favored A1 and A3 ARs affinities (range 170–950 nM). In contrast, the presence of a phenoxy group at the 5 position reduced affinity at all the ARs.
Different binding affinities were observed when an acyl chain was introduced at the N7 position. In fact, a combination of an acyl chain at the N7 position with a methylthio (11) or a phenoxy (17) groups at the 5 position afforded good affinity at the A2A AR (range 180–400 nM), while the affinities at the other AR subtypes was poor. However, the presence of a dimethylamino group at the 5 position (23) was detrimental to affinity at all the AR subtypes.
Incorporation of an arylcarbamoyl moiety at the N5 position of the pyrazolo-triazolopyrimidine nucleus (e.g., compound 2) enhanced hA3 AR affinity. In general, the affinity at the A1 AR was poor (range from 3 μM to >10 μM) independent of the substitution at the 5 position (12, 13, 18, 19, 24, 25). However, phenoxy (18, 19) or dimethylamino (24) groups at the 5 position enhanced the affinity at the A2A subtype (range 39–580 nM).
Thus, most of these derivatives were nearly inactive at the hA3 AR, with the exception of compounds 12, 18, and 24, which showed hA3 affinity in the high nanomolar range (range 311–633 nM) and were characterized by the presence of an unsubstituted phenylcarbamoyl moiety at the N7 position, independently of the substitution at the N5 position. The low affinity at the hA3 AR, was quite surprising considering that triazolotriazine derivatives were simplified analogs of the pyrazolo-triazolopyrimidine antagonists of the hA3 AR. Nevertheless, a careful structural analysis of these two classes of compounds, by comparing the derivatives 24 and 2, clearly indicated that the dimethylamino group on the triazolotriazine nucleus is not favored. In fact, the dimethylamino group seemed not to correspond to the N8-methyl group of derivative 2, but was similar to its N7 pattern of substitution, which was extensively demonstrated to be inactive at the hA3 AR.13–15
Regarding the activity of this series at the hA2B AR, most of compounds were almost inactive at this receptor subtype. Only two compounds, bearing a phenoxy group at the 5 position (5, 18), showed promising activity in the adenylyl cyclase assays at the A2B AR, with an EC50 ranging from 3.4 to 8.8 μM. In particular, derivative 5, which contained a free amino group at the 7 position, was the most potent at the hA2B AR, and could represent a starting point for new non-xanthine hA2B AR antagonists. Nevertheless, the high potency of this compound at the A2A AR (18.3 nM) clearly indicated that further investigation would be needed in order to delineate the activities at these two ARs. A summary of the most relevant structure–activity features of the novel triazolotriazine analogs has been reported in Chart 4.
Chart 4.

Summary of the most relevant SAR features of the novel triazolotriazine analogs.
2.3. Molecular modeling
It was clear from the data analysis that it was very difficult to define a robust SAR profile of this new class of compounds. For this reason and with the aim to better understand these pharmacological results, molecular modeling and docking studies have been performed in parallel at the A2A and A3 ARs. Therefore, we built models of the rA2A and hA3 receptors by homology modeling, using as template the crystal structure of hA2A receptor (PDB code: 3EML);39 methodological details are summarized in Section 4. Figure 1 shows the alignment of the three amino acid sequences (hA2A/rA2A sequence identity 82%; hA2A/hA3 sequence identity 41%).
Figure 1.

Multiple sequence alignment of hA2A receptor, rat A2A receptor and hA3 receptor. Conserved residues in all the sequences are identified by stars. Differences in sequences of rA2A and hA3 compared to hA2A are shown in boldface type.
Then, we performed docking studies to recognize the hypothetical binding motif of the newly synthesized 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives. We compared our docking simulation results with the docking poses of ZM241385 at both receptors. As reported in Section 4, four different programs have been used to calibrate our docking protocol using the crystallographic pose of ZM241385 into human A2A as reference. Based on the lowest average ligand RMSD value obtained from the different docking algorithms, we decided to use GOLD as docking program for the pose inspection of the novel 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives.
As shown in Figure 2, the binding motif of ZM241385 at the rA2A receptor was similar to the crystallographic pose on the hA2A receptor; this was consistent with the binding affinity of ZM-241385 at the two receptors (Ki = 0.21–0.50 nM and Ki = 0.7–1.0 nM, respectively).40,41 Consistently, all the residues belonging to the binding pocket are conserved in both receptors. The numbering of the amino acids in parenthesis follows the numbering convention of Ballesteros and Weinstein: according to this scheme, each amino acid identifier starts with the helix number, followed by the position relative to a reference residue among the most conserved amino acid in that helix, with the reference residue arbitrarily assigned the number 50.42
Figure 2.

Structure superimposition of crystallographic pose (in magenta) and docking pose (in green) of ZM241385 inside the rA2A AR binding site. Side chains of some amino acids important for ligand recognition and H-bonding interactions are highlighted. Hydrogen atoms are not displayed.
From the analysis of docking of ZM241385 in the rA2A AR, it appeared that the bicyclic triazolotriazine core was anchored through an aromatic stacking interaction with Phe163 (EL2), an aliphatic hydrophobic interaction with Ile269 (7.39), and a hydrogen bonding interaction with Asn248 (6.55). The exocyclic amino group, linked to the bicyclic core of ZM241385, interacts with two polar residues, Asn248 (6.55) and Glu164 (EL2). The phenyl ring forms hydrophobic interactions with Pro262 (7.32) and Met265 (7.35). The phenylethylamine substituent was directed towards the more solvent exposed extracellular region (EL2 and EL3) rather than towards the transmembrane domains of the receptor. The furan ring was located deep in the ligand binding cavity and directed towards TM5 and TM7; it forms hydrophobic interactions with the highly conserved Trp241 (6.48), an important residue in receptor activation,43 His245 (6.52) and Leu244 (6.51).
The triazolotriazine derivatives with a free amino group at the 7 position (5–7) show a similar binding motif to ZM-241385 inside the transmembrane region of the rA2A receptor, as reported in Figure 3. In particular, the three H-bonding interactions with Asn248 (6.55) and Glu164 (EL2) are conserved.
Figure 3.

Structure superimposition of docked conformations of compound 5 (in red, Ki rA2AAR = 18.3 ± 3.4) and compound 18 (in magenta, Ki rA2AAR = 38.9 ± 3.5) inside the rA2A AR binding pocket. Side chains of some amino acids important for ligand recognition and H-bonding interactions are highlighted. Hydrogen atoms are not displayed.
Among these, compound 5 (Ki rA2AAR = 18.3 ± 3.4) was the most potent because the phenoxy group forms strong hydrophobic interactions with side chains of the following residues: Leu162 (EL2), Phe163 (EL2), Pro262 (7.32), Met265 (7.35), Tyr266 (7.36) and Ile269 (7.39). Calculated log P values of compounds 6 and 7 (1.01 and −0.24, respectively) could explain the different affinities at the receptor, even though the two R1 substituents were not involved in particular interactions with residues of the binding pocket. The hydrogen bonding network with Asn248 (6.55) and Glu164 (EL2) seems to be critical both for the recognition of these antagonist structures and for receptor selectivity versus hA3. In particular, Glu164 (EL2) of rA2A receptor subtype (Glu169 in hA2A) was not present in the corresponding position of hA3 receptor, where this amino acid was replaced by valine (Val169); in fact molecular docking results on hA3 suggest that compounds 5, 6 and 7 (Ki hA3AR = 489 ± 63, 2580 ± 780, 7270 ± 2230, respectively) form only two (with Asn250) of the three hydrogen bonding interactions (data not shown).
Among other compounds (8–25) the most potent at the rA2A receptor was 18 (Ki rA2AAR = 38.9 ± 3.5). As shown in Figure 3, because of the presence of the phenylcarbamoyl moiety at N7, the binding pose of compound 18 results to be quite different compared with the unsubstituted derivatives (ZM241385, 5–7). In fact, the R substituent was directed towards TM2 and TM3 rather than towards TM5 and TM6 and was located in a hydrophobic pocket delimited by Ala56 (2.57), Ile57 (2.58), Phe59 (2.60), Ala60 (2.61), Ile63 (2.64), Phe77 (3.28), Ala78 (3.29), Phe80 (3.31) and Val81 (3.32). Nevertheless, compound 18 formed also the same hydrophobic interactions as compound 5 and ZM241385, but only one hydrogen bonding interaction with Asn248 (6.55). Moreover the bicyclic triazolotriazine core of the disubstituted ligand was aligned with the same region of the other two compounds.
In the hA3 receptor, the presence of the less bulky side chain of Val169 (EL2) allows the phenylcarbamoyl moiety of compound 18 to direct towards TM5 and TM6 and so it looses the interactions with the residues of the hydrophobic pocket. Moreover, all the 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives were located deeper in the ligand binding cavity of hA3; consequently they lose the π–π stacking interaction with Phe168 (EL2).
With respect to the R1 moieties, the SAR at rA2A was similar for compounds 8–25 and compounds 5–7. It seems that the presence of too bulky substituents at the N7 position was not well tolerated because of unfavourable steric interactions.
To analyze in a more quantitative way the possible ligand–receptor recognition mechanism, the individual electrostatic ( ) and hydrophobic ( ) contributions to the interaction energy (ΔEint) of each receptor residue has been calculated (see Section 4 for more details). Analyzing the results of this study (collected in Figs. 4 and 5) it is clear that, from the electrostatic point of view, two of the most critical residues affecting the affinity at ARs seem to be the asparagine 6.55 (Asn253 in hA2A, Asn248 in rA2A and Asn250 in hA3) and the glutamic acid located within EL2 of both human and rat A2A (Glu169 and Glu164, respectively) but mutated in valine in hA3 (Val169). In particular, Asn 6.55 is responsible of two stabilizing interactions with ZM241385 in both human and rat A2A and this is supported by the electrostatic contribution of around −13 kcal/mol to the whole interaction energy, in particular −9.5 kcal/mol in hA2A and −17.9 kcal/mol in rA2A (Fig. 4). In addition, the glutamic acid on EL2 can strongly interact through an additional hydrogen-bond with the exocyclic amino group of ZM-241385 as supported by the stabilizing electrostatic contribution of around −13 kcal/mol to the whole interaction energy, in particular −9.0 kcal/mol in hA2A and −17.6 kcal/mol in rA2A (Fig. 4). Consistently, this specific interaction is missing in hA3.
Figure 4.

Electrostatic interaction energy (in kcal/mol) between the ligand and each single amino acid involved in ligand recognition calculated from: (A) crystallographic binding mode of ZM241385 inside the hA2A receptor (PDB code: 3EML); (B) hypothetical binding mode of ZM241385 inside the rA2A receptor obtained after docking simulations; (C) hypothetical binding mode of compound 5 inside the rA2A receptor obtained after docking simulations; and (D) hypothetical binding mode of compound 18 inside the rA2A receptor obtained after docking simulations.
Figure 5.

Hydrophobic interaction energy (in arbitrary hydrophobic unit) between the ligand and each single amino acid involved in ligand recognition calculated from: (A) crystallographic binding mode of ZM241385 inside the hA2A receptor (PDB code: 3EML); (B) hypothetical binding mode of ZM241385 inside the rA2A receptor obtained after docking simulations; (C) hypothetical binding mode of compound 5 inside the rA2A receptor obtained after docking simulations; and (D) hypothetical binding mode of compound 18 inside the rA2A receptor obtained after docking simulations.
Interestingly, compound 5 presents a very similar electrostatic energy contributions to ZM241385 supporting the hypothesis of a common TM binding motif. Conversely, compound 18 completely abolishes the capability to interact with both asparagine 6.55 and glutamic acid on EL2 due to the presence of phenylcarbamoyl moiety that forces the triazolotriazine moiety to flip 180° (around its parallel TM axis) inside the TM binding cleft. However, the lack of these two stabilizing interactions seems to be balanced by the presence of several additional hydrophobic interactions as mapped in Figure 5.
In fact, besides the three hydrophobic contributions mediated by the conserved phenylalanine on EL2 (Phe168 in hA2A, Phe163 in rA2A and Phe168 in hA3), the leucine 6.51 (Leu249 in hA2A, Leu244 in rA2A and Leu246 in hA3) and the tryptophan 6.48 (Trp246 in hA2A, Trp241 in rA2A and Trp243 in hA3), the phenylcarbamoyl moiety at N7 is surround by several hydrophobic side chains such as, for example for the rA2A, Ala56 (2.57), Ile57 (2.58), Phe59 (2.60), Ala60 (2.61), Ile63 (2.64), Phe77 (3.28), Ala78 (3.29), Phe80 (3.31) and Val81 (3.32).
3. Conclusion
The present study has led to the identification of a new class of promising AR antagonists, the 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives, which were designed as simplified analogues of our previously reported hA2A and hA3 antagonists. To depict the binding motif of these new AR antagonists, a novel model of the rA2A and hA3 receptor, based on the recently published structure of the human A2A receptor, was built. These new AR models provide a self-consistent framework that rationalizes the available SAR data. Moreover, a very preliminary hypothesis, concerning the specific roles of a few crucial amino acids in affecting the molecular mechanism of both ligand-entering process and TM-recognition process, has been proposed.
4. Experimental section
4.1. Chemistry
4.1.1. General
Reactions were routinely monitored by thin-layer chromatography (TLC) on silica gel (precoated F254 Merck plates). Infrared spectra (IR) were measured on a Jasco FT-IR instrument. 1H NMR were determined in CDCl3 or DMSO-d6 solutions with a Varian Gemini 200 spectrometer, peaks positions are given in parts per million (δ) downfield from tetramethylsilane as internal standard, and J values are given in hertz. Light petroleum ether refers to the fractions boiling at 40–60 °C. Melting points were determined on a Buchi-Tottoli instrument and are uncorrected. Flash chromatography was performed using Merck 60–200 mesh silica gel. Elemental analyses were performed by the microanalytical laboratory of Dipartimento di Chimica, University of Trieste, and were within ±0.4% of the theoretical values for C, H and N.
4.1.1.1. 7-Amino-5-dimethylammino-2-(2-furyl)-[1,2,4]triazol-o[1,5-a]-[1,3,5] triazine (7)
To a solution of phenoxy derivative 5 (0.3 g, 1.01 mmol) in ethanol (5 mL) was added a large excess (5 equiv) of an ethanolic solution (33%) of dimethylamine (0.7 mL). The resulting solution was transferred to a sealed tube and heated at 120 °C for 4 h. Then the solvent was removed at reduced pressure and the crude residue purified by flash chromatography (EtOAc–light petroleum 1:1) to afford the desired product (7) as a pale yellow solid in a good yield (80%). Mp 260–262 °C (EtOAc–light petroleum); IR (KBr): 3300, 1620, 1510 cm−1; 1H NMR (DMSO-d6) δ: 3.1 (s, 6H); 6.7 (dd, 1H, J = 2, J = 4), 7.1 (d, 1H, J = 2); 7.9 (d, 1H, J = 4); 8.2 (br s, 2H). MW 245.24. Anal. Calcd for C10H11N7O: C, 48.98; H, 4.52; N, 39.98. Found: C, 49.15; H, 4.50; N, 39.77.
4.1.2. General procedure for the preparation of the 7-(aralkyl-carbonyl)amino-2-(2-furyl)-5-substituted[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives (8–11, 14–17, 20–23)
A solution of amino derivative (5–7) (1 mmol), appropriate acyl chloride (1.2 equiv) and triethylamine (1.2 equiv) in dry dioxane (10 mL) was poured at reflux under argon atmosphere for 12 h. The solvent was removed in vacuo, and the crude residue purified by flash chromatography (EtOAc–light petroleum 3:7) to afford the desired compound.
4.1.2.1. 7-Diphenyl-acetylamino-2-(2-furyl)-5-methylthio[1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (8)
Yield 65%; pale yellow solid; mp 172–173 °C (EtOAc–light petroleum); IR (KBr): 1680, 1520, 1420 cm−1; 1H NMR (DMSO-d6) δ: 2.53 (s, 3H); 5.1 (s, 1H); 6.7 (dd, 1H, J = 2, J = 4); 7.1 (d, 1H, J = 2);7.2–7.6 (m, 10H); 7.9 (d, 1H, J = 4); 9.87 (br s, 1H). MW 442.49. Anal. Calcd for C23H18N6O2S: C, 62.43; H, 4.10; N, 18.99. Found: C, 62.66; H, 4.18; N, 19.13.
4.1.2.2. 2-(2-Furyl)-5-methylthio-7-phenylacetylamino-[1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (9)
Yield 75%; brown solid; mp 70–72 °C (EtOAc–light petroleum); IR (KBr): 1680, 1510, 1450 cm−1; 1H NMR (DMSO-d6) δ: 2.51 (s, 3H); 4.0 (s, 2H); 6.7 (dd, 1H, J = 2, J = 4); 7.1 (d, 1H, J = 2); 7.2–7.6 (m, 5H); 7.9 (d, 1H, J = 4); 11.8 (br s, 1H). MW 366.40. Anal. Calcd for C17H14N6O2S: C, 55.73; H, 3.85; N, 22.94. Found: C, 55.63; H, 3.79; N, 22.90.
4.1.2.3. 2-(2-Furyl)-7-[4-(methoxyphenyl)acetyl]amino-5-methylthio [1,2,4]triazolo[1,5-a][1,3,5]triazine (10)
Yield 78%; pale yellow; mp 138–141 °C (EtOAc–light petroleum); IR (KBr): 1680, 1525, 1455 cm−1; 1H NMR (DMSO-d6) δ: 2.5 (s, 3H); 3.8 (s, 3H); 4.3 (s, 3H); 4.5 (s,2H); 6.7 (dd, 1H, J = 2, J = 4); 6.9 (d, 2H, J = 9); 7.1 (d, 1H, J = 2); 7.2 (d, 2H, J = 9); 7.9 (d, 1H, J = 4); 11.8 (br s, 1H). MW 396.42. Anal. Calcd for C18H16N6O3S: C, 54.54; H, 4.07; N, 21.20. Found: C, 54.70; H, 4.11; N, 21.08.
4.1.2.4. 2-(2-Furyl)-5-methylthio-7-n-pentylcarbonylamino- [1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (11)
Yield 70%; white solid; mp 125–127 °C (EtOAc–light petroleum); IR (KBr): 1675, 1520, 1450 cm−1; 1H NMR (DMSO-d6) δ: 0.91 (t, 3H, J = 7); 1.01–1.2 (m, 4H); 1.4–1.61 (m, 2H); 2.5 (s, 3H); 3.32 (t, 2H, J = 7); 6.7 (dd, 1H, J = 2, J = 4); 7.1 (d, 1H, J = 2); 7.9 (d, 1H, J = 4); 10.93 (br s, 1H). MW 346.41. Anal. Calcd for C15H18N6O2S: C, 52.01; H, 5.24; N, 24.26. Found: C, 52.15; H, 5.20; N, 24.07.
4.1.2.5. 7-Diphenylacetylamino-2-(2-furyl)-5-phenoxy[1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (14)
Yield 67%; pale yellow solid; mp 136–139 °C (EtOAc–light petroleum); IR (KBr): 1680, 1520, 1425 cm−1; 1H NMR (DMSO-d6) δ: 5.12 (s, 1H); 6.73 (dd, 1H, J = 2, J = 4), 7.1 (d, 1H, J = 2); 7.2–7.6 (m, 15H); 7.9 (d, 1H, J = 4); 10.15 (br s, 1H). MW 488.50. Anal. Calcd for C28H20N6O3: C, 68.84; H, 4.13; N, 17.20. Found: C, 68.91; H, 4.21; N, 17.08.
4.1.2.6. 2-(2-Furyl)-5-phenoxy 7-phenylacetylamino- [1,2,4]tria-zolo[1,5-a]-[1,3,5]triazine (15)
Yield 65%; white solid; mp 208–210 °C (EtOAc–light petroleum); IR (KBr): 1680, 1520, 1425 cm−1; 1H NMR (DMSO-d6) δ: 4.2 (s, 2H); 6.7 (dd, 1H, J = 2, J = 4), 7.1 (d, 1H, J = 2,); 7.2–7.6 (m, 10H); 7.9 (d, 1H, J = 4); 11.01 (br s, 1H). MW 412.40. Anal. Calcd for C22H16N6O3: C, 64.07; H, 3.91; N, 20.38. Found: C, 64.23; H, 3.88; N, 20.28.
4.1.2.7. 2-(2-Furyl)-7-[4-(methoxyphenyl)acetyl]amino-5-phen-oxy[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (16)
Yield 70%; white solid; mp 178–181 °C (EtOAc–light petroleum); IR (KBr): 1680, 1525, 1455 cm−1; 1H NMR (DMSO-d6) δ: 3.8 (s, 3H); 4.2 (s, 2H); 6.73 (dd, 1H, J = 2, J = 4), 7.12 (d, 1H, J = 2); 7.2–7.45 (m, 7H); 7.62 (d, 2H, J = 9); 7.9 (d, 1H, J = 4); 10.77 (br s, 1H). MW 442.43. Anal. Calcd for C23H18N6O4: C, 62.44; H, 4.10; N, 19.00. Found: C, 62.28; H, 4.15; N, 18.89.
4.1.2.8. 2-(2-Furyl)-7-n-pentylcarbonylamino-5-phenoxy[1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (17)
Yield 71%; white solid; mp 157–159 °C (EtOAc–light petroleum); IR (KBr): 1685, 1515, 1450 cm−1; 1H NMR (DMSO-d6) δ: 0.93 (t, 3H, J = 7); 1.05–1.19 (m, 4H); 1.43–1.65 (m, 2H); 3.35 (t, 2H, J = 7); 6.68 (dd, 1H, J = 2, J = 4), 7.21 (d, 1H, J = 2); 7.35–7.63 (m, 5H); 7.94 (d, 1H, J = 4); 10.58 (br s, 1H). MW 392.41. Anal. Calcd for C20H20N6O3: C, 61.21; H, 5.14; N, 21.42. Found: C, 61.53; H, 5.23; N, 21.58.
4.1.2.9. 5-Dimethylamino-7-diphenylacetylamino-2-(2-furyl)-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (20)
Yield 70%; yellow solid; mp 105–107 °C (EtOAc–light petroleum); IR (KBr): 1680, 1520, 1425 cm−1; 1H NMR (DMSO-d6) δ: 3.13 (s, 6H); 4.32 (s, 1H); 6.68 (dd, 1H, J = 2, J=4), 7.21 (d, 1H, J = 2); 7.32–7.65 (s, 10H); 7.88 (d, 1H, J = 4); 10.12 (br s, 1H). MW 439.47. Anal. Calcd for C24H21N7O2: C, 65.59; H, 4.82; N, 22.31. Found: C, 65.77; H, 4.82; N, 22.43.
4.1.2.10. 5-Dimethylamino-2-(2-furyl)-7-phenylacetylamino-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (21)
Yield 75%; yellow solid; mp 226–228 °C (EtOAc–light petroleum); IR (KBr): 1680, 1525, 1455 cm−1; 1H NMR (DMSO-d6) δ: 3.15 (s, 6H); 4.28 (s, 2H); 6.69 (dd, 1H, J = 2, J = 4), 7.08 (d, 1H, J = 2); 7.4–7.55 (m, 5H); 7.85 (d, 1H, J = 4); 11.02 (br s, 1H). MW 363.37. Anal. Calcd for C18H17N7O2: C, 59.50; H, 4.72; N, 26.98. Found: C, 59.68; H, 4.71; N, 26.75.
4.1.2.11. 5-Dimethylamino-2-(2-furyl)-7-[4-(methoxyphenyl)-acetyl]amino-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (22)
Yield 73%; pale yellow solid; mp 168–171 °C (EtOAc–light petroleum); IR (KBr): 1680, 1530, 1460 cm−1; 1H NMR (DMSO-d6) δ: 3.09 (s, 6H); 3.88 (s, 3H); 4.45 (s, 2H); 6.73 (dd, 1H, J = 2, J = 4), 7.12 (d, 1H, J = 2); 7.18 (d, 2H, J = 9); 7.47 (d, 2H, J = 9); 7.85 (d, 1H, J = 4); 10.43 (br s, 1H). MW 393.41. Anal. Calcd for C19H19N7O3: C, 58.01; H, 4.87; N, 24.92. Found: C, 57.78; H, 4.74; N, 24.83.
4.1.2.12. 5-Dimethylamino-2-(2-furyl)-7-n-pentylcarbonylami-no-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (23)
Yield 68%; white solid; mp 147–148 °C (EtOAc–light petroleum); IR (KBr): 1685, 1515, 1450 cm−1; 1H NMR (DMSO-d6) δ: 0.89 (t,3H, J = 7); 0.99–1.05 (m, 4H); 1.32–1.55 (m,2H); 3.14 (s, 6H); 3.29 (t, 2H, J = 7); 6.72 (dd, 1H, J = 2, J = 4); 7.15 (d, 1H, J = 2); 7.86 (d, 1H, J = 4); 10.04 (br s, 1H). MW 343.38. Anal. Calcd for C16H21N7O2: C, 55.96; H, 6.16; N, 28.55. Found: C, 56.13; H, 6.23; N, 28.73.
4.1.3. General procedure for the preparation of the 7-(arylcarbamoyl)amino-2-(2-furyl)-5-substituted)[1,2,4]triazolo[1,5-a]-[1,3,5]triazine derivatives (12, 13, 18, 19, 24, 25)
A solution of amino compound (1 mmol) (5–7), appropriate iso-cyanate (1.2 equiv) in dry dioxane was refluxed under argon for 18 h. The solvent was removed in vacuo, and the crude product was purified by flash chromatography (EtOAc–light petroleum 4:6) to afford the final compound (12, 13, 18, 19, 24, 25).
4.1.3.1. 2-(2-Furyl)-5-methylthio-7-(phenylcarbamoyl)amino-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (12)
Yield 75%; yellow solid; mp 165–167 °C (EtOAc–light petroleum); IR (KBr): 1660, 1510, 1450 cm−1; 1H NMR (DMSO-d6) δ: 2.54 (s, 3H); 6.65 (dd, 1H, J = 2, J = 4); 7.13 (d, 1H, J = 2); 7.25–7.54 (m, 5H); 7.92 (d, 1H, J = 4); 8.76 (br s, 1H); 11.55 (br s, 1H). MW 367.39. Anal. Calcd for C16H13N7O2S: C, 52.31; H, 3.57; N, 26.29. Found: C, 52.45; H, 3.63; N, 26.61.
4.1.3.2. 2-(2-Furyl)-7-[4-methyl(phenylcarbamoyl)]amino-5-methylthio [1,2,4]triazolo[1,5-a]-[1,3,5]triazine (13)
Yield 69%; yellow solid; mp 169–171 °C (EtOAc–light petroleum); IR (KBr): 1665, 1525, 1450 cm−1; 1H NMR (DMSO-d6) δ: 2.28 (s, 3H); 2.55 (s, 3H); 6.66 (dd, 1H, J = 2, J = 4); 7.07 (d, 1H, J = 2); 7.23 (d, 2H, J = 9); 7.65 (d, 2H, J = 9); 7.83 (d, 1H, J = 4); 8.32 (br s, 1H); 11.03 (br s, 1H). MW 381.41. Anal. Calcd for C17H15N7O2S: C, 53.53; H, 3.96; N, 25.71. Found: C, 53.57; H, 3.90; N, 25.59.
4.1.3.3. 2-(2-Furyl)-5-phenoxy-7-(phenylcarbamoyl)amino[1,2,4]-triazolo[1,5-a]-[1,3,5]triazine (18)
Yield 65%; pale yellow solid; mp 196–198 °C (EtOAc–light petroleum); IR (KBr): 1658, 1509, 1455 cm−1; 1H NMR (DMSO-d6) δ: 6.62 (dd, 1H, J = 2, J = 4), 7.03 (d, 1H, J = 2); 7.2–7.6 (m, 10H); 7.85 (d, 1H, J = 4); 8.77 (br s, 1H); 10.78 (br s, 1H). MW 414.39. Anal. Calcd for C21H15N7O3: C, 61.01; H, 3.66; N, 23.72. Found: C, 61.25; H, 3.72; N, 23.85.
4.1.3.4. 2-(2-Furyl)-7-[4-methyl(phenylcarbamoyl)]amino-5-phenoxy[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (19)
Yield 73%; pale yellow solid; mp 203–205 °C (EtOAc–light petroleum); IR (KBr): 1668, 1530, 1465 cm−1; 1H NMR (DMSO-d6) δ: 2.31 (s, 3H); 6.71 (dd, 1H, J = 2, J = 4), 7.12 (d, 1H, J = 2); 7.23–7.45 (m, 7H); 7.57 (d, 2H, J = 9); 7.86 (d, 1H, J = 4); 8.45 (br s, 1H); 10.08 (br s, 1H). MW 427.42. Anal. Calcd for C22H17N7O3: C, 61.82; H, 4.01; N, 22.94. Found: C, 62.01; H, 4.13; N, 23.07.
4.1.3.5. 5-Dimethylamino-2-(2-furyl)-7-(phenylcarbamoyl)amino-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (24)
Yield 80%; white solid; mp 270–274 °C (EtOAc–light petroleum); IR (KBr): 1665, 1515, 1460 cm−1; 1H NMR (DMSO-d6) δ: 3.14 (s, 6H); 6.70 (dd, 1H, J = 2, J = 4), 7.09 (d, 1H, J = 2); 7.34–7.58 (m, 5H); 7.89 (d, 1H, J = 4); 8.32 (br s, 1H); 10.65 (br s, 1H). MW 364.40. Anal. Calcd for C17H16N8O2: C, 56.04; H, 4.43; N, 30.75. Found: C, 56.21; H, 4.38; N, 30.58.
4.1.3.6. 5-Dimethylamino-2-(2-furyl)-7-[4-methyl(phenylcar-bamoyl)]amino-[1,2,4]triazolo[1,5-a]-[1,3,5]triazine (25)
Yield 83%; white solid; mp 263–266 °C (EtOAc–light petroleum); IR (KBr): 1665, 1520, 1455 cm−1; 1H NMR (DMSO-d6) δ: 2.24 (s, 3H); 3.16 (s, 6H); 6.69 (dd, 1H, J = 2, J = 4), 7.11 (d, 1H, J = 2); 7.24 (d, 2H, J = 9); 7.58 (d, 2H, J = 9); 7.86 (d, 1H, J = 4); 8.76 (br s, 1H); 11.03 (br s, 1H). MW 378.39. Anal. Calcd for C18H18N8O2: C, 57.74; H, 4.79; N, 29.61. Found: C, 57.35; H, 4.77; N, 29.78.
4.2. Biological activity
4.2.1. Radioligand binding to rA1 and rA2A ARs
Procedures for preparation of rat brain membranes were reported previously.44,45 For binding experiments, membranes were frozen and stored at −20 °C for ≤2 months. Adenosine deaminase (ADA) was from Boehringer Mannheim (Indianapolis, IN). [3H]R-PIA was from Amersham (Arlington Heights, IL), and [3H]CGS 21680 was from DuPont NEN (Boston, MA).
Binding of [3H]R-PIA to the A1 AR from rat cortical membranes and of [3H]CGS 21680 to the A2A AR from rat striatal membranes was performed as described previously.46 Adenosine deaminase (ADA, 2 units/mL) was present during the preparation of brain membranes. Additional ADA was not added during incubation with the radioligand.
4.2.2. Radioligand binding to hA3 ARs
[125I]-AB-MECA was utilized in radioligand binding assays to membranes prepared from CHO cells expressing recombinant hA3 ARs, as previously described.36 ADA (3 units/mL) was present during the preparation of the membranes, in a preincubation of 30 min at 30 °C, and during the incubation with the radioligands. All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%. Incubations were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each. At least six different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in Graph-Pad (Prism, San Diego, CA), were converted to dissociation constants (Ki) as described.47 Hill coefficients of the tested compounds were in the range of 0.8–1.1.
4.2.3. Adenylyl cyclase activity
Due to the lack of a suitable radioligand the affinity of antagonists and the relative potency of agonists at A2B. ARs was determined in adenylyl cyclase experiments. The procedure was carried out as described previously37,38 with minor modifications. Membranes were incubated with about 150,000 cpm of [α-32P]ATP for 20 min in the incubation mixture as described37,38 without EGTA and NaCl. For agonists the EC50-values for the stimulation of adenylyl cyclase were calculated with the Hill equation. Hill coefficients in all experiments were near unity. IC50 values for concentration-dependent inhibition of NECA-stimulated adenylyl cyclase caused by antagonists were calculated accordingly. Ki values for antagonists were then calculated with the Cheng and Prusoff equation.48
4.3. Computational methodologies
All modeling studies were carried out on a 20 CPU (Intel Core™2 Quad CPU 2.40 GHz) linux cluster. Homology modeling, energy calculation, and analyses of docking poses were performed using the Molecular Operating Environment (MOE, version 2008.10) suite.49 The software package MOPAC (version 7),50 implemented in MOE suite, was utilized for all quantum mechanical calculations. Docking simulations were performed using GOLD suite.51 Log P values were calculated using ACDLABS 10.0.52
4.3.1. Homology model of rA2A AR and hA3 AR
Based on the assumption that GPCRs share similar TM boundaries and overall topology, homology models of the ARs were constructed. First, the amino acid sequences of TM helices of rA2A and hA3 receptor were aligned with those of hA2A AR,39 guided by the highly conserved amino acid residues, including the DRY motif (Asp3.49, Arg3.50, and Tyr3.51) and three proline residues (Pro4.60, Pro6.50, and Pro7.50) in the TM segments of GPCRs. The same boundaries were applied for the TM helices of rA2A and hA3 ARs as they were identified from the X-ray crystal structure for the corresponding sequences of hA2A AR,39 the CR coordinates of which were used to construct the seven TM helices for the rA2A and hA3 ARs. The loop domains were constructed by the loop search method implemented in MOE on the basis of the structure of compatible fragments found in the Protein Data Bank. In particular, loops were modeled first in random order. For each loop, a contact energy function analyzes the list of candidates collected in the segment searching stage, taking into account all atoms already modeled and any atoms specified by the user as belonging to the model environment. These energies were then used to make a Boltzmann-weighted choice from the candidates, the coordinates of which were then copied to the model. Any missing side chain atoms were modeled using the same procedure. Side chains are modeled using a library of rotamers generated by systematic clustering of the Protein Data Bank data, using the same procedure. Side chains belonging to residues whose backbone coordinates were copied from a template were modeled first, followed by side chains of modeled loops. Outgaps and their side chains were modeled last. Special caution has to be given to the second extracellular loop (EL2) because amino acids of this loop could be involved in direct interactions with the ligands. A driving force to this peculiar fold of the EL2 loop might be the presence of a disulfide bridge between cysteines in TM3 and EL2. The constraints were applied before the construction of the homology model, in particular during the sequences alignment, selecting the cysteine residues involved in the disulfide bridge in hA2A to be constrained with the corresponding cysteines in hA3 sequence. In particular, Cys166 (EL2) in hA2A was constrained with Cys166 (EL2) in hA3 and Cys77 (3.25) in hA2A with Cys83 (3.25) in hA3. When performing the alignment, MOE-Align attempts to minimize the number of constraint violations. Then, after running the homology modeling, the presence of the conserved disulfide bridge in the model was manually checked. Since this covalent link was conserved in all ARs modeled in the current study, the EL2 loop was modeled using a hA2A-like constrained geometry around the EL2-TM3 disulfide bridge. In rA2A receptor all the three disulfide bridge that characterize the template were conserved; for this reason the loop was modeled constraining all its residues. After the heavy atoms were modeled, all hydrogen atoms were added and were then minimized with MOE using the AMBER9953 force field. The minimizations were carried out by the 1000 steps of steepest descent followed by conjugate gradient minimization until the rms gradient of the potential energy was less than 0.1 kcal mol−1 Å−1. We used Protonate 3D methodology, part of the MOE suite, for protonation state assignment by selecting a protonation state for each chemical group that minimizes the total free energy of the system (taking titration into account).54
Protein stereochemical evaluation was then performed using several tools (Ramachandran and Chi plots measure phi/psi and chi1/chi2 angles, clash contacts reports) implemented in MOE suite.49
4.3.2. Molecular docking of rA2A AR and hA3 AR antagonists
Ligand structures were built using MOE-builder tool, part of the MOE suite,49 and were subjected to MMFF94x55 energy minimization until the rms of conjugate gradient was 0.05 kcal mol−1 Å−1. Charges were calculated using ESP methodology.
Four different programs have been used to calibrate our docking protocols: MOEDOCK,49 GOLD,51 GLIDE,56 and PLANTS.57
Therefore ZM241385 was re-docked to the crystal structure of the hA2A AR (PDB code: 3EML) with different docking algorithms and scoring functions (see Table 2).
Table 2.
Comparison of different molecular docking protocols
| Docking protocol | Best RMSD (Å) | Best ranked pose RMSD (Å) | Mean poses RMSD (Å) | Number of poses with RMSD <2.5 Å |
|---|---|---|---|---|
| MOE tabù search | 1.61 | 3.35 | 5.65 | 4/25 |
| MOE simulated annealing | 2.17 | 4.36 | 6.47 | 1/25 |
| MOE genetic algorithm | 2.25 | 9.06 | 6.66 | 2/25 |
| GOLD (goldscore) | 0.63 | 1.95 | 1.20 | 25/25 |
| GOLD (chemscore) | 1.31 | 3.90 | 3.13 | 11/25 |
| GOLD (asp) | 0.61 | 4.96 | 1.50 | 23/25 |
| GLIDE | 0.79 | 2.71 | 6.82 | 7/25 |
| PLANTS (chemplp) | 0.93 | 1.98 | 6.96 | 3/25 |
| PLANTS (plp) | 0.84 | 1.93 | 6.70 | 6/25 |
| PLANTS (plp95) | 1.97 | 11.80 | 8.22 | 4/25 |
Each docking was performed automatically to the binding site of the hA2A AR without any constraints and without the presence of water molecules. For all the different docking simulations, the center of the docking box or of the docking sphere was set in the same point (got from the experimental pose of ZM241385 inside the crystal structure of hA2A AR) and the number of independent docking runs was set to 25. Then RMSD values between predicted and crystallographic positions of ZM241385 were calculated.
As shown in Table 2, for each docking result there is at least one pose in good agreement with the experimental binding mode (RMSD value <2.5 Å). These poses with lowest RMSD value differ from the crystallographic pose of ZM241385 mainly for the position of the phenylethylamine chain, while the bicyclic triazolotriazine core is almost aligned. However, the mean RMSD value is quite high for the most part of the docking protocols tested except for GOLD. Docking performed with GOLD gives the lowest RMSD value, the lowest mean RMSD value and the highest number of poses with RMSD value <2.5 Å.
Based on the best docking performance, all antagonist structures were docked into the hypothetical TM binding site of the hA3 AR and the rA2A AR models, by using the dock tool part of the GOLD suite. Searching is conducted within a user-specified docking sphere, using the Genetic Algorithm protocol and the Gold Score scoring function. GOLD performs a user-specified number of independent docking runs (25 in our specific case) and writes the resulting conformations and their energies in a molecular database file. The resulting docked complexes were subjected to MMFF94x energy minimization until the rms of conjugate gradient was <0.1 kcal mol−1 Å−1. Charges for the ligands were imported from the MOPAC output files using PM3/ESP methodology.
Prediction of antagonist–receptor complex stability (in terms of corresponding pKi value) and the quantitative analysis for non-bonded intermolecular interactions (H-bonds, transition metal, water bridges, hydrophobic, electrostatic) were calculated and visualized using several tools implemented in MOE suite.39
Electrostatic and hydrophobic contributions to the binding energy of individual amino acids have been calculated based on the AMBER99 force field as implemented in MOE suite.39 To estimate the electrostatic contributions, atomic charges for the ligands were calculated using PM3/ESP methodology; instead partial charges for protein amino acids were calculated based on the AMBER99 force field.
Acknowledgments
The molecular modeling work coordinated by S.M. was carried out with financial support from the University of Padova, Italy, and the Italian Ministry for University and Research (MIUR), Rome, Italy. S.M. is also very grateful to Chemical Computing Group for the scientific and technical partnership. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- GPCRs
G protein-coupled receptors
- AR
adenosine receptor
- ADA
adenosine deaminase
- cAMP
cyclic adenosine monophosphate
- h
human
- SAR
structure–affinity relationship
- r
rat
- DPCPX
8-cyclopentyl-1,3-dipropyl-xanthine
- CGS 21680
2-[4-(2-carboxyethyl)phenethyl]amino-5′-(N-ethylcarbamoyl)adenosine
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- NECA
5′-(N-ethyl-carboxamido)adenosine
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- TM
transmembrane
- RMSD
root mean square deviation
- EL2
second extracellular loop
- MOE
molecular operating environment
- ZM241385
4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}{1,3,5,}triazin-5-yl amino]ethyl)phenol
References and notes
- 1.Downey JM, Cohen MV, Ytrehus K, Liu Y. Ann NY Acad Sci. 1994;723:82. [PubMed] [Google Scholar]
- 2.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Mol Pharmacol. 1997;52:846. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson KA, von Lubitz DKJE, Daly JW, Fredholm BB. Trends Pharmacol Sci. 1996;17:108. doi: 10.1016/0165-6147(96)10002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fredholm BB. Int Rev Neurobiol. 1997;40:259. [PubMed] [Google Scholar]
- 5.Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Trends Pharmacol Sci. 1998;19:148. doi: 10.1016/s0165-6147(98)01179-1. [DOI] [PubMed] [Google Scholar]
- 6.Fozard JR, McCarthy C. Curr Opin Invest Drugs. 2002;3:69. [PubMed] [Google Scholar]
- 7.Richardson PJ, Kase H, Jenner PG. Trends Pharmacol Sci. 1997;18:338. doi: 10.1016/s0165-6147(97)01096-1. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro JA, Sebastiao AM, de Mendonca A. Prog Neurobiol. 2002;68:377. doi: 10.1016/s0301-0082(02)00155-7. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, Burbiel JC, Maass A, Muller CE. Expert Opin Emerg Drugs. 2003;8:537. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 10.Fredholm BB. Drug News Perspect. 2003;16:283. doi: 10.1358/dnp.2003.16.5.829316. [DOI] [PubMed] [Google Scholar]
- 11.Moro S, Spalluto G, Jacobson KA. Trends Pharmacol Sci. 2005;26:44. doi: 10.1016/j.tips.2004.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:364. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 13.Moro S, Gao ZG, Jacobson KA, Spalluto G. Med Res Rev. 2006;26:131. doi: 10.1002/med.20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Monopoli A, Ongini E, Varani K, Borea PA. J Med Chem. 2002;45:115. doi: 10.1021/jm010924c. [DOI] [PubMed] [Google Scholar]
- 15.Baraldi PG, Cacciari B, Spalluto G, Bergonzoni M, Dionisotti S, Ongini E, Varani K, Borea PA. J Med Chem. 1998;41:2126. doi: 10.1021/jm9708689. [DOI] [PubMed] [Google Scholar]
- 16.Baraldi PG, Cacciari B, Spalluto G, Pineda de las Infantas y Villatoro MJ, Zocchi C, Dionisotti S, Ongini E. J Med Chem. 1996;39:1164. doi: 10.1021/jm950746l. [DOI] [PubMed] [Google Scholar]
- 17.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Klotz K-N, Leung E, Varani K, Gessi S, Merighi S, Borea PA. J Med Chem. 1999;42:4473. doi: 10.1021/jm991114s. [DOI] [PubMed] [Google Scholar]
- 18.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Moro S, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA. J Med Chem. 2000;43:4768. doi: 10.1021/jm001047y. [DOI] [PubMed] [Google Scholar]
- 19.Baraldi PG, Cacciari B, Moro S, Spalluto G, Pastorin G, Da Ros T, Klotz K-N, Varani K, Gessi S, Borea PA. J Med Chem. 2002;45:770. doi: 10.1021/jm0109614. [DOI] [PubMed] [Google Scholar]
- 20.Maconi A, Pastorin G, Da Ros T, Spalluto G, Gao ZG, Jacobson KA, Baraldi PG, Cacciari B, Varani K, Borea PA. J Med Chem. 2002;45:3579. doi: 10.1021/jm020974x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastorin G, Da Ros T, Spalluto G, Deflorian F, Moro S, Cacciari B, Baraldi PG, Gessi S, Varani K, Borea PA. J Med Chem. 2003;46:4287. doi: 10.1021/jm030852k. [DOI] [PubMed] [Google Scholar]
- 22.Pinna A, Volpini R, Cristalli G, Morelli M. Eur J Pharmacol. 2005;512:157. doi: 10.1016/j.ejphar.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 23.Vu CB, Pan D, Peng B, Kumaravel G, Phadke D, Engber T, Huamg C, Reilly J, Tam S, Petter RC. Bioorg Med Chem Lett. 2004;14:4831. doi: 10.1016/j.bmcl.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 24.Dowling JE, Vessels JT, Haque S, Chang HX, van Vloten K, Kumaravel G, Engber TM, Jin X, Phadke D, Wang J, Ayyub E, Petter RC. Bioorg Med Chem Lett. 2005;15:4809. doi: 10.1016/j.bmcl.2005.07.052. [DOI] [PubMed] [Google Scholar]
- 25.Gang Y, Haque S, Sha L, Kumaravel G, Wang J, Engber TM, Whalley ET, Conlon PR, Chang H, Kiesman WF, Petter RC. Bioorg Med Chem Lett. 2005;15:511. doi: 10.1016/j.bmcl.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 26.Vu CB, Shields P, Peng B, Kumaravel G, Jin X, Phadke D, Wang J, Engber T, Ayyub E, Petter RC. Bioorg Med Chem Lett. 2004;14:4835. doi: 10.1016/j.bmcl.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 27.Peng B, Kumaravel G, Yao G, Sha L, Van Vlijmen H, Bohnert T, Huang C, Vu CB, Ensinger CL, Chang H, Engber TM, Whalley E, Petter RC. J Med Chem. 2004;47:6218. doi: 10.1021/jm0494321. [DOI] [PubMed] [Google Scholar]
- 28.Vu CB, Pan D, Kumaravel G, Smits G, Jin X, Phadke D, Engber T, Huang C, Reilly J, Tam S, Grant D, Hetu G, Petter RC. J Med Chem. 2005;48:2009. doi: 10.1021/jm0498396. [DOI] [PubMed] [Google Scholar]
- 29.Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, Collis MG. Br J Pharmacol. 1995;115:1096. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeZwart M, Vollinga RC, Beukers MW, Sleegers DF, von Frijtag Drabbe Kunzel JK, De Groote M, IJzerman AP. Drug Dev Res. 1999;48:95. [Google Scholar]
- 31.Ji XD, Jacobson KA. Drug Des Discovery. 1999;16:217. [PMC free article] [PubMed] [Google Scholar]
- 32.Caulkett PWR, Jones G, McPartlin M, Renshaw ND, Stewart SK, Wright B. J Chem Soc, Perkin Trans I. 1995:801. [Google Scholar]
- 33.Schwabe U, Trost T. Naunyn-Schmiedeberg’s Arch Pharmacol. 1980;313:179. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 34.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. J Pharmacol Exp Ther. 1989;251:888. [PubMed] [Google Scholar]
- 35.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Proc Natl Acad Sci USA. 1993;90:10365. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol Pharmacol. 1994;45:978. [PMC free article] [PubMed] [Google Scholar]
- 37.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 38.Klotz KN, Cristalli G, Grifantini M, Vittori S, Lohse MJ. J Biol Chem. 1985;260:14659. [PubMed] [Google Scholar]
- 39.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunha RA, Constantino MD, Ribeiro JA. Br J Pharmacol. 1997;122:1279. doi: 10.1038/sj.bjp.0701507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ongini E, Dionisotti S, Gessi S, Irenius E, Fredholm BB. Naunyn- Schmiedeberg’s Arch Pharmacol. 1999;359:7. doi: 10.1007/pl00005326. [DOI] [PubMed] [Google Scholar]
- 42.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
- 43.Audet M, Bouvier M. Nat Chem Biol. 2008;4:397. doi: 10.1038/nchembio.97. [DOI] [PubMed] [Google Scholar]
- 44.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. J Med Chem. 1994;37:636. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. Mol Pharmacol. 1994;45:1101. [PMC free article] [PubMed] [Google Scholar]
- 46.van Bergen A, van Galen PJM, Stiles GL, Jacobson KA. ACS 206th National Meeting; Chicago, IL. August, 1993; Abstract MEDI217. [Google Scholar]
- 47.Linden JJ. Cyclic Nucleotide Res. 1982;8:163. [PubMed] [Google Scholar]
- 48.Cheng YC, Prusoff HR. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 49.MOE (Molecular Operating Environment), version 2008.10; software available from Chemical Computing Group Inc. 1010 Sherbrooke Street West, Suite 910, Montreal, Quebec, Canada H3A 2R7: http://www.chemcomp.com. [Google Scholar]
- 50.Stewart JJP. MOPAC. Vol. 7. Fujitsu Limited; Tokyo, Japan: 1993. [Google Scholar]
- 51.GOLD suite, version 4.0.1; software available from Cambridge Crystallographic Data Centre Cambridge Crystallographic Data Centre. 12 Union Road Cambridge CB2 1EZ UK: http://www.ccdc.cam.ac.uk. [Google Scholar]
- 52.ACDLABS, version 10.0; software available from Advanced Chemistry Development, Inc. 110 Yonge Street, 14th floor, Toronto, Ontario, Canada M5C 1T4: [Google Scholar]
- 53.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179. [Google Scholar]
- 54.Labute P. Proteins. 2009;75:187. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halgren T. J Comput Chem. 1996;17:490. [Google Scholar]
- 56.Halgren TA, Myrphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT. J Med Chem. 2004;47:1739. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 57.Korb O, Stützle T, Exner TE. J Chem Inf Model. 2009;49:84. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]