

Abstract
Gaucher disease results from the deficiency of the lysosomal enzyme glucocerebrosidase (EC 3.2.1.45). Although the functional gene for glucocerebrosidase (GBA) and its pseudogene (psGBA), located in close proximity on chromosome 1q21, have been studied extensively, the flanking sequence has not been well characterized. The recent identification of human metaxin (MTX) immediately downstream of psGBA prompted a closer analysis of the sequence of the entire region surrounding the GBA gene. We now report the genomic DNA sequence and organization of a 75-kb region around GBA, including the duplicated region containing GBA and MTX. The origin and endpoints of the duplication leading to the pseudogenes for GBA and MTX are now clearly established. We also have identified three new genes within the 32 kb of sequence upstream to GBA, all of which are transcribed in the same direction as GBA. Of these three genes, the gene most distal to GBA is a protein kinase (clk2). The second gene, propin1, has a 1.5-kb cDNA and shares homology to a rat secretory carrier membrane protein 37 (SCAMP37). Finally, cote1, a gene of unknown function lies most proximal to GBA. The possible contributions of these closely arrayed genes to the more atypical presentations of Gaucher disease is now under investigation.
[The sequence data described in this paper have been submitted to the GenBank data library under accession no. AF023268.]
Gaucher disease, the inherited deficiency of the enzyme glucocerebrosidase (EC3.2.1.45), is the most common inherited lysosomal hydrolase deficiency. The gene for glucocerebrosidase (GBA) is located on chromosome 1q21 (Ginns et al. 1985) and is comprised of 11 exons (Horowitz et al. 1989). A highly homologous pseudogene (psGBA) is located nearby (Choudary et al. 1985), and has contributed significantly to the origin of mutations in GBA (Tsuji et al. 1987). Cormand et al. (1997) have recently provided a localization of this region relative to six markers from the Généthon human linkage map. Analyses of the mutations present in patients have revealed both single missense mutations (Beutler and Gelbart 1997) and other recombinant alleles, including several mutations that originate from the pseudogene sequence (Eyal et al. 1990; Latham et al. 1990). Patients have also been described with alleles resulting from a fusion between GBA and psGBA (Zimran et al. 1990).
Many attempts have been made to correlate patient genotypes with the clinical presentation of Gaucher disease. Although there is some predictive value of certain alleles for either mild or severe disease (Zimran et al. 1989; Beutler and Grabowski 1995), no specific symptom complex can be correlated with a unique genotype (Sidransky et al. 1994; NIH Technology Assessment Panel 1996). Based on clinical presentation, Gaucher disease has been divided into three types. Type 1 patients have very heterogeneous presentations, ranging from asymptomatic adults to young children with severe hepatosplenomegaly and bone involvement. Type 2 is invariably fatal, with infants classically developing symptoms at 2–6 months and dying by 2 years of age (Frederickson and Sloan 1972). More recently, the severe phenotype of a knockout mouse model of Gaucher disease (Tybulewicz et al. 1992; Willemsen et al. 1995) prompted the recognition of a subset of severely affected type 2 patients who present and die in the perinatal period (Sidransky et al. 1992). Type 3 includes patients with varying degrees of neurological impairment that develops in childhood or early adulthood.
A recent attempt to generate a point mutation mouse model of Gaucher disease led to the discovery of a novel gene, metaxin (MTX), which in the mouse is contiguous to and transcribed convergently to GBA. MTX shares a bidirectional promoter with the gene for thrombospondin 3 (Thbs3). The insertion of a neomycin resistance cassette in the 3′ flanking region of GBA resulted in a knockout of the murine MTX (Bornstein et al. 1995). Metaxin is a component of the protein translocation apparatus of the mitochondrial outer membrane (Armstrong et al. 1997). Homozygosity for the MTX knockout results in an embryonic lethal phenotype. Human MTX is located downstream of psGBA and a pseudogene for metaxin (psMTX) was subsequently identified downstream of GBA in the intergenic region (Long et al. 1996). The region downstream of psGBA encodes for MTX, Thbs3 (Vos et al. 1992; Adolph et al. 1995), and polymorphic epithelial mucin 1 (Muc1) (Ligtenberg et al. 1990; Vos et al. 1995).
In this study we report the sequence of 75 kb of DNA flanking GBA and establish a more detailed organization of the locus.
RESULTS
The location of λ and plasmid subclones derived from a 410-kb yeast artificial chromosome (YAC) containing the GBA locus is shown in Figure 1. Southern blot analyses of DNA from one patient demonstrated a GBA–psGBA fusion gene and suggested that the functional gene is located 16 kb upstream to the pseudogene (Zimran et al. 1990). We have sequenced the entire intergenic region and confirm that the two sequences are located 16 kb apart. A comparison of sequences upstream to both GBA and psGBA has delineated the duplication that gave rise to psGBA and permitted an approximation of the age of evolution of the duplication.
Figure 1.

Map of clones and subclones on chromosome 1q21. (A) The YAC clone MNG1 was subcloned to cosmid YC11 or YAC MNG4. The λ subclones were derived from MNG4. The sequences of cosmid and λ subclones were compared to the sequences of genes in the GenBank database. The shaded boxes represent the genes at this locus. Genes shown above the line are transcribed left to right; MTX is transcribed in the opposite direction. (psMTX is not transcribed.) (B) Expanded map of λ clones, showing the EcoRI plasmid subclones used for sequencing.
Computer analyses of the 75 kb of sequence were performed using GRAIL (gene recognition analysis internet link) (Uberbacher and Mural 1991) to locate potential exonic regions and BLAST-N (Altschul et al. 1990) to identify sequence homology to other genes. Three genes have been detected in the 32-kb region upstream of glucocerebrosidase. One gene, propin1, has 65% sequence identity to rat secretory carrier membrane protein 37 (SCAMP37) (Brand and Castle 1993), whereas a second gene, cote1, has no close homology to any genes characterized in the GenBank database (Fig. 2A). Primers corresponding to putative exonic sequences identified by GRAIL were used to generate probes for isolating human DNA clones for these two genes from a brain cDNA library. A third gene, which was previously characterized as a protein kinase, clk2, and mapped to chromosome 1 (Hanes et al. 1994), has also been located upstream of GBA. Thus, the GBA locus and its flanking regions consist of at least seven functional genes and two pseudogenes, all of which, except metaxin, are transcribed in the same direction. A BLASTN search for EST homologies revealed segments corresponding to portions of clk2, propin1, and cote1, including several homologous to intronic regions of clk2.
Figure 2.

Schematic representation of the 75 kb of sequence surrounding the GBA gene. (A) Order of the seven genes and two pseudogenes at the locus. (B) Intron/exon organization of clk2, propin1, and cote1. Genes shown in boxes above the line are transcribed left to right. MTX is transcribed right to left.
cote1
cote1, the gene located nearest to GBA, is between 6 and 14 kb upstream of human GBA. Initially a 1.8-kb cDNA for cote1 was isolated from a human hippocampal library. Hybridization of this cDNA to a human multiple tissue Northern blot (Clontech) revealed a 3.0- to 3.4-kb transcript in eight different tissues (Fig. 3). In addition, an ∼6-kb transcript was present in brain tissue. A Northern blot of eight brain regions (Clontech) indicated that the larger transcript was only present in the cerebellum. Reexamination of the human genomic sequence farther upstream of the 1.8-kb cDNA revealed additional potential exons. Although the entire cDNA is predicted to be ∼3.15 kb and to contain 12 exons (Fig. 2B), no full-length cote1 human cDNA has been obtained.
Figure 3.
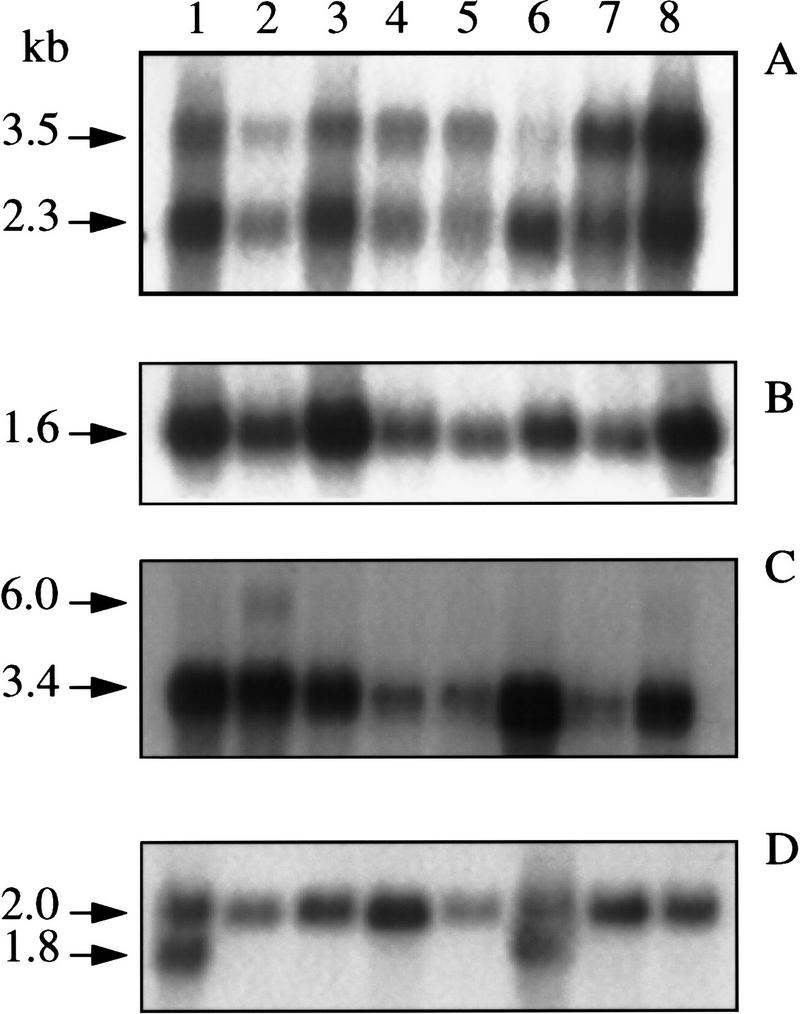
Human multitissue Northern blot hybridized to clk2 cDNA (A), propin1 cDNA (B), cote1 cDNA (C), and β-actin cDNA (D). Lanes 1–8 are heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas, respectively.
propinl
GRAIL analyses of the original YAC sequence initially identified two exons of a second upstream gene, propin1, and probes from these exons were used to isolate a cosmid clone that extended farther upstream. A 1.5-kb cDNA, corresponding to a genomic sequence spanning ∼6.4 kb in length and composed of nine exons (Fig. 2B), was isolated from a pSport plasmid human brain library, using exon-derived PCR-amplified probes. RACE (rapid amplification of cDNA ends) PCR placed the 5′ end of the transcript at least 68 bp farther upstream than the longest isolated cDNA clone. propin1 is located between 15 and 21 kb upstream of GBA and has 65.9% and 54.9% identity to the rat SCAMP37 cDNA-coding region and amino acid sequence, respectively (Brand and Castle 1993). Because rat SCAMP37 belongs to a family of proteins, the relatively low sequence homology suggests that the human gene may be orthologous to another member of the group.
Northern blot analyses using a human propin1 cDNA probe demonstrated a 1.5- to 1.6-kb transcript in eight human tissues (Fig. 3) and in eight brain regions (Clontech). There is a SP1-binding element at the beginning of exon 1 and two in intron 1. The 3′ end of the propin1 transcript and the 5′ end of cote1 are ∼500 bp apart.
Species Conservation
To determine the evolutionary conservation of these two new genes, the human cDNAs were hybridized to a Southern blot containing DNA from various vertebrates and budding yeast. Most of the exons of both human propin1 and cote1 are represented by a single EcoRI restriction fragment that is about the same size for each probe (data not shown). The entire region containing propin1, cote1, and GBA is syntenic in mouse and human (S. Winfield, unpubl.). These findings reflect a conservation of the organization of propin1, cote1, and GBA and their proximity across species.
clk2 kinase
A BLAST search of the sequence 5′ to propin1 identified a third gene, clk2 kinase (Hanes et al. 1994). The 13 exons of this gene are located between 21.5 and 32 kb 5′ to GBA (Fig. 2B). This gene, as well as propin1 and cote1, is transcribed in the same orientation as GBA. The 5′ end of clk2 kinase is within a CpG island, whereas the 3′ end is <600 bp upstream of propin1.
When a clk2 kinase probe, generated by PCR amplification of human brain cDNA using primers from exons 5 and 13, was hybridized to a multiple tissue Northern blot, bands of 2.3 and 3.5 (Fig. 3) were observed in each lane. The relative intensity of the two mRNAs varied from tissue to tissue (Figure 3). These transcripts differed in size from those isolated from leukocyte mRNA as described by Hanes et al. (1994).
Duplication of GBA and MTX
A duplication occurred at the human GBA locus, giving rise to pseudogenes for glucocerebrosidase and metaxin. The identification of the endpoints of the original duplication and an estimate of the age of the duplication were obtained from a comparison of the sequence in the intergenic region between GBA and psGBA with the sequence upstream of GBA.
Alignment of >4 kb of sequence downstream of both GBA and psGBA exhibited >97% homology. This region corresponds to most of MTX and psMTX (Fig. 2). The 3′ end of the duplication is within exon 2 of metaxin. Although psMTX is missing all of exon 1 and part of exon 2, the remaining exonic sequences of MTX andpsMTX show 99% homology.
The duplicated region, which extends ∼5.5 kb 5′ to GBA and 11.8 kb upstream of psGBA, can be aligned with 88% homology if several Alu inserts are removed from each region and if a 6.1-kb segment is removed from the 5′ flanking sequence of the pseudogene (Fig. 4). The 5′ end of this homology reflects the endpoint of the duplication and coincides with an Alu sequence poly(A) tail.
Figure 4.

Schematic illustration of the alignment of 5′ flanking regions of GBA and psGBA. Arrows represent Alu sequences. A large insert of 6113 bp contains eight complete Alu sequences and several partial Alu sequences. The remaining sequence has been aligned with 88% identity.
Other Features of the GBA Locus
There are at least 50 Alu sequences within the 75 kb of DNA characterized at this locus, with 15 complete and several partial Alu sequences in the 16 kb between GBA and psGBA (Fig. 4). Many of these Alu sequences have been inserted into the intergenic region and into introns of GBA (Horowitz et al. 1989) following the duplication event. Both a dinucleotide and a tetranucleotide repeat sequence have been identified in the regions flanking GBA.
DISCUSSION
Sequence analysis of the region surrounding the GBA gene reveals three genes located within 32 kb upstream of GBA. The physical relationship between GBA and psGBA, and between MTX and psMTX, as well as a delineation of the duplication that gives rise to the pseudogenes for both GBA and MTX, has been established. This region on chromosome 1q21 contains seven functional genes and two pseudogenes.
The presence of these closely clustered genes may have important implications for the study of patients with Gaucher disease. To date, the analysis of mutations in the GBA gene has not been adequate to explain the vast phenotypic heterogeneity encountered in the disorder. It is possible that there may be modifying genes located outside of the GBA region. However, for some time it has been appreciated that some mutant GBA alleles arose from recombination or fusion events occurring at the locus. The possibility that flanking genes may also be altered during these recombination events is particularly intriguing.
Alterations in genes near GBA could potentially contribute to the phenotypic heterogeneity observed in Gaucher patients, as has been seen in Hunter Syndrome, where patients with atypical symptoms have deletions encompassing portions of other genes near the human iduronate-2-sulfatase locus (Timms et al. 1997).
The delineation of the limits of the recombination event giving rise to the psGBA and psMTX pseudogenes, described by Long et al. (1996), was based on the homology on the 3′ ends of GBA and psGBA. Based on the alignment of the 5′ flanking sequences of GBA and psGBA, an alternative site for this recombination located 24 bp farther 5′ is also possible.
Alignment of the duplicated regions permits an estimate of the timing of the duplication. Because both the human and the rhesus monkey contain the duplication, we suggest that it occurred prior to the divergence of the Great Apes and Old World monkeys 25 million years ago. Based on Miyamoto’s calculation of time since divergence of primate species, the duplication event occurred ∼36–48 million years ago (Miyamoto et al. 1988). Because the Alu sequence present in both copies of the duplication is a member of the Sx family, which is ∼40 million years old (Makalowski et al. 1994), the duplication event occurred no more than 40 million years ago. The recombination events at the GBA locus represent another example of regional duplications occurring during human evolution that can contribute to human pathology (Timms et al. 1997).
Transcripts from all three of the newly identified genes upstream of GBA are observed in all tissues studied (Fig. 3). cote1, the gene nearest to GBA, lacks homology to any known gene, and its function is not known. The largest open reading frame encodes 669 amino acids with two potential N-glycosylation sites, a leucine zipper, and multiple potential phosphorylation sites and N-myristoylation sites. Because of the close proximity of cote1 to GBA, studies of its expression in patients with Gaucher disease are a priority.
Although propin1 has some homology to SCAMP37, its function is also unknown. It retains specific motifs observed in the rat SCAMP37 amino acid sequence, including an α-helical leucine zipper and one zinc finger domain. SCAMPs are components of post-Golgi membranes that function as recycling carriers to the cell surface. They have broad tissue distribution, subcellular localization, and are present in multiple pathways. If propin1 does encode a SCAMP, it is intriguing to speculate that such a protein could be involved in the intracellular transport of lysosomal glucocerebrosidase.
The human clk2 kinase gene that we have identified within this region had previously been localized to chromosome 1 and has characteristics of a serine/threonine kinase. In our study, Northern blot analyses of human tissues clearly demonstrate two transcripts hybridizing to a clk2 kinase cDNA probe, but it is not clear whether the larger species is an alternatively spliced clk2 transcript or results from the cross hybridization of another protein.
Our current study demonstrates that the region surrounding the GBA is far more complex than previously appreciated, containing seven genes and two pseudogenes. Transgenic and knockout mice are being generated in hope that they may give insight into the function of cote1, propin1, and clk2. Common locus control, regulatory elements, or overlapping transcripts could coordinately affect expression of several of these genes and, in the case of Gaucher disease, may influence the expression level of glucocerebrosidase. The potential involvement of contiguous gene effects in explaining the more unusual phenotypes encountered among Gaucher patients requires further study.
METHODS
Isolation of Genomic Clones
The Center for Genetics in Medicine (CGM) YAC library (Burke and Olson 1991) was screened by PCR amplification of ordered arrays of pooled clones (Green and Olson 1990) using a primer pair specific for the human GBA gene. A YAC clone, CGY2981, contained both GBA and psGBA. A his3 mutation was introduced into the yeast from strain MB11, and the his5 mutation removed by standard mating procedures (Sherman 1991). The resulting strain, MNG1, was transformed with fragmentation vectors pBP108 and pBP109 (Pavan et al. 1991) to reduce the insert size. One of the derivatives, MNG4, with an insert of ∼85 kb, was confirmed to contain both GBA and psGBA and, following partial Sau3AI digestion, was subcloned to Lambda Dash (Stratagene, La Jolla, CA). The full-length YAC clone, MNG1, following partial Sau3AI digestion, was also subcloned into SuperCos (Stratagene) .
Several λ subclones that hybridized to a human 1.6-kb GBA cDNA were subcloned into pGEM7zf+ (Promega, Madison, WI) for sequencing. Plasmid subclones were ordered using sequence across the junction points in the λ DNA. A cosmid subclone of the YAC containing 16 kb of DNA upstream of GBA was also subcloned to pGEM7zf+ and pGEM11zf+ for sequencing (Fig. 1).
Complementary DNA Clones
A human hippocampal cDNA library (Lambda ZapII) was obtained from Stratagene. The human brain plasmid library in pCMV–SPORT was from Life Technologies (Gaithersburg, MD).
RACE PCR was used to determine the 5′ ends of cDNAs. PCR amplification was carried out using human brain cDNA ligated to an anchor (Clontech) or on cDNA isolated from the pSPORT human brain library, with anchor or vector primers and nested gene-specific primers.
DNA Sequence Analysis
Plasmid and cosmid DNA were prepared by either standard alkaline lysis (Maniatis et al. 1982), Perfect Prep kits (5 Prime → 3 Prime, Boulder, CO), or in some cases by CsCl gradient purification. Sequencing was performed on alkaline-denatured, double-stranded template using the Sequenase Reagent kit (U.S. Biochemical, Inc., Cleveland, OH) or on λ subclones using the Circumvent ThermoCycle Dideoxy DNA Sequencing kit (New England Biolabs, Beverly, MA) according to manufacturer’s protocols. PCR products, cosmids, and subclones were sequenced using a Perkin-Elmer Applied Biosystems model 373A automated sequencer with the FS enzyme system and dye terminator chemistry according to the manufacturer’s protocols (Perkin Elmer, Foster City, CA). Oligonucleotides were synthesized on an Expedite (Perseptive Biosystems, Framingham, MA) or Cyclone Synthesizer (Milligen Biosearch, Milford, MA) and used without purification. All sequence was determined in both directions, entered into PCGene, and analyzed by GRAIL (Uberbacher and Mural 1991) to determine potential coding areas. A BLAST-N (Altschul et al. 1990) comparison of putative exon sequences with the GenBank database was used to identify homologies. The BLAST-X (Altschul et al. 1990) program was also used to compare the predicted amino acid sequence to the protein database.
Northern and Zooblot Hybridization
Multiple human tissue Northern blots and multiple species (Zoo) blots were obtained from Clontech Laboratories. Hybridization was carried out according to the manufacturer’s protocol using cDNA from clk2 kinase, propin1, cote1, and β-actin as probes.
Acknowledgments
We thank Collaborative Research, Inc., for providing the YAC clone, Dr. Roger Reeves for the YAC fragmenting vectors, and Dr. Miles Brennan for the yeast strain MB11. We also acknowledge Dr. Wojciech Makalowski for his help with Alu characterization, and Elizabeth Alzona and Kay Kuhns for their assistance in the preparation of this manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sidranse@irp.nimh.nih.gov; FAX (301) 402-6438.
REFERENCES
- Adolph KW, Long GL, Winfield S, Ginns EI, Bornstein P. Structure and organization of the human thrombospondin 3 gene (Thbs3) Genomics. 1995;27:329–336. doi: 10.1006/geno.1995.1050. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Armstrong LC, Komiya T, Bergman BE, Mihara K, Bornstein P. Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J Biol Chem. 1997;272:6510–6518. doi: 10.1074/jbc.272.10.6510. [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. 1997;23:2–7. doi: 10.1006/bcmd.1997.0114. [DOI] [PubMed] [Google Scholar]
- Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basis of inherited disease. New York, NY: McGraw-Hill; 1995. pp. 2641–2670. [Google Scholar]
- Bornstein P, McKinney CE, LaMarca ME, Winfield S, Shingu T, Devarayalu S, Vos HL, Ginns EI. Metaxin, a gene contiguous to both thrombospondin 3 and glucocerebrosidase, is required for embryonic development in the mouse: Implications for Gaucher disease. Proc Natl Acad Sci. 1995;92:4547–4551. doi: 10.1073/pnas.92.10.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand SH, Castle JD. SCAMP 37, a new marker within the general cell surface recycling system. EMBO J. 1993;12:3753–3761. doi: 10.1002/j.1460-2075.1993.tb06053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DT, Olson MV. Preparation of clone libraries in yeast artificial-chromosome vectors. In: Guthrie C, Fink GR, editors. Methods in enzymology. Guide to yeast genetics and molecular biology. San Diego, CA: Academic Press; 1991. pp. 251–270. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Ginns EI, Barranger JA. Molecular cloning and analysis of the human β-glucocerebrosidase gene. DNA. 1985;4:74. [Google Scholar]
- Cormand B, Montfort M, Chabás L, Vilageliu L, Grinberg D. Genetic fine localization of the β-glucocerebrosidase (GBA) and prosaposin (PSAP) genes: Implications for Gaucher disease. Hum Genet. 1997;100:75–79. doi: 10.1007/s004390050468. [DOI] [PubMed] [Google Scholar]
- Eyal N, Wilder S, Horowitz M. Prevalent and rare mutations among Gaucher patients. Gene. 1990;96:277–283. doi: 10.1016/0378-1119(90)90264-r. [DOI] [PubMed] [Google Scholar]
- Frederickson DS, Sloan HR. Glucosylceramide lipidoses: Gaucher’s disease. In: Stanbury JB, Wyngarden JB, Frederickson DS, editors. The metabolic basis of inherited disease. New York, NY: McGraw-Hill; 1972. pp. 730–759. [Google Scholar]
- Ginns EI, Choudary PV, Tsuji S, Martin B, Stubblefield B, Sawyer J, Hozier J, Barranger JA. Gene mapping and leader polypeptide sequence of human glucocerebrosidase: Implications for Gaucher disease. Proc Natl Acad Sci. 1985;82:7101–7105. doi: 10.1073/pnas.82.20.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ED, Olson MV. Systematic screening of yeast artificial-chromosome libraries by use of the polymerase chain reaction. Proc Natl Acad Sci. 1990;87:1213–1217. doi: 10.1073/pnas.87.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes JJ, von der Kammer H, Klaudiny JJ, Scheit KH. Characterization by cDNA cloning of two new human protein kinases: Evidence by sequence comparison for a new family of mammalian protein kinases. J Mol Biol. 1994;244:665–672. doi: 10.1006/jmbi.1994.1763. [DOI] [PubMed] [Google Scholar]
- Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics. 1989;4:87–96. doi: 10.1016/0888-7543(89)90319-4. [DOI] [PubMed] [Google Scholar]
- Latham T, Grabowski GA, Theophilus BDM, Smith FI. Complex alleles of the acid β-glucocerebrosidase gene in Gaucher disease. Am J Hum Genet. 1990;47:79–86. [PMC free article] [PubMed] [Google Scholar]
- Ligtenberg MJL, Vos HL, Gennissen MC, Hilkens J. Episialin, a carcinoma-associated mucin, is generated by a polymorphic gene encoding splice variants with alternative amino termini. J Biol Chem. 1990;265:5573–5578. [PubMed] [Google Scholar]
- Long GL, Winfield S, Adolph KW, Ginns EI, Bornstein P. Structure and organization of the human metaxin gene (MTX) and pseudogene. Genomics. 1996;33:177–184. doi: 10.1006/geno.1996.0181. [DOI] [PubMed] [Google Scholar]
- Makalowski W, Mitchell GA, Labuda D. Alu sequences in the coding regions of mRNA: A source of protein variability. Trends Genet. 1994;10:188–193. doi: 10.1016/0168-9525(94)90254-2. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- Miyamoto MM, Koop BF, Slightom JL, Goodman M, Tennant MR. Molecular systematics of higher primates: Genealogical relations and classification. Proc Natl Acad Sci. 1988;85:7627–7631. doi: 10.1073/pnas.85.20.7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH Technology Assessment Panel on Gaucher Disease. Gaucher disease: Current issues in diagnosis and treatment. J Amer Med Assoc. 1996;275:548–553. [PubMed] [Google Scholar]
- Pavan WJ, Hieter P, Sears D, Burkhoff A, Reeves RH. High-efficiency yeast artificial chromosome fragmentation vectors. Gene. 1991;106:125–127. doi: 10.1016/0378-1119(91)90576-w. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. In: Guthrie C, Fink GR, editors. Methods in enzymology. Guide to yeast genetics and molecular biology. San Diego, CA: Academic Press; 1991. pp. 3–21. [Google Scholar]
- Sidransky E, Sherer DM, Ginns EI. Gaucher disease in the neonate: A distinct Gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr Res. 1992;32:494–498. doi: 10.1203/00006450-199210000-00023. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Bottler A, Stubblefield B, Ginns EI. DNA mutational analysis of type 1 and type 3 Gaucher patients: How well do mutations predict phenotype? Hum Mutat. 1994;3:25–28. doi: 10.1002/humu.1380030105. [DOI] [PubMed] [Google Scholar]
- Timms KM, Bondeson M-L, Ansari-Lari MA, Lagerstedt K, Muzny DM, Dugan-Rocha SP, Nelson DL, Pettersson U, Gibbs RA. Molecular and phenotypic variation in patients with severe Hunter syndrome. Hum Mol Genet. 1997;6:479–486. doi: 10.1093/hmg/6.3.479. [DOI] [PubMed] [Google Scholar]
- Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, Ginns EI. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316:570–575. doi: 10.1056/NEJM198703053161002. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VLJ, Tremblay ML, LaMarca ME, Willemsen R, Stubblefield B, Winfield S, Zablocka B, Sidransky E, Martin BM, Huang SP, Mintzer KA, Westphal H, Mulligan RC, Ginns EI. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature. 1992;357:407–410. doi: 10.1038/357407a0. [DOI] [PubMed] [Google Scholar]
- Uberbacher EC, Mural RJ. Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proc Natl Acad Sci. 1991;88:11261–11265. doi: 10.1073/pnas.88.24.11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos HL, Devarayalu S, de Vries Y, Bornstein P. Thrombospondin 3 (Thbs3), a new member of the thrombospondin gene family. J Biol Chem. 1992;267:12192–12196. [PubMed] [Google Scholar]
- Vos HL, Mockensturm-Wilson M, Rood PML, Maas AMCE, Duhig T, Gendler SJ, Bornstein P. A tightly organized conserved gene cluster on mouse chromosome 3 (E3-F1) Mamm Genome. 1995;6:820–822. doi: 10.1007/BF00539013. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Tybulewicz V, Sidransky E, Eliason WK, Martin BM, LaMarca ME, Reuser AJJ, Tremblay M, Westphal H, Mulligan RC, Ginns EI. A biochemical and ultrastructural evaluation of the type 2 Gaucher mouse. Mol Chem Neuropath. 1995;24:179–192. doi: 10.1007/BF02962142. [DOI] [PubMed] [Google Scholar]
- Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E. Prediction of severity of Gaucher’s disease by identification of mutations at DNA level. Lancet. 1989;2:349–352. doi: 10.1016/s0140-6736(89)90536-9. [DOI] [PubMed] [Google Scholar]
- Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E. A glucocerebrosidase fusion gene in Gaucher disease. Implications for the molecular anatomy, pathogenesis, and diagnosis of this disorder. J Clin Invest. 1990;85:219–222. doi: 10.1172/JCI114415. [DOI] [PMC free article] [PubMed] [Google Scholar]