

Abstract
Neuronal development involves several discrete morphological steps requiring migration of newborn neurons to characteristic locations, extension of axons and dendrites into proper target regions, and formation of synapses with appropriate partners. Small GTPases such as Rac1, are believed to be critical regulators of these processes. We have previously reported that Rac1 is highly expressed in mouse hippocampus, where NMDA receptor activation causes Rac1 to translocate to the membrane in a manner similar to that observed in other non-neuronal cells. Additionally Rac1 has been seen to play a role in activation of signal transduction pathways associated with hippocampal learning and memory. Because of the established role of LTP and LTD in learning and memory processes, in this study we investigate whether Rac1 plays also an active and critical role in these types of long-term synaptic plasticity. We found that activation of Rac1 is associated with long-term plasticity, both LTP and LTD. Rac1 appears to have a transient role during the induction of NMDA receptor-dependent LTP, but does not have an effect on LTP maintenance and expression. Similar results were found for NMDA receptor-dependent induction of LTD, while mGluR-dependent LTD was shown to be significantly altered but not abolished. The results of these experiments provide essential knowledge regarding the signaling mechanisms that underlie synaptic plasticity, as well as learning and memory processes, which in turn offers insights into the basis of diseases involving memory impairment, such as Fragile X syndrome, Alzheimer’s disease, William’s syndrome, Angelman syndrome (AS), and schizophrenia.
Keywords: Rac1, plasticity, LTP, LTD, NSC23766, EHT1864
1. INTRODUCTION
The role of the small GTPases as signaling molecules has been studied extensively in many cell types, but their importance is still being discovered in the field of neuroscience. The Rho subfamily of GTPases are involved primarily in the regulation of cytoskeletal organization in response to extracellular growth factors as well as in diverse cellular events such as membrane trafficking, transcriptional regulation, and cell growth control and development (Van Aelst and D'Souza-Schorey, 1997). One member of the Rho GTPase subfamily, Rac1, has been implicated in regulation of the mitogenic response, superoxide generation, regulation of transcription (Ridley, 2001a), morphological changes necessary for migration (Ridley, 2001b), activation of the release sites for exocytosis (Humeau et al., 2002) and specific cytoskeleton rearrangements that play a crucial role in cell motility and cytokinesis (Van Aelst and D'Souza-Schorey, 1997). In the CNS, neuronal development involves several discrete morphological steps requiring migration of newborn neurons to characteristic locations, extension of axons and dendrites into proper target regions, and formation of synapses with appropriate partners. The small GTPases including Rac1, are believed to be critical regulators of these processes (Luo, 2000;Sin et al., 2002;Dickson, 2001;Nikolic and Chernoff, 2002;Caron, 2003), participating in neuronal morphogenesis, migration, polarity, axon growth and guidance, dendrite elaboration, plasticity and synapse formation (Luo, 2000;Waetzig and Herdegen, 2002;Dickson, 2001;Nikolic and Chernoff, 2002). Previous work in our laboratory demonstrated that Rac1 is highly expressed in mouse hippocampus and that NMDA receptor activation in hippocampal slices causes Rac1 to translocate from the cytosol to the membrane and become activated (Rac1-GTP) (Tejada-Simon et al., 2006). Additionally, NMDA receptor-dependent activation of Rac1 appears to be associated with hippocampus-dependent contextual fear conditioning tasks in the adult mice (Martinez et al., 2007). These observations suggest that Rac1 localization and function are connected to cognitive tasks via NMDA receptor activation following hippocampus-dependent learning in the behaving animal.
The most commonly studied form of long-term plasticity in area CA1 is dependent on the activation of the NMDA subtype of glutamate receptors (Collingridge et al., 1983, Harris et al., 1994). If NMDA receptor activation is necessary for long-term plasticity such as long-term potentiation (LTP) and long-term depression (LTD) and translocation and activation of Rac1 is linked to NMDA receptor activation, then it is reasonable to hypothesize that translocation and activation of Rac1 may occur during plasticity. In this manuscript, to assess the contribution and importance of Rac1 to long-term plasticity in the mouse hippocampus, we investigated the electrophysiological consequences of Rac1 inhibition using a pharmacological approach. We found that Rac1 plays a role in plasticity at hippocampal synapses during LTP and LTD. Together our data suggest that Rac1 is associated and necessary for LTP- and LTD-inducing stimulation in the adult mouse brain.
2. MATERIAL AND METHODS
2.1. Preparation of hippocampal slices and brain homogenates
All animal experiments were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals. Additionally, animals were handled and sacrificed in compliance with institutional regulation and policies. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Houston. All efforts were made to minimize animal suffering and to reduce the number of animals used. Male C57BL6 mice (8–12 weeks old) were sacrificed by cervical dislocation under gentle anesthesia. Brains were rapidly removed and briefly submerged in ice-cold cutting saline solution (5mM glucose, 110mM sucrose, 60mM NaCl, 28mM NaHCO3, 3mM KCl, 1.25mM NaH2PO4, 7mM MgCl2, and 0.5mM CaCl2, 0.6mM ascorbate). All solutions were saturated with 95% O2/5% CO2.
For hippocampal slices, 400 µm thick slices were prepared using a 1000Plus Vibratome sectioning system (Vibratome Co., St. Louis, Missouri). Slices were allowed to equilibrate in a 50% cutting saline solution and 50% artificial cerebrospinal fluid solution (ACSF, 25mM glucose, 125mM NaCl, 2.5mM KCl, 1.25mM NaH2PO4, 25 NaHCO3, 1mM MgCl2, and 2mM CaCl2) saturated with 95% O2/5% CO2 at room temperature for 30 minutes. After the experiments, hippocampi were carefully handled and when appropriate hippocampal CA1 region was sectioned using a microscope with cutting solution-soaked filter paper mounted on a glass platform resting on ice.
For hippocampal homogenates, area CA1 was isolated immediately and homogeneized in homogenizing buffer complete (HBC; 10mM Hepes, 1mM EDTA, 1mM EGTA, 150mM NaCl, 50mM NaF, 10 mM sodium pyrophosphate, 1mM sodium orthovanadate, 10 µg/ml leupeptin, 2 µg/ml aprotinin, 1 µM microcystin-LR, and 200nM calyculin A). The samples were then centrifuged at 1500g for 10min at 4 °C to eliminate debris. Total protein concentrations were determined (Bradford, 1976).
2.2. Electrophysiology
Transverse hippocampal slices (400 µm) were prepared from age-matched animals as described previously. Slices were transferred to an interface chamber (Harvard Apparatus, Holliston, MS) at 30–32°C, perfused with oxygenated ACSF (perfusion rate: 1–2ml/min) and allowed to recover for a minimum of 1 hr prior to recording. For Rac1 inhibitor treatments, the drug was perfused during induction or maintenance phase as indicated. Extracellular field recordings were obtained from area CA1. A bipolar enamel-coated platinum stimulating electrode was placed in CA3 Schaffer collateral/commissural fibers and a glass recording electrode (resistance 1–4 MΩ) filled with ACSF was placed into stratum radiatum of area CA1. Synaptic responses to electrical stimulation were collected every 20 sec and averaged over 2min using a stimulus intensity that produces 30–50% of the maximum initial slope of the extracellular field excitatory postsynaptic potential (fEPSP). Baseline fEPSPs were monitored for at least 20 min before the application of any drug. For LTP induction in area CA1, baseline fEPSPs were monitored for at least 20 min before delivering four HFS trains (1 sec at 100 Hz) with an inter-train interval of 5 min with responses recorded for about 2 hrs after HFS. For TBS, we stimulated the Schaffer collateral pathway with three theta bursts with a one minute inter-burst interval and recorded signals from area CA1 stratum radiatum. One theta burst was comprised of five trains with 200ms intervals and each train consisting of 4 pulses (100Hz). The stimulus intensity of the pulses was matched to that used during baseline recordings.
NMDAR-dependent LTD was induced with low-frequency stimulation (LFS, 900 pulses at 1Hz for 15min). mGluR-LTD was induced by 100µM (S)-3,5-dihydroxyphenylglycine, DHPG (Tocris, Ellisville, MO) in the presence of 100µM ((2R)-amino-5-phosphonovaleric acid (AP-5 or APV, Tocris, Ellisville, MO) in ACSF applied for 5min after 20min of stable baseline. Data points were normalized to the baseline mean prior to delivery of HFS, LFS or drug and is collected using pClamp version 10 (Molecular Devices, Sunnyvale, CA). Baseline stability was tested by recording signals for ≥1.5hrs using the same stimulus value.
2.3. Rac1 activation assay
Rac1 activation was determined using an affinity purification assay. To determine expression levels of Rac1-GTP, 10 µg of GST tagged PAK-PBD protein beads (Millipore, Temecula, CA) were mixed with 200 µg protein of fresh hippocampal homogenates containing phosphatase and protease inhibitor cocktail (EMD, San Diego, CA). Fifty µl of HBC buffer were added to increase the reaction volume. Samples were placed on a rocker for 2 hours at 4°C. PAK-PBD-GST beads then were pelleted by centrifugation at 14,000g for 10 minutes at 4°C. The supernatant was carefully removed without disturbing the pelleted beads, followed by three more washes and centrifugations. The final pellet was resuspended in 50 µl Laemmli buffer. Samples were placed in a 100°C water bath for 3 minutes to remove Rac1-GTP from the beads. Amount of Rac1 protein was then analyzed by electrophoresis and immunoblot analysis.
2.4. Electrophoresis and Immunoblot analysis
Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, 12%) followed by electrophoretic transfer to a polyvinylidene difluoride (PVDF) membrane, and incubated in Tris-buffered saline with Tween 20 (TTBS, 50 mM Tris-HCl [pH 7.5–8.0], 150 mM NaCl, and 0.1% Tween 20) containing 5% non-fat milk for 1 hour at room temperature. Membranes were incubated with the desired antibody (Rac1, 1:10.0000 from Millipore; total PAK1 and phosphorylated PAK1 1:1000, from Cell Signaling) overnight at 4°C. Blots were washed three times for 15 min in TTBS, followed by incubation in the corresponding secondary antibody (horseradish peroxidase-conjugated goat anti-mouse IgG [1:5,000] from Promega, Madison, WI for Rac1; anti-rabbit for total PAK1 and phosphorylated PAK1). This was followed by washing three times in 1 X TTBS and visualized using enhanced chemiluminescence (ECL, Amersham Biosciences, Piscataway, NJ) and by exposing the blots to X-Omat Blue film (Kodak, Rochester, NY) for ~ 15 s to 1 min. Densitometric analysis of all samples were performed using NIH Image J software.
2.5. Data analysis
Statistical analysis was performed using Student’s t tests for normally distributed variables to determine the significant differences between control and treatment. A p value of less than 0.05 was considered statistically significant. Data for electrophysiological experiments comparing the initial slope of the fEPSP at various time points before and after LTP/LTD-inducing stimulation was evaluated statistically with a two-way analysis of variance over all post-tetanus time points, with p < 0.05 as significance criteria.
3. RESULTS
3.1. Induction of long-term potentiation in the hippocampus is coupled to activation of the small GTPase Rac1 in area CA1
It is believed that long-term plasticity is triggered by synaptic activation, dependent on the activation of the NMDA receptor and critical for learning and memory as well as pathological conditions. There are two major forms of long-term synaptic plasticity in the mammalian brain, long-term potentiation (LTP) and long-term depression (LTD). Our laboratory previously reported that in hippocampal slices, pharmacological activation of the NMDA receptor leads to membrane translocation and activation of Rac1 (Tejada-Simon et al., 2006). We further reported that activation of Rac1 is correlated with associative learning (Martinez et al., 2007). Since induction of LTP by HFS has been associated with activation of NMDA receptors, we evaluated whether induction of LTP is also related with activation of Rac1 and whether Rac1 is necessary for LTP. First, LTP was induced in hippocampal slices with four trains of high frequency stimulation (Figure 1A). At various time points after the last stimulation as indicated during the experiment, hippocampal slices were collected and the activation state of Rac1 was determined. LTP induction was associated with a transient activation of Rac1 that lasted for at least 2 hours after the last train of stimulation. Activation of Rac1 correlated with the activation of one of its effectors, PAK1, which has been reported to have a critical role in the actin remodeling process directed by Rac1 and important for neuronal plasticity and function (Figure 1C, 1D). Since this form of LTP is NMDA receptor-dependent, induction of LTP was impaired by blockade of NMDA receptor with amino-phosphonovaleric acid (APV), an agent that selectively blocks the NMDA subtype of glutamate receptors without altering baseline synaptic transmission (Figure 1B). This pharmacologic blockade of NMDA receptors abolished not only induction of LTP but also Rac1 activation (Figure 1C). These results suggest that Rac1 is associated to LTP and that activation of Rac1 during LTP is a NMDA receptor-dependent phenomenon.
Figure 1. Activation of Rac1 is associated with LTP.

A. Hippocampal slices were stimulated at CA3 afferents and responses were recorded from area CA1 dendrites. Four trains of high frequency stimulation (HFS, 1 sec at 100 Hz spaced 5 min apart; indicated by arrows) induces LTP in hippocampal slices. Right: shows fEPSP sample traces recorded during baseline (dashed line, a) and 2hrs after fourth HFS (solid line, b). Scale 1mV/5ms. n=7. B. LTP is associated with NMDA receptor activation. Wildtype hippocampal slices were subjected to four trains of HFS (1 sec at 100 Hz spaced 5 min apart; indicated by arrows) in presence of APV (100 µM), an NMDA receptor antagonist that blocks LTP (n=6). C. At various time points (2, 6, 10 and 120 min) after the fourth stimulation leading to LTP, hippocampal slices were collected, area CA1 dissected, homogenized and assayed for Rac1 activation. Representative western blot depicting expression of Rac1-GTP, total Rac1, phospho-PAK1 and total PAK1 in stimulated (+) and unstimulated (−) slices subjected to HFS while treated or not with APV (100 µM). n=6. D. Data quantification (mean ± SEM, n=6) representing expression of Rac1-GTP and phospho-PAK1 after HFS. * denotes statistically significance respect to unstimulated slices (p<0.05, Student t-test).
3.2. Pharmacological inhibition of Rac1 impairs induction of hippocampal long-term plasticity in a dose-dependent manner
The involvement of Rac1 in long-term plasticity was further tested in hippocampal slices by independent perfusion of two different compounds that inhibit Rac1. This approach offers us a selective pharmacologic tool to study the need for Rac1 activity in long-term plasticity. One of these drugs is NSC23766, a small molecule that has been shown to act as a Rac1 specific inhibitor in non-neuronal cell cultures by interfering with certain Rac1 GEFs, impairing GDP/GTP exchange (Gao et al., 2004). The second pharmacological agent is EHT1864, defined also as a small molecule that is able to inhibit specifically Rac1, inhibiting both guanine nucleotide association and nucleotide exchange (Shutes et al., 2007). It is believed that NSC23766 works by blocking certain GEF-Rac1 interactions, while EHT1864 promotes GTP nucleotide unloading.
To first establish whether synaptic transmission is affected by these drugs, using constant stimulus intensity we determined the effects of drug application on baseline synaptic transmission by monitoring EPSPs before and after incubation of slices with the Rac1 inhibitors. Consistent results were found for both Rac1 inhibitors, indicating that basal synaptic transmission is not critically affected by the application of inhibitors (Figure 2A, 2B).
Figure 2. Activation of Rac1 is necessary for induction of long-term potentiation (LTP).
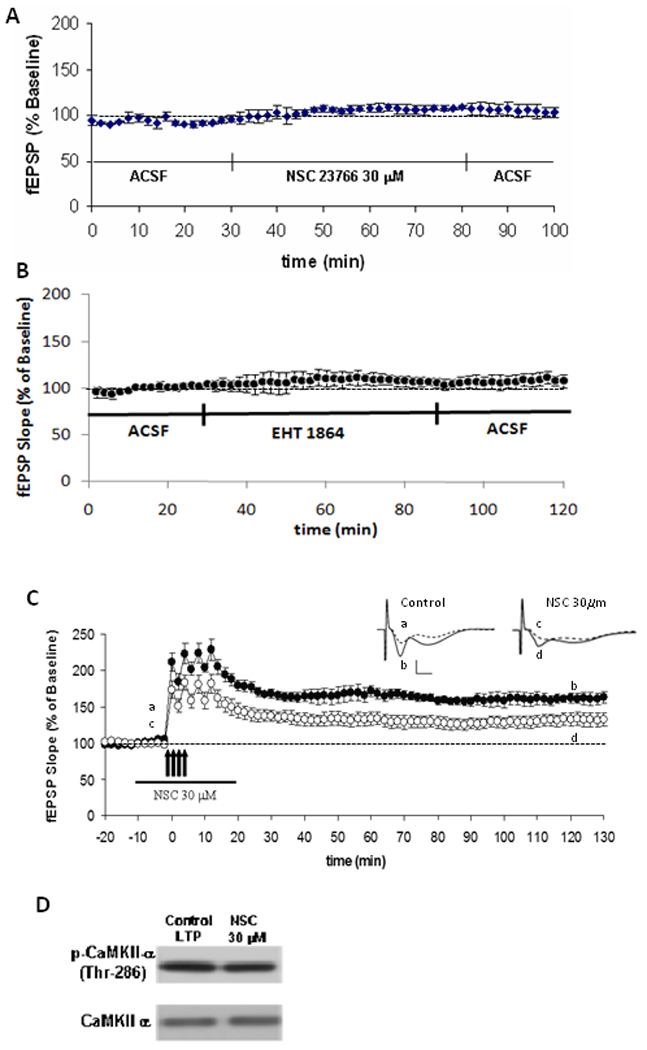
Effect of (A) NSC23766 and (B) EHT 1864 on baseline synaptic transmission. Baseline responses were recorded for 30 min before slices were perfused for 50 min with NSC23766 (30 µM) or EHT1864 (60 µM) (n=12). Responses were recorded for an additional 20–30 min after washout of the drug. C. Four trains of HFS (1 sec at 100 Hz; indicated by arrows) spaced 5 min apart induced LTP in vehicle-treated (ACSF) control hippocampal slices (●), but was attenuated (p<0.05) when HFS was delivered in the presence of NSC23766 (30 µM; [○]). Inset: Sample traces recorded during baseline (dashed lines, a, c) and 2hrs post-HFS (solid lines, b, d). Scale 1mV/5ms. D. Representative immunoblots depicting expression of phospho-CaMKII and total CaMKII in vehicle- and NSC23766-treated slices after HFS stimulation. (n=7).
To specifically study the role and importance of Rac1 in long-term plasticity in the brain, NMDA receptor-dependent synaptic transmission in the CA1 hippocampal subregion was evaluated via field potential recording (fEPSPs) evoked by stimulation of Schaffer collateral/commissural fibers in hippocampal slices (8–12 weeks old mice) treated with Rac1 inhibitors. We found that inhibition of Rac1 by NSC23766 during the induction phase resulted in significantly impaired LTP (Figure 2C, p<0.05). This observed inhibition of LTP by Rac1 inhibitors could be due to off-target effects of the drug rather than because Rac1 activation is prevented. Thus, we determined whether the inhibitors perhaps blocked other required molecules for LTP, such as Calcium/Calmodulin-dependent Kinase II (CaMKII), a key regulator of synaptic strength that is persistently activated by stimuli that elicit LTP. Monitoring CaMKII by immunoblot, our results indicate that the phosphorylation and total protein levels of CaMKII do not vary when slices are treated with Rac1 inhibitors (Figure 2D). Phosphorylation of CaMKII determines whether long-term plasticity is induced. Therefore, in hippocampal slices treated with Rac1 inhibitors, a constant phosphorylation state of this protein after LTP induction, as compared to untreated slices, indicates that this kinase is functional as well as the upstream NMDA receptors that activate it.
The results of the previous experiment indicate that Rac1 modulates the induction of LTP, but it does not address the possible role for Rac1 in the maintenance of LTP. To test this possibility, we induced LTP as described previously and added NSC23766 about 20 min after the last LTP-inducing HFS. Pre-established LTP was unaffected by the addition of NSC23766 when monitored continuously after the addition of the drug (Figure 3, p>0.05). These results indicate that Rac1 modulates the induction, but not the maintenance of LTP. In an attempt to decipher whether the effect produced by NSC23766 was reversible, hippocampal slices were incubated with this Rac1 inhibitor before and during the induction phase of LTP, resulting in impaired potentiation as previously shown. Subsequently and approximately 15–20 minutes after the last train of HFS, the drug was removed and slices were incubated in ACSF. More than two hours after the initial induction protocol, slices were subjected to another set of 4 trains of HFS. Field potential recording (fEPSPs) evoked by stimulation of Schaffer collateral fibers in hippocampal slices were again evaluated. Thus, using this protocol designed to allow the washout of the drug followed by another round of stimulation, we found a significant and lasting increase in fEPSP slope produced by this second set of HFS (Figure 4). These observations suggest that the inactivation of Rac1 is likely transient and reversible. Altogether these data indicate that pharmacological inhibition of Rac1, while leaving basal synaptic transmission intact, specifically blocks induction of LTP in the hippocampus. This effect seems to be fully reversible and not disruptive for the brain tissue.
Figure 3. Effect of Rac1 inhibition on the maintenance of LTP in area CA1.
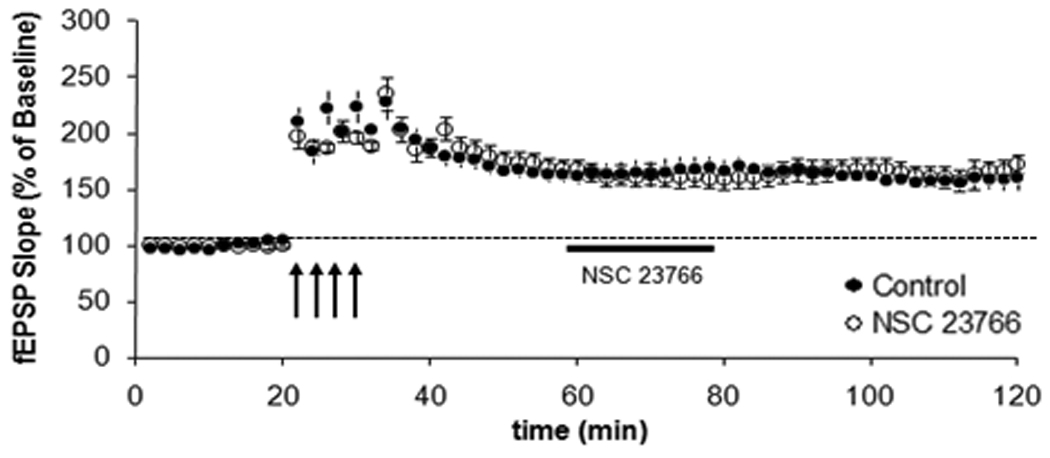
Stable baseline responses were recorded for 20 min before LTP was induced in hippocampal slices with four 100 Hz 1-sec trains of HFS (L-LTP) given 5 min apart (●). Approximately 20 min after the last train of HFS, NSC23766 (30 µM, [○]) was washed into the chamber and the potentiation monitored for one more hour. During the maintenance phase, perfusion of slices with NSC23766 did not have an effect on LTP (p>0.05, n=7).
Figure 4. Impairment of LTP by pharmacological inhibition of Rac1 is reversible.

Hippocampal slices were incubated with NSC23766 (30 µM, [○]) during LTP induction, followed by withdrawal of the drug 15–20 min after the last train of HFS. Perfusion of ACSF allowed the drug to be washed out of the recording chamber. Two hours after the initial induction protocol, slices were subjected to a second set of four trains of HFS and fEPSPs evaluated from the Schaffer collateral fibers (n=6).
Next we determined whether a stronger depotentiation could be achieved by suppressing Rac1 to a greater extent. To this end, LTP was induced in hippocampal slices with four 1-sec trains of HFS (L-LTP) given 5 min apart. During the induction phase, slices were perfused with NSC 23766 or EHT1864 at different concentrations. Our results indicate that impairment of LTP by pharmacological inhibition of Rac1 is dose-dependent, with the highest doses bringing fEPSPs to baseline levels (Figure 5A, 5B, p<0.05). Impaired LTP correlates with significant decreases in levels of active Rac1 (Figure 5C, 5D, p<0.05). Both inhibitors have very similar and comparable effects.
Figure 5. Impairment of LTP by pharmacological inhibition of Rac1 is dose dependent.

A. LTP was induced in hippocampal slices with four 1-sec trains of HFS (L-LTP) given 5 min apart. During the induction phase, perfusion of slices with the Rac1 inhibitor, NSC23766, at different concentrations as indicated in the figure (from 30 to 120 µM) altered LTP induction in a dose dependent fashion. Insets represent sample traces recorded during baseline (dashed lines, a, c, e, g) and 2hrs post-HFS (solid lines, b, d, f, h) for every concentration tested (p<0.05, n=8 per concentration). B. LTP was induced in hippocampal slices as described above. During the induction phase, perfusion of slices with another Rac1 inhibitor, EHT1864, at different concentrations as indicated in the figure (60 and 100 µM) equally altered LTP induction in a dose dependent manner. Insets represent sample traces recorded during baseline (dashed lines, a, c, e) and 2hrs post-HFS (solid lines, b, d, f) for every concentration tested (p<0.05, n=8 per concentration). C. Representative immunoblot showing expression of Rac1-GTP in stimulated (+) and unstimulated (−) hippocampal slices treated or not with different concentrations of NSC23766. D. Group data (mean ± SEM, n=5) representing expression of Rac1-GTP in stimulated (+) and unstimulated (−) hippocampal slices treated or not with different concentrations of NSC23766. * denotes statistically significance respect to unstimulated slices (p<0.05, Student t-test).
Furthermore, to mimic more physiological conditions, we used theta burst stimulation (TBS) protocol to induce LTP. TBS is proposed to mimic hippocampus neuronal firing patterns generated by mice during exploratory and learning behavior. Three theta bursts (five trains with 200ms intervals, each train consisting of 4–100 Hz pulses) with a one minute interburst interval were used to induce LTP in area CA1 stratum radiatum. During the induction phase, slices were perfused with either NSC23766 or EHT1864. Our results indicate that pharmacological inhibition of Rac1 by NSC23766 also impairs TBS induced L-LTP (Figure 6A, p<0.05). However perfusion of EHT1864 did not have an effect on TBS induced L-LTP (Figure 6B, p<0.05).
Figure 6. Activation of Rac1 is necessary for TBS induced LTP.

Hippocampal slices were stimulated at CA3 afferents and responses were recorded from area CA1 dendrites. A. Three theta bursts indicated by arrows (one theta burst = five trains consisting of 4–100 Hz pulses, 200 ms intervals with a one minute inter-burst interval) induced TBS-LTP in vehicle-treated (ACSF) control hippocampal slices (●), but was attenuated when delivered in the presence of 30 µM NSC23766 (○) (p<0.05, n=7). B. TBS-LTP was not altered when hippocampal slices were perfused with 60 µM EHT1864 (○). Insets: Sample traces recorded during baseline (dashed lines, a, c) and 2hrs post-TBS (solid lines, b, d). Scale 1mV/5ms.
Another major form of long-term synaptic plasticity in the mammalian brain is long-term depression (LTD). LTD expression involves also formation and elimination of synaptic structures and appears to have also a role in learning and memory. Thus, it is reasonable to hypothesize that LTD-associated structural changes might be mediated by proteins that modulate the remodeling of the actin cytoskeleton, such as Rac1. As for LTP, some forms of LTD are triggered by synaptic activation and dependent on the activation of the NMDA receptor. Consequently, we next examined whether Rac1 is also involved in modulation of LTD by inducing depotentiation in hippocampal slices treated with Rac1 inhibitors. NMDA receptor-dependent LTD was induced by low frequency stimulation (LFS) delivered at 1 Hz for 15 min. This LTD protocol produced a significant lasting decrease in fEPSP slope in control slices perfused with ACSF. However, perfusion of slices with either NSC23766 (30 µM; Figure 7A, p<0.05) or EHT1864 (60 µM; Figure 7B, p<0.05) significantly impaired LTD induction.
Figure 7. Rac1 is associated with long-term depression (LTD).


A, B. LTD was induced in hippocampal slices with LFS delivered at 1 Hz for 15 min. Perfusion of slices with either NSC23766 (30 µM [A]) or EHT1864 (60 µM [B]) altered LTD induction. Insets represent sample traces recorded during baseline (dashed lines, a, c) and 1hr post-LFS (solid lines, b, d). (p<0.05), n=6. Control slices (●, ▲), NSC23766 treated slices (○), EHT1864 treated slices (△). C, D. LTD was induced in hippocampal slices with 100 µM DHPG for 5 min. All experiments were performed in the presence of the N-methyl-D-aspartate inhibitor APV (100 µM). Perfusion of slices with NSC23766 (30 µM [C]) or EHT1864 (60 µM [D]) altered LTD induction. Insets represent sample traces recorded during baseline (dashed lines, a, c) and 1hr post-DHPG (solid lines, b, d). (p<0.05, n=6). Control slices (●), NSC23766 treated slices (○), EHT1864 treated slices (△).
A second major form of LTD requires mGluR receptors. This form of LTD can be chemically induced by application of the group I mGluR agonist 3,5-dihydroxypehnylglycine (DHPG). LTD was induced in hippocampal slices with 100 µM DHPG for 5 min. All experiments were performed in the presence of the N-methyl-D-aspartate inhibitor APV (100 µM). In this case, perfusion of slices with NSC23766 (30 µM) or EHT1864 (60 µM), even though it induced LTD, did not provoke the long-lasting depression of synaptic transmission seen in slices perfused with ACSF and used as controls (Figure 7C and 7D). Our results show that upon pharmacological inhibition of Rac1, mGluR-dependent LTD was altered but at a lower level than the effect reported for NMDA receptor-dependent LTD.
4. DISCUSSION
Rac1 signaling pathway has been implicated in learning (Diana et al., 2007; Haditsch et al., 2009; Martinez et al., 2007), as well as some forms of cognitive disorders and X-linked mental retardation syndromes (Chelly and Mandel, 2001). These cognitive disorders are associated with abnormalities in dendritic spine structure, which is dependent of the actin cytoskeleton regulated by Rac1. Previous finding from studies of animals in which LTP and LTD were impaired as the result of either genetic defects, lesions or pharmacological treatments, suggest that LTP and LTD may be involved in certain types of hippocampal-dependent memory (Nakao et al., 2002; Naie and Manahan-Vaughan 2005; Schmitt et al., 2005). Thus, in this study we focused on elucidating the function of Rac1 in long-term synaptic plasticity. We considered these experiments to be important in view of different psychiatric disorders featuring cognitive impairment, structural deficiencies and aberrant plasticity, where, experimentally, a link between these phenomena still needs to be established. For example, there is strong evidence that mGluR-LTD is involved in the pathogenesis of Fragile X syndrome (FXS), as hippocampal plasticity is abnormal in a mouse model for this cognitive disorder (Huber et al., 2002). These mice, as well as FXS patients also present abnormally long, thin dendritic spines (Comery et al., 1997; Irwin et al., 2000), as well as defects in cognition (Comery et al., 1997; Galvez and Greenough, 2005; Irwin et al., 2000; Purpura, 1974) that could be caused by the aberrant expression of LTD reported. Likewise, alterations in neuroplasticity as well as behavioral abnormalities have been seen in other psychiatric disorders such as depression and schizophrenia (Elhardt et al., 2010).
Our work herein shows that the small GTPase Rac1 has a critical role in long-term synaptic plasticity. We have shown that LTP induction results in rapid Rac1 and PAK1 activation, which can be blocked by the inhibition of NMDA receptor activation. Pharmacological inhibition or Rac1 showed a pronounced deficit in long-term potentiation as well as long-term depression deficit at CA1 neurons, without altering receptor activation and calcium influx (as indicated by unaffected CaMKII), events acting upstream of Rac1 in the induction of long-term plasticity.
Studies by our laboratory and others have employed a genetic approach, investigating synaptic plasticity in Rac1 deficient mice (Haditsch et al., 2009). The results of these studies are in agreement with our pharmacology experiments, suggesting that Rac1 is associated and necessary for the induction of long-term plasticity at CA1 area synapses.
For the first time to our knowledge, two agents that specifically inhibit Rac1 have been used in electrophysiological experiments to study the role of Rac1 in long-term plasticity in the mouse brain. Independent studies showed that their effects were dose dependent and reversible. It is believed that these two drugs affect the activation of Rac1 in different but possibly complementary ways; one by hindering the interaction of Rac1 with its guanine exchange factors (GEFs) and two by unloading GTP from the activated form of Rac1. GEF proteins typically have the highest affinity for unloaded GTPases (Rac1-GDP), thus these two drugs may have competing mechanisms. Theoretically, GEF affinity could be increased by EHT1864 unloading the nucleotide GTP. Alternatively, NSC23766 could have greater affinity for Rac1 after the nucleotide has been unloaded. If this is true, the concomitant use of both inhibitors could produce a higher and prolonged effect regarding Rac1 outcome in synaptic plasticity. Studies are on the way to study possible synergism of these drugs.
ACKNOWLEDGEMENTS
This work was supported by generous grants from the FRAXA Research Foundation, the Jérôme LeJeune Foundation and the National Institutes of Health Grant NS48037 (M.V.T.S). We thank Dr. Lindsay Schwarz for her thoughtful commentaries and help editing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal.Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Caron E. Rac signalling: a radical view. Nat Cell Biol. 2003;5:185–187. doi: 10.1038/ncb0303-185. [DOI] [PubMed] [Google Scholar]
- Chelly J, Mandel JL. Monogenic causes of X-linked mental retardation. Nat. Rev. Gen. 2001;2:669–680. doi: 10.1038/35088558. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana G, Valentini G, Travaglione S, Falzano L, Pieri M, Zona C, Meschini S, Fabbri A, Fiorentini C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA. 2007;104(2):636–641. doi: 10.1073/pnas.0610059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Rho GTPases in growth cone guidance. Curr Opi Neurobiol. 2001;11:103–110. doi: 10.1016/s0959-4388(00)00180-x. [DOI] [PubMed] [Google Scholar]
- Elhardt M, Martinez LA, Tejada-Simon MV. Neurochemical, behavioral and architectural changes after chronic inactivation of NMDA receptors in mice. Neurosci. Lett. 2010;468(2):166–171. doi: 10.1016/j.neulet.2009.10.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am.J. Med. Genet. 2005;135:155–160. doi: 10.1002/ajmg.a.30709. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA. 2004;101(20):7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, Palmer TD. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol. Cell. Neurosci. 2009;41:409–419. doi: 10.1016/j.mcn.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl. Acad. Sci USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Popoff MR, Kojima H, Doussau F, Poulain B. Rac GTPase plays an essential role in exocytosis by controlling the fusion competence of release sites. J Neurosci. 2002;22:7968–7981. doi: 10.1523/JNEUROSCI.22-18-07968.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nature Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Martinez LA, Klann E, Tejada-Simon MV. Translocation and activation of Rac in the hippocampus during associative contextual fear learning. Neurobiol. Learn. Mem. 2007;88(1):104–113. doi: 10.1016/j.nlm.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Naie K, Manahan-Vaughan DP. Pharmacological antagonism of metabotropic glutamate receptor 1 regulates lont-term potentiation and spatial reference memory in the dentate gyrus of freely moving rats via N-methyl-D-aspartate and metabotropic glutamate receptor-dependent mechanisms. Eur. J. Neurosci. 2005;21:411–421. doi: 10.1111/j.1460-9568.2005.03864.x. [DOI] [PubMed] [Google Scholar]
- Nakao K, Ikegaya Y, Yamada MK, Nishiyama N, Natsuki N. Hippocampal long-term depression as an index of satial working memory. Eur. J. Neurosci. 2002;16:970–974. doi: 10.1046/j.1460-9568.2002.02159.x. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Chernoff J. High midsummer for small GTPases. Trends Cell Biol. 2002;12:495–497. doi: 10.1016/s0962-8924(02)02365-6. [DOI] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine "Dysgenesis" and mental retardation. Science. 1974;186:1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001a;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001b;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Schmitt WB, Sprengel R, Mack V, Draft RW, Seeburg PH, Deacon RM, Rawlins JN, Bannerman DM. Restoration of spatial working memory by genetic rescue of GluR-A-deficient mice. Nat. Neurosci. 2005;8:270–272. doi: 10.1038/nn1412. [DOI] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419:475–480. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Villasana LE, Serrano F, Klann E. NMDA receptor activation induces translocation and activation of Rac in mouse hippocampal area CA1. Biochem Bioph Res Comm. 2006;343:504–512. doi: 10.1016/j.bbrc.2006.02.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes & Develop. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T. A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J Biol Chem. 2003;278:567–572. doi: 10.1074/jbc.M207391200. [DOI] [PubMed] [Google Scholar]