

Abstract
Alu insertion polymorphisms (polymorphisms consisting of the presence/absence of an Alu element at a particular chromosomal location) offer several advantages over other nuclear DNA polymorphisms for human evolution studies. First, they are typed by rapid, simple, PCR-based assays; second, they are stable polymorphisms—newly inserted Alu elements rarely undergo deletion; third, the presence of an Alu element represents identity by descent—the probability that different Alu elements would independently insert into the exact same chromosomal location is negligible; and fourth, the ancestral state is known with certainty to be the absence of an Alu element. We report here a study of 8 loci in 1500 individuals from 34 worldwide populations. African populations exhibit the most between-population differentiation, and the population tree is rooted in Africa; moreover, the estimated effective time of separation of African versus non-African populations is 137,000 ± 15,000 years ago, in accordance with other genetic data. However, a principal coordinates analysis indicates that populations from Sahul (Australia and New Guinea) are nearly as close to the hypothetical ancestor as are African populations, suggesting that there was an early expansion of tropical populations of our species. An analysis of heterozygosity versus genetic distance suggests that African populations have had a larger effective population size than non-African populations. Overall, these results support the African origin of modern humans in that an earlier expansion of the ancestors of African populations is indicated.
The Alu family of short interspersed elements is one of the most successful mobile genetic elements, having arisen to a copy number in excess of 500,000 within primate genomes in the last 65 million years (for recent reviews, see Okada 1991; Schmid and Maraia 1992; Deininger and Batzer 1993, 1995). Alu repeats are thought to be ancestrally derived from the 7SL RNA gene and mobilize through an RNA polymerase III-derived transcript in a process termed retroposition. Each Alu sequence is ∼300bp in length; therefore, Alu repeats comprise ∼5% of the human genome.
Alu sequences can be divided into different subfamilies or clades of related elements based on commonly shared diagnostic mutations. Here, we use the new standardized nomenclature to refer to various Alu subfamilies (Batzer et al. 1996a). Two of the most recently formed subfamilies of Alu elements within the human genome have been termed Ya5 and Ya8 (Batzer et al. 1990; Batzer and Deininger 1991). Members of the Ya8 Alu subfamily are characterized by all five of the Ya5 diagnostic mutations, as well as three additional diagnostic mutations. Because both the Ya5 and Ya8 Alu subfamilies share a number of diagnostic mutations we refer to this lineage collectively as Ya5/8 (Batzer et al. 1996b). The Ya5/8 Alu subfamily lineage is comprised of 500–2000 elements that are restricted to the human genome (Arcot et al. 1996), although a few Ya5 Alu family members have been found in chimpanzees (for review, see Deininger and Batzer 1995). In parallel, a second subfamily that is an independent derivative of the Y lineage of Alu sequences, termed Yb8, has also expanded to ∼500 copies within the human genome (Batzer et al. 1995, 1996a).
Some Ya5/8 and Yb8 Alu elements have retroposed so recently that they are polymorphic for presence/absence at a specific location within the human genome (Batzer and Deininger 1991; Batzer et al. 1991, 1995; Kass et al. 1994; Hammer 1995). The distribution of these elements varies in different human population groups (Batzer and Deininger 1991; Batzer et al. 1991, 1994, 1995, 1996b; Perna et al. 1992; Kass et al. 1994; Hammer 1995). These polymorphic Ya5/8 and Yb8 Alu insertions serve as a unique set of nuclear DNA markers for the study of human evolution, as they are stable polymorphisms that are identical by descent. In addition, the ancestral state of each Alu insertion is known, facilitating accurate rooting of population networks (Batzer et al. 1994).
Previously, we have reported on 4 Alu insertion polymorphisms in 16 worldwide populations; here, we analyze the distribution of eight polymorphic Alu insertions in a survey of 1500 individuals from 34 worldwide population groups. Our results indicate that these polymorphic Alu insertions have an African origin, although populations from Sahul (Australia and New Guinea) are also close to the hypothetical ancestral population, possibly indicating an early expansion of human populations in the tropics. Using a model specific for Alu insertion polymorphisms, we estimate that the effective separation time between African and non-African populations was relatively recent, in accordance with estimates from other genetic data. We also find that African populations probably have had a larger effective population size than non-African populations; overall, our results are in agreement with an earlier expansion of populations in Africa, which corresponds to an African origin of modern humans.
RESULTS AND DISCUSSION
Population Statistics
An average of 1500 individuals from 34 worldwide populations (Fig. 1) were typed for each of the 8 Alu insertion polymorphisms (Table 1). The allele frequencies and sample sizes for each population are shown in Table 2. All loci were polymorphic in all populations, with the exception of D1 in Nigerians, FXIIIB in CAR Pygmies, and A25 in the Moluccans, which were all fixed for the absence of the Alu element (Table 2). Significant departures from Hardy–Weinberg equilibrium expectations were observed for 10 of 269 comparisons. Because ∼13 comparisons would be expected to be significant at the 5% level by chance alone and because none of the significant departures cluster by locus or by population, we consider these to represent normal statistical fluctuations. The probabilities of these χ2 outliers range from Pr = 0.04 to Pr = 0.003.
Figure 1.
Map of sample localities.
Table 1.
Population Statistics for 8 Alu Insertion Polymorpisms in 34 Populations
| a. Loci | |||
| Locus | No. of individuals | Heterozygosity (±s.e.)a | Gst |
|---|---|---|---|
| TPA25 | 1525 | 0.485 ± 0.003 | 0.079 |
| PV92 | 1432 | 0.490 ± 0.003 | 0.212 |
| APO | 1501 | 0.267 ± 0.009 | 0.066 |
| ACE | 1528 | 0.493 ± 0.002 | 0.092 |
| FXIIIB | 1396 | 0.494 ± 0.002 | 0.282 |
| D1 | 1434 | 0.405 ± 0.007 | 0.075 |
| A25 | 1593 | 0.275 ± 0.009 | 0.061 |
| B65 | 1593 | 0.497 ± 0.001 | 0.089 |
| Average | 1500 | 0.426 ± 0.035 | 0.128 |
| b. Geographic regions | ||||
| Regionb | No. of populations | No. of individuals | Heterozygosity (±s.e.)a | Gst |
|---|---|---|---|---|
| Africa | 6 | 176 | 0.402 ± 0.030 | 0.088 |
| Europe | 7 | 334 | 0.396 ± 0.052 | 0.011 |
| Western Asia | 7 | 262 | 0.414 ± 0.041 | 0.036 |
| Southeast Asia | 7 | 359 | 0.377 ± 0.042 | 0.058 |
| Sahul | 3 | 185 | 0.308 ± 0.039 | 0.001 |
| Americas | 4 | 184 | 0.381 ± 0.049 | 0.026 |
| World | 34 | 1500 | 0.426 ± 0.035 | 0.128 |
Mean of the population heterozygosites.
As indicated by color in Fig. 1.
Table 2.
Alu Insertion Frequencies in 34 Human Populations
| Na | ACE | TPA25 | PV92 | APO | FXIIIB | D1 | A25 | B65 | |
|---|---|---|---|---|---|---|---|---|---|
| Alaska Natives | 41 | 0.58 | 0.30 | 0.62 | 0.92 | 0.92 | 0.42 | 0.15 | 0.45 |
| Australia | 69 | 0.91 | 0.13 | 0.15 | 0.87 | 0.65 | 0.04 | 0.35 | 0.39 |
| Bretons | 54 | 0.48 | 0.56 | 0.27 | 0.90 | 0.40 | 0.39 | 0.16 | 0.56 |
| China | 49 | 0.67 | 0.35 | 0.86 | 0.82 | 0.71 | 0.17 | 0.10 | 0.35 |
| European–American | 57 | 0.51 | 0.56 | 0.18 | 0.94 | 0.47 | 0.44 | 0.20 | 0.56 |
| Filipino | 47 | 0.53 | 0.63 | 0.80 | 0.98 | 0.72 | 0.36 | 0.14 | 0.57 |
| French | 53 | 0.48 | 0.56 | 0.23 | 0.99 | 0.42 | 0.46 | 0.16 | 0.57 |
| French Acadian | 46 | 0.51 | 0.43 | 0.18 | 0.92 | 0.48 | 0.42 | 0.12 | 0.53 |
| Greek Cypriot | 48 | 0.39 | 0.53 | 0.25 | 0.95 | 0.62 | 0.27 | 0.12 | 0.65 |
| Greenland Natives | 41 | 0.55 | 0.33 | 0.61 | 0.94 | 0.79 | 0.45 | 0.17 | 0.19 |
| India–Christian | 27 | 0.60 | 0.57 | 0.48 | 0.67 | 0.61 | 0.28 | 0.14 | 0.31 |
| India–Hindu | 28 | 0.52 | 0.34 | 0.52 | 0.85 | 0.66 | 0.10 | 0.05 | 0.35 |
| India–Muslim | 26 | 0.52 | 0.41 | 0.30 | 0.86 | 0.66 | 0.32 | 0.12 | 0.40 |
| Java | 32 | 0.86 | 0.39 | 0.84 | 0.78 | 0.92 | 0.42 | 0.06 | 0.58 |
| !Kung | 40 | 0.29 | 0.17 | 0.20 | 0.88 | 0.17 | 0.16 | 0.61 | 0.50 |
| Malaysian | 47 | 0.64 | 0.50 | 0.72 | 0.76 | 0.73 | 0.27 | 0.02 | 0.42 |
| Mayan | 51 | 0.68 | 0.65 | 0.79 | 0.94 | 0.90 | 0.45 | 0.21 | 0.27 |
| Moluccan | 48 | 0.67 | 0.56 | 0.69 | 0.76 | 0.78 | 0.19 | 0.00 | 0.26 |
| Mvskoke | 50 | 0.70 | 0.49 | 0.57 | 0.96 | 0.79 | 0.46 | 0.21 | 0.48 |
| Nguni | 43 | 0.40 | 0.21 | 0.24 | 0.60 | 0.12 | 0.27 | 0.41 | 0.60 |
| Nigerian | 11 | 0.27 | 0.41 | 0.09 | 0.50 | 0.08 | 0.00 | 0.22 | 0.83 |
| Pakistan | 42 | 0.44 | 0.51 | 0.30 | 0.72 | 0.23 | 0.17 | 0.07 | 0.37 |
| PNG–Coastal | 48 | 0.66 | 0.16 | 0.36 | 0.66 | 0.30 | 0.17 | 0.02 | 0.27 |
| PNG–Highland | 68 | 0.74 | 0.16 | 0.24 | 0.68 | 0.30 | 0.01 | 0.04 | 0.18 |
| Pushtoon | 50 | 0.52 | 0.55 | 0.33 | 0.86 | 0.57 | 0.27 | 0.18 | 0.49 |
| Pygmy–CAR | 17 | 0.12 | 0.21 | 0.26 | 0.74 | 0.00 | 0.47 | 0.35 | 0.78 |
| Pygmy–Zaire | 17 | 0.32 | 0.24 | 0.35 | 0.85 | 0.03 | 0.59 | 0.53 | 0.82 |
| Sotho | 48 | 0.38 | 0.33 | 0.29 | 0.68 | 0.18 | 0.31 | 0.39 | 0.48 |
| Swiss | 43 | 0.37 | 0.45 | 0.20 | 0.94 | 0.48 | 0.34 | 0.12 | 0.58 |
| Taiwan | 46 | 0.50 | 0.64 | 0.90 | 0.93 | 0.97 | 0.38 | 0.22 | 0.54 |
| Tamill | 47 | 0.69 | 0.56 | 0.56 | 0.81 | 0.61 | 0.34 | 0.17 | 0.55 |
| Tenggaras | 90 | 0.64 | 0.38 | 0.50 | 0.78 | 0.81 | 0.19 | 0.05 | 0.40 |
| Turkish Cypriot | 33 | 0.33 | 0.58 | 0.33 | 0.98 | 0.39 | 0.35 | 0.09 | 0.64 |
| UAE | 42 | 0.33 | 0.44 | 0.30 | 0.97 | 0.39 | 0.08 | 0.12 | 0.41 |
(N) Number of individuals.
The average heterozygosity for each locus was substantial, with several values approaching the theoretical maximum heterozygosity of 0.5 for a biallelic locus (Table 1a). Although we expected that heterozygosity would be high, because these loci were ascertained on the basis that they were known to be polymorphic, an analysis of the distribution of allele frequencies at loci ascertained in a similar manner indicates that ascertainment bias alone does not completely account for the observed frequency spectrum; the distributions also contain information on the demographic history of human populations (Sherry et al. 1997).
The Gst values (a measure of the amount of subpopulation differentiation) were also substantial (Table 1a), and were statistically significant for each locus by a χ2 contingency analysis (not shown), indicating that there are significant differences in the frequency of the Alu element across these human populations at each of these eight loci. The average Gst value was 0.128, comparable to other studies of nuclear DNA polymorphisms in human populations (Nei and Roychoudhury 1982; Jorde et al. 1995), and means that ∼13% of the total variance in allele frequency differences at these eight loci is found between populations and 87% is found within populations.
For comparing patterns of variation among populations, we grouped the 34 populations into 6 geographic regions, consisting of Africa, Europe, Western Asia, Southeast Asia, Sahul (Australia and New Guinea), and the Americas (Table 1b). The average heterozygosity for each region was substantial, ranging from a low of 0.308 in Sahul to a high of 0.414 in Western Asia (Table 1b). The Gst value was calculated for the differentiation among the subpopulations within each region and was greatest in Africa (Table 1b). With respect to these loci, African populations show the most between-population diversity of any geographic region of the world, although it should be kept in mind that this finding is based on just eight loci.
Population Relationships
We used both tree reconstruction and principal coordinates (PC) analysis to investigate population relationships. Tree reconstruction methods depict population relationships as a series of bifurcations, which are commonly interpreted as representing population splits; however, it is important to realize that clusters of populations in such trees could arise from migration instead of from shared ancestry. A neighbor-joining tree depicting the population relationships (Fig. 2) shows some concordance with geography. All of the African populations cluster together, as do the Sahulian and the European populations. The Southeast Asian and American populations are intermingled in one cluster. This may reflect inadequate sampling of populations from the Americas in this study, as a more extensive study of 5 of these loci in 24 native American populations does show more clustering of native American populations separate from Southeast Asian populations (Novick et al. 1997). The Western Asian populations are not clustered together on the tree, which could reflect either genetic contributions from both eastern Asia and Europe to these geographically intermediate populations, or an inability to accurately resolve population relationships with just eight loci.
Figure 2.
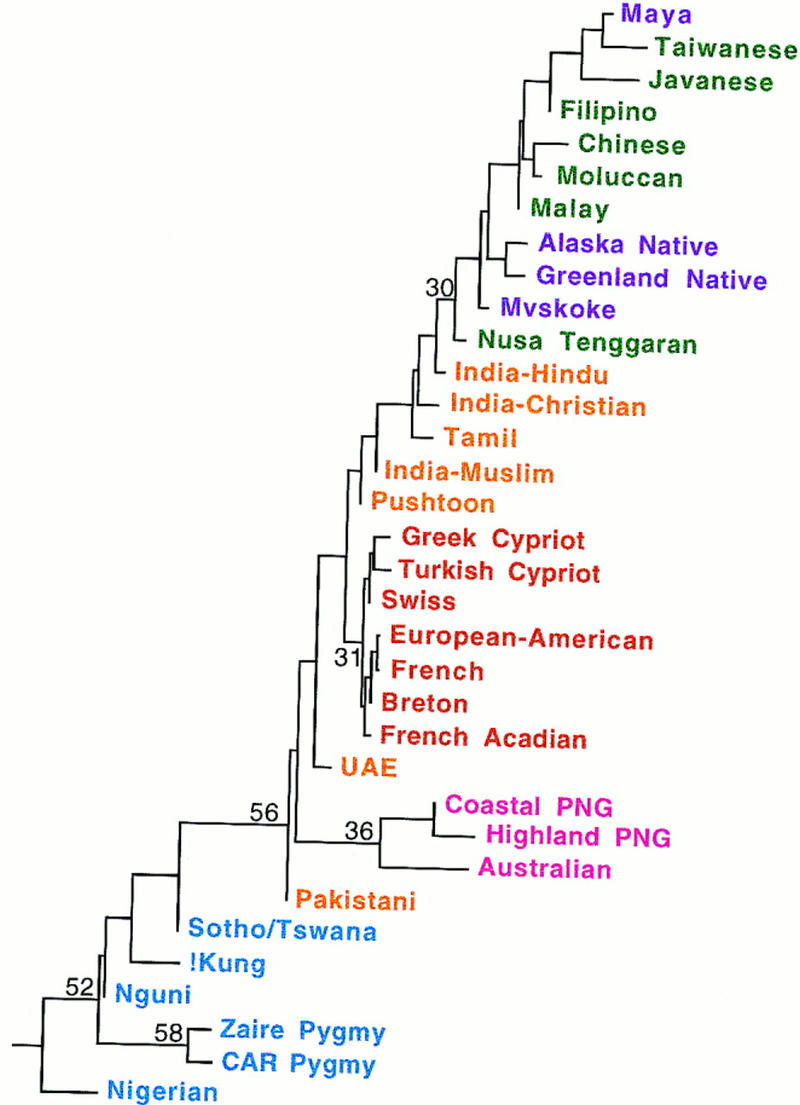
Neighbor-joining tree of population relationships. This tree is rooted where a hypothetical ancestral population—in which the frequency of the Alu element at each of the eight loci is set to 0.0—attaches to the unrooted network. Numbers indicate (in per cent) the fraction of 500 bootstrap replicates that supported a particular grouping.
To root the tree, we included a hypothetical ancestral population in which the frequency of the Alu element at each locus was set to zero. The root of the tree is within the cluster of African populations (Fig. 2), in agreement with an analysis of other gene frequency data that used the chimpanzee as an outgroup (Nei and Takezaki 1996). Apparently the Alu insert frequencies in African populations have undergone the least amount of change from the ancestral state. Assuming that allele frequency change at these loci occurs primarily by drift, which in turn is influenced by population size, this suggests that African and non-African populations have a different demographic history. One possible scenario involves an early expansion of the ancestors of African populations, thereby “freezing” allele frequencies when they are more similar to the root, followed by a later expansion of the ancestors of non-African populations, when allele frequencies would have drifted farther from the root. Alternatively, non-African populations may have been derived from African populations by a bottleneck event, which would have accelerated drift in the non-African populations.
Bootstrap resampling (Felsenstein 1985) was used to assess the strength of support of the data for the branching structure of the tree. A total of 500 bootstrap replications were performed, of which 52% placed the root as shown (Fig. 2). An additional 12% of the bootstrap replications placed the root somewhere else within the cluster of African populations, which would still be consistent with an African origin. The bootstrap values for various other clusters in the tree ranged from 26% to 58% (Fig. 2). None of these values reach statistical significance, which is not surprising with just eight loci; however, the bootstrap is known to underestimate the true level of statistical support (Sitnikova et al. 1995); hence, there may be more support for the clusters in the tree than the bootstrap values alone would indicate. Overall, these results are concordant with numerous other genetic polymorphisms that appear to have their source in Africa and/or indicate an early, separate expansion of African populations (Stoneking 1993; Bowcock et al. 1994; Goldstein et al. 1995; Armour et al. 1996; Tishkoff et al. 1996) and that have been taken as evidence for an African origin of our species.
A PC analysis (Cavalli-Sforza et al. 1994) of the allele frequencies at the eight polymorphic Alu insertion loci was also performed (Fig. 3). The first two coordinates, which provide the most information for a two-dimensional depiction of population relationships, account for 73% of the variance in the data. The same relationships that were evident in the neighbor-joining tree (Fig. 2) are also present here (Fig. 3); namely, geographic clusters of African and European populations are evident, whereas the New World populations are intermingled with the Southeast Asian populations. Two Sahulian populations cluster together in the analysis while the third is intermingled with the more dispersed Western Asian populations. Furthermore, the spread of populations within each geographic cluster is qualitatively similar to the Gst results (Table 1b); African, Southeast Asian, and Western Asian populations have the highest Gst values and exhibit the greatest spread. However, whereas the ancestral population in the tree (lacking the Alu element at each locus) is closest to the African populations, the African and Sahulian populations appear to be equally close to the ancestral population in the PC plot. This is because African populations are most similar to the ancestral population in the first PC, but Sahulian populations are most similar in the second PC.
Figure 3.
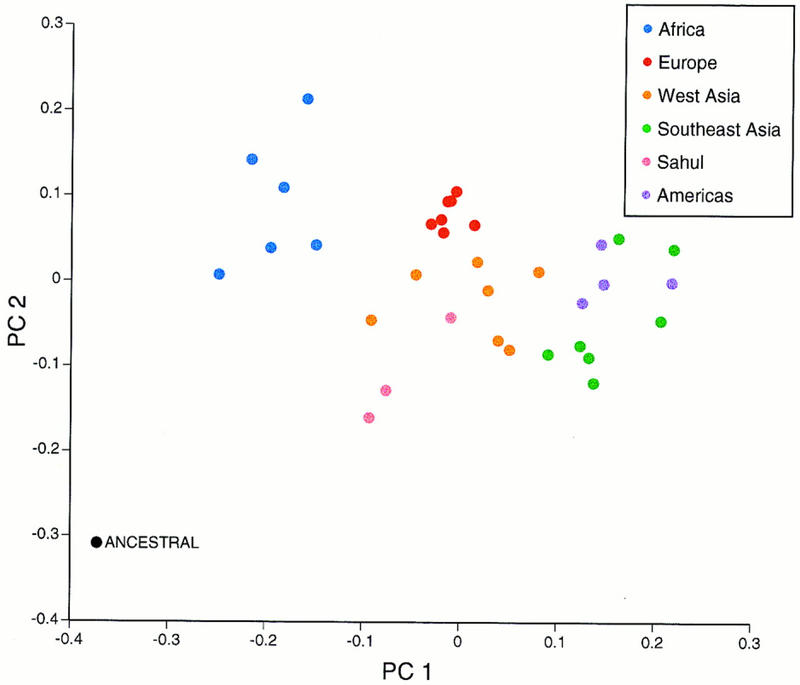
Plot of the first two principal coordinates of the allele frequencies at the eight polymorphic Alu insertion loci. The same relative population relationships were observed when the hypothetical ancestral population (consisting of allele frequencies of zero for the presence of the Alu element at each locus) was removed from the analysis (data not shown), indicating that the inclusion of the ancestral population is not distorting the population relationships.
A previous PC analysis of a subset of these data (Batzer et al. 1994) by Harpending et al. (1996) also found African and Sahulian populations to be closest to the root. Harpending et al. suggest this indicates an earlier expansion of the ancestors of tropical populations, followed by a later expansion of the ancestors of more peripheral populations such as Europeans, Asians, and Amerindians. However, because the African and Sahulian populations are closest to the root along different principal coordinates, which are orthogonal, African and Sahulian populations resemble the root in different ways. One way this situation could arise is if the earliest division of human populations was into a western (African) and eastern (Sahulian) component, with independent expansions following a period of separation.
Most genetic data do not link African and Sahulian populations (Cavalli-Sforza et al. 1994), possibly because the ancestral state for most genetic data is unknown. One notable exception is an analysis of human and chimpanzee restriction fragment length polymorphisms, which identified the most probable ancestral allelic states and demonstrated that African and Sahulian populations had the highest frequencies of ancestral alleles (Mountain and Cavalli-Sforza 1994). Analyses of morphological variation in skulls (Howells 1973) and teeth (Stringer et al. 1997) also associate African and Sahulian populations, although this is thought to reflect selection acting in similar ways on these tropical populations (Cavalli-Sforza et al. 1994). Analyses of additional genetic loci for which the ancestral state is known are needed to verify this preliminary association of tropical populations based on Alu insertion polymorphisms.
Estimated Effective Separation Time of African vs. Non-African Populations
An estimate of the time of separation of African and non-African populations can be obtained from the amount of genetic distance that has accumulated between them, assuming that this genetic distance has accumulated in the absence of any further migration. For human populations this is a dubious assumption at best, and it is not clear what meaning one should attach to “separation times” between human populations. Nevertheless, separation times between African and non-African populations have been estimated from various types of genetic data (Nei and Roychoudhury 1982; Bowcock et al. 1994; Goldstein et al. 1995; Armour et al. 1996; Knight et al. 1996; Tishkoff et al. 1996), and our purpose in doing so here is to see whether the Alu insertion polymorphisms are concordant with other nuclear DNA loci. Because we are not attaching any specific meaning to the actual estimated separation time between African and non-African populations, we will henceforth refer to this as the effective separation time, to emphasize that this would be a true separation time only if African and non-African populations had remained completely isolated after this time. We used the model of Tachida and Iizuka (1993), in which the effective separation time is a function of Nei’s genetic distance (estimated for an Alu subfamily), the time when the Alu subfamily began expanding from a single master copy (tb), and the time when Alu subfamily expansion ended (te). For the Ya5/8 Alu subfamily, we used Tachida and Iizuka’s estimate (1993) of tb = 4.3 million years ago, and we assumed that te = 0 (i.e., this subfamily is still expanding) because recent insertion events have been observed (for review, see Deininger and Batzer 1995). The genetic distance estimate must include both polymorphic and monomorphic loci (i.e., loci for which the Alu element is present on every chromosome) of the Alu subfamily, as an estimate of genetic distance based solely on polymorphic loci will be biased (Tachida and Iizuka 1993). Random screening of human-specific Alu elements indicates that ∼20% are polymorphic (Arcot et al. 1996); hence, we estimate that for the 8 polymorphic loci in our study, we need to include 32 monomorphic loci in the genetic distance calculation. The resulting average genetic distance between African and non-African populations is 0.0246, which from equation 33 of Tachida and Iizuka (1993) corresponds to an effective separation time of 137,000 ± 15,000 years ago.
The estimate of tb = 4.3 million years to get the above effective separation time between African and non-African populations may be too recent, as a few Ya5/8 elements are known in chimpanzees and gorillas, which diverged from humans 4–6 million years ago. If one uses instead tb = 5 million years, then the separation time becomes 159,000 years ago, whereas tb = 6 million years gives a separation time of 187,000 years ago; these are not substantially different from the above estimate of 137,000 years ago for the effective separation of African and non-African populations. These estimates of the effective separation time of African and non-African populations, based specifically on a model for polymorphic Alu insertion loci, are in good agreement with studies of other nuclear DNA polymorphisms (Nei and Roychoudhury 1982; Bowcock et al. 1994; Goldstein et al. 1995; Armour et al. 1996; Knight et al. 1996; Tishkoff et al. 1996). Thus, we conclude that the Alu loci do not differ in this respect from other nuclear loci.
Heterozygosity vs. Distance from the Centroid and Effective Population Size
Previous work has demonstrated that in a structured population a simple linear relationship is expected between the mean heterozygosity of a population and the genetic distance of that population from the centroid (Harpending and Ward 1982), and empirical studies have shown this relationship to hold (Harpending and Ward 1982; Crawford et al. 1989; McComb et al. 1995). A plot of heterozygosity versus distance from the centroid for the 34 populations in this study (Fig. 4) shows a good fit between the observed relationship and that predicted by the model, except that all of the African populations have a greater heterozygosity than predicted. In previous studies of genetic data, populations exhibiting greater heterozygosity than predicted by the model were thought to be experiencing higher rates of gene flow from other populations, which would elevate heterozygosity (Harpending and Ward 1982; Batzer et al. 1996b). However, high rates of gene flow should also reduce population differentiation, but the Gst analysis (Table 1b) indicates that African populations show the largest between-population differences.
Figure 4.
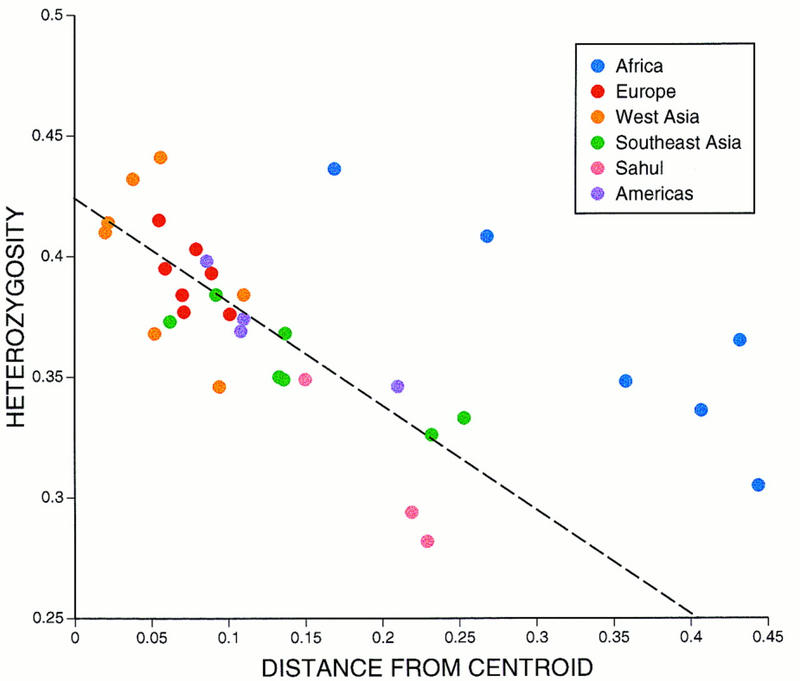
Plot of heterozygosity vs. distance from the centroid. The broken line is the expected relationship predicted by the model of Harpending and Ward (1982), according to the formula hi = H(1 − ri), where ri is the distance from the centroid and hi and H are the heterozygosities of population i and the total population, respectively.
The model outlined above assumes no systematic bias in the ascertainment of polymorphic loci. Previous studies showing elevated heterozygosities in Europeans because of ascertainment bias should not conform to this model. However, the Alu insertion loci analyzed here were either derived from the literature or the genome of an African–American individual. Therefore the ascertainment bias in these polymorphisms is minimal and satisfies the assumptions of the model. We think that the most likely explanation for the elevated heterozygosity in African populations may be that the model depicted by the broken line in Figure 4 assumes that all populations are of the same relative effective size. If African populations have been larger than non-African populations, then the heterozygosity of African populations would also be increased (Relethford and Harpending 1995). We note that this explanation implies a greater effective population size across Africa, as we analyzed a linguistically and geographically diverse sample of African populations, all of which deviated from the model (Fig. 4).
Other groups have previously suggested that their results could be explained by either an African origin or a larger African population size (Armour et al. 1996; Tishkoff et al. 1996); however, our results provide the first direct evidence in support of the latter hypothesis. A similar analysis of worldwide craniometric variation also found an elevated heterozygosity in African populations when population sizes were assumed to be equal (Relethford and Harpending 1994). This analysis also showed that the difference in size between African and non-African populations could be estimated by manipulating the relative population sizes to give the best fit to the Harpending–Ward model; we are currently exploring how best to do this for the large number of populations in the present analysis.
Previous studies have found greater genetic diversity in African populations for mitochondrial DNA (mtDNA) (Cann et al. 1987; Vigilant et al. 1991), Y-DNA (Hammer 1995), and nuclear DNA (Bowcock et al. 1994; Armour et al. 1996; Tishkoff et al. 1996) and have claimed that such greater African diversity reflects a greater antiquity of African populations and, hence, an African origin of modern humans. Others have pointed out that this greater African diversity could instead reflect a larger effective size for African populations (Relethford and Harpending 1995; Rogers and Jorde 1995; Armour et al. 1996; Harpending et al. 1996; Tishkoff et al. 1996), and this study supports this argument. A greater genetic diversity in African populations therefore does not necessarily imply an African origin of modern humans, but neither does it contradict this, as African populations could have been both larger than non-African populations and the source of modern humans. Moreover, Harpending et al. (1996) have equated the hypothesis of an African origin of modern humans with a demographic scenario in which the ancestors of African populations expanded earlier than the ancestors of non-African populations. The fact that African populations are closest to the ancestral condition for these Alu insertion polymorphisms, and exhibit a greater effective population size, is in excellent agreement with this scenario. Analyses of additional loci for which the ancestral state is known should continue to prove instructive in understanding human origins.
METHODS
Population Samples
The following previously described populations were studied: Nigerian (Batzer et al. 1994), Central African Republic (CAR) Pygmy (Bowcock et al. 1987), Zaire Pygmy (Bowcock et al. 1987), !Kung (Soodyall et al. 1996), Sotho/Tswana (Soodyall et al. 1996), Nguni (Soodyall et al. 1996), European–American (Batzer et al. 1996b), Swiss (Batzer et al. 1996b), Breton (Batzer et al. 1996b), French (Batzer et al. 1996b), French Acadian (Batzer et al. 1996b), Greek Cypriot (Batzer et al. 1994), Turkish Cypriot (Batzer et al. 1994), Pushtoon (Melton et al. 1995), Tamil (Melton et al. 1995), Chinese (Melton et al. 1995), Taiwanese (Melton et al. 1995), Filipino (Melton et al. 1995), Malaysian (Melton et al. 1995), Javanese (Melton et al. 1995), Moluccan (Perna et al. 1992), Nusa Tenggaran (Perna et al. 1992), Australian (Perna et al. 1992), Coastal Papua New Guinean (PNG) (Perna et al. 1992), Highland Papua New Guinean (Perna et al. 1992), Alaskan Native (Batzer et al. 1994), Greenland Native (Batzer et al. 1996b), Mvskoke (Weiss et al. 1993), and Mayan (Weiss et al. 1993). United Arab Emirates (UAE) samples were collected in Dubai. Pakistani samples were collected from native Pakistani individuals working in Dubai. Indian–Christian, –Muslim, and –Hindu samples were collected from Madras in southern India. DNA from Pakistani, Indian and UAE samples was prepared from blood stains using an IsoQuick nucleic acid extraction kit (MicroProbe Corporation, Bothell, WA).
Typing of Alu Insertion Polymorphisms
PCR amplification conditions, genotyping by agarose gel electrophoresis, and primer sequences for all eight loci (TPA25, PV92, FXIIIB, APO, ACE, D1, A25 and B65) were described previously (Arcot et al. 1995a,b; Batzer et al. 1996b). The TPA25, FXIIIB, ACE, and APO loci are also known as PLAT, F13B, APOAI, and DCPI, respectively. All 8 loci are present in humans and absent from orthologous positions within the genomes of (at least) 15 nonhuman primate genomes (Arcot et al. 1995a,b; Batzer et al. 1996b). The observed numbers of each genotype for each locus and population are available upon request from either M. Stoneking or M.A. Batzer.
Data Analysis
Unbiased estimates of heterozygosity (and associated standard error) and Gst values, corrected for sample size, were calculated using equations in Nei (1987). The GENDIST program in PHYLIP 3.5 (Felsenstein 1993) was used to calculate Nei’s genetic distance (Nei 1972) between each pair of populations for the eight polymorphic loci, and the NEIGHBOR program was used to construct a neighbor-joining tree (Saitou and Nei 1987) from these genetic distances. Programs in PHYLIP 3.5 were also used to perform 500 bootstrap replications of the tree. Principal coordinate analysis was performed using a program provided by H. Harpending (Pennsylvania State University, University Park). Population separation times were estimated using the model of Tachida and Iizuka (1993), as described in more detail above. The standard error of the population separation time estimate was computed by jack-knifing (Sokal and Rohlf 1981) Nei’s distance, and the resulting population separation times, over the eight polymorphic loci. The model of Harpending and Ward (1982) was used to assess the relative amount of gene flow experienced by each population. In this model, which utilizes the standardized variance–covariance (R) matrix of allele frequencies, a simple linear relationship is expected between the heterozygosity of each population and the distance of the population from the centroid (the arithmetic mean of the allele frequencies): ri = (pi − P)2/(P)(1 − P), where ri is the distance from the centroid and pi and P are the frequency of the Alu insertion in population i and in the total population, respectively. The above equation was used to compute the distance from the centroid for each locus separately, and these values were then averaged over the eight loci.
Acknowledgments
We thank K. Bhatia, A.S.M. Sofro, K. Weiss, J. Martinson, B.J.B. Keats, P.A. Ioannou, J.-P. Moisan, S.M. Milligan, M. Hochmeister, and W.D. Scheer for samples; L. Ludvico for technical assistance; and H. Harpending for valuable discussion and assistance with the principal coordinates analysis. This work was supported by grants from the National Science Foundation (SBR-9423118 to M.S.), the National Institutes of Health (RO1 HG00770 to P.L.D.), and the U.S. Department of Energy (DOE) (LDRD 94-LW-103 to M.A.B.). Work at Lawrence Livermore National Laboratory was conducted under the auspices of the U.S. DOE (contract W-7405-ENG-48).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mbatze@lsumc.edu; FAX (504) 568-6037.
REFERENCES
- Arcot SS, Fontius JJ, Deininger PL, Batzer MA. Identification and analysis of a “young” polymorphic Alu element. Biochem Biophys Acta. 1995a;1263:99–102. doi: 10.1016/0167-4781(95)00080-z. [DOI] [PubMed] [Google Scholar]
- Arcot SS, Wang Z, Weber JL, Deininger PL, Batzer MA. Alu repeats: A source for the genesis of primate microsatellites. Genomics. 1995b;29:136–144. doi: 10.1006/geno.1995.1224. [DOI] [PubMed] [Google Scholar]
- Arcot SS, Adamson AW, Lamerdin JE, Kanagy B, Deininger PL, Carrano AV, Batzer MA. Alu fossil relics—Distribution and insertion polymorphism. Genome Res. 1996;6:1084–1092. doi: 10.1101/gr.6.11.1084. [DOI] [PubMed] [Google Scholar]
- Armour JAL, Anttinen T, May CA, Vega EE, Sajantila A, Kidd JR, Kidd KK, Bertranpetit J, Paabo S, Jeffreys AJ. Minisatellite diversity supports a recent African origin for modern humans. Nature Genet. 1996;13:154–160. doi: 10.1038/ng0696-154. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. A human-specific subfamily of Alu sequences. Genomics. 1991;9:481–487. doi: 10.1016/0888-7543(91)90414-a. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Kilroy GE, Richard PE, Shaikh TH, Desselle TD, Hoppens CL, Deininger PL. Structure and variability of recently inserted Alu family members. Nucleic Acids Res. 1990;18:6793–6798. doi: 10.1093/nar/18.23.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Gudi VA, Mena JC, Foltz DW, Herrera RJ, Deininger PL. Amplification dynamics of human-specific (HS) Alu family members. Nucleic Acids Res. 1991;19:3619–3623. doi: 10.1093/nar/19.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Stoneking M, Alegria-Hartman M, Bazan H, Kass DH, Shaikh TH, Novick GE, Ioannou PA, Scheer WD, Herrera RJ, Deininger PL. African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci. 1994;91:12288–12292. doi: 10.1073/pnas.91.25.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Rubin CM, Hellmann-Blumberg U, Alegria-Hartman M, Leeflang EP, Stern JD, Bazan HA, Shaikh TH, Deininger PL, Schmid CW. Dispersion and insertion polymorphism in two small subfamilies of recently amplified human Alu repeats. J Mol Biol. 1995;247:418–427. doi: 10.1006/jmbi.1994.0150. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL, Hellmann-Blumberg U, Jurka J, Labuda D, Rubin CM, Schmid CW, Zietkiewicz E, Zukerkandl E. Standardized nomenclature for Alu repeats. J Mol Evol. 1996a;42:3–6. doi: 10.1007/BF00163204. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Arcot SS, Phinney JW, Alegria-Hartman M, Kass DH, Milligan SM, Kimpton C, Gill P, Hochmeister M, Ioannou PA, Herrera RJ, Boudreau DA, Scheer WD, Keats BJB, Deininger PL, Stoneking M. Genetic variation of recent Alu insertions in human populations. J Mol Evol. 1996b;42:22–29. doi: 10.1007/BF00163207. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Bucci C, Hebert JM, Kidd JR, Kidd KK, Friedlaender JS, Cavalli-Sforza LL. Study of 47 DNA markers in five populations from four continents. Gene Geogr. 1987;1:47–64. [PubMed] [Google Scholar]
- Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. High resolution of human evolutionary trees with polymorphic microsatellites. Nature. 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton, N.J: Princeton University Press; 1994. [Google Scholar]
- Crawford MH, Dykes DD, Polesky HF. Genetic structure of Mennonite populations of Kansas and Nebraska. Hum Biol. 1989;61:493–514. [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Evolution of retroposons. Evol Biol. 1993;27:157–196. [Google Scholar]
- ————— . SINE master genes and population biology. In: Maraia RJ, editor. The impact of short interspersed elements (SINEs) on the host genome. Georgetown, TX: R.G. Landes; 1995. pp. 43–60. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- ————— . PHYLIP, version 3.5c. Seattle, WA: University of Washington; 1993. [Google Scholar]
- Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc Natl Acad Sci. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF. A recent common ancestry for human Y chromosomes. Nature. 1995;378:376–378. doi: 10.1038/378376a0. [DOI] [PubMed] [Google Scholar]
- Harpending HC, Ward RH. Chemical systematics and human populations. In: Nitecki M, editor. Biochemical aspects of evolutionary biology. Chicago, IL: University of Chicago Press; 1982. pp. 213–256. [Google Scholar]
- Harpending H, Relethford J, Sherry ST. Methods and models for understanding human diversity. In: Boyce AJ, Mascie-Taylor CGN, editors. Molecular biology and human diversity. Cambridge, UK: Cambridge University Press; 1996. pp. 283–299. [Google Scholar]
- Howells WW. Cranial variation in man: A study by multivariate analysis of patterns of difference among recent human populations. Pap Peabody Mus Archaeol Ethnol Harv Univ. 1973;67:1–259. [Google Scholar]
- Jorde LB, Bamshad MJ, Watkins WS, Zenger R, Fraley AE, Krakowiak PA, Carpenter KD, Soodyall H, Jenkins T, Rogers AR. Origins and affinities of modern humans: A comparison of mitochondrial and nuclear genetic data. Am J Hum Genet. 1995;57:523–538. doi: 10.1002/ajmg.1320570340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass DH, Aleman C, Batzer MA, Deininger PL. Identification of a human specific Alu insertion in the factor XIIIB gene. Genetica. 1994;94:1–8. doi: 10.1007/BF01429214. [DOI] [PubMed] [Google Scholar]
- Knight A, Batzer MA, Stoneking M, Tiwari HK, Scheer WD, Herrera RJ, Deininger PL. DNA sequences of Alu elements indicate a recent replacement of the human autosomal genetic complement. Proc Natl Acad Sci. 1996;93:4360–4364. doi: 10.1073/pnas.93.9.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb J, Blagitko N, Comuzzie AG, Schanfield MS, Sukernik RI, Leonard WR, Crawford MH. VNTR DNA variation in Siberian indigenous populations. Hum Biol. 1995;67:217–229. [PubMed] [Google Scholar]
- Melton T, Peterson R, Redd AJ, Saha N, Sofro ASM, Martinson J, Stoneking M. Polynesian genetic affinities with Southeast Asian populations as identified by mtDNA analysis. Am J Hum Genet. 1995;57:403–414. [PMC free article] [PubMed] [Google Scholar]
- Mountain JL, Cavalli-Sforza LL. Inference of human evolution through cladistic analysis of nuclear DNA restriction polymorphisms. Proc Natl Acad Sci. 1994;91:6515–6519. doi: 10.1073/pnas.91.14.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Genetic distance between populations. Am Nat. 1972;106:283–292. [Google Scholar]
- ————— . Molecular evolutionary genetics. New York, NY: Columbia University Press; 1987. [Google Scholar]
- Nei M, Roychoudhury AK. Genetic relationship and evolution of human races. Evol Biol. 1982;14:1–59. [Google Scholar]
- Nei M, Takezaki N. The root of the phylogenetic tree of human populations. Mol Biol Evol. 1996;13:170–177. doi: 10.1093/oxfordjournals.molbev.a025553. [DOI] [PubMed] [Google Scholar]
- Novick, G.E., C.C. Novick, J. Yunis, E. Yunis, P. Antunez de Mayolo, W.D. Scheer, P.L. Deininger, M. Stoneking, D.S. York, M.A. Batzer, and R.J. Herrera. 1997. Polymorphic Alu insertions and the Oriental origin of Native American populations. Hum. Biol. (in press). [PubMed]
- Okada N. SINEs. Curr Opin Genet Dev. 1991;1:498–504. doi: 10.1016/s0959-437x(05)80198-4. [DOI] [PubMed] [Google Scholar]
- Perna NT, Batzer MA, Deininger PL, Stoneking M. Alu insertion polymorphism: A new type of marker for human population studies. Hum Biol. 1992;64:641–648. [PubMed] [Google Scholar]
- Relethford JH, Harpending HC. Craniometric variation, genetic theory, and modern human origins. Am J Phys Anthropol. 1994;95:249–270. doi: 10.1002/ajpa.1330950302. [DOI] [PubMed] [Google Scholar]
- ————— Ancient differences in population size can mimic a recent African origin of modern humans. Curr Anthropol. 1995;36:667–674. [Google Scholar]
- Rogers AR, Jorde LB. Genetic evidence on modern human origins. Hum Biol. 1995;67:1–36. [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schmid CW, Maraia R. Transcriptional regulation and transpositional selection of active SINE sequences. Curr Opin Genet Dev. 1992;2:874–882. doi: 10.1016/s0959-437x(05)80110-8. [DOI] [PubMed] [Google Scholar]
- Sherry, S.T., H.C. Harpening, M.A. Batzer, and M. Stoneking. 1997. Alu evolution in human populations: Using the coalescent to estimate effective population size. Genetics (in press). [DOI] [PMC free article] [PubMed]
- Sitnikova T, Rzhetsky A, Nei M. Interior-branch and bootstrap tests of phylogenetic trees. Mol Biol Evol. 1995;12:319–333. doi: 10.1093/oxfordjournals.molbev.a040205. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry. New York, NY: W.H. Freeman & Co.; 1981. [Google Scholar]
- Soodyall H, Vigilant L, Hill AV, Stoneking M, Jenkins T. mtDNA control-region sequence variation suggests multiple independent origins of an “Asian-specific” 9-bp deletion in sub-Saharan Africans. Am J Hum Genet. 1996;58:595–608. [PMC free article] [PubMed] [Google Scholar]
- Stoneking M. DNA and recent human evolution. Evol Anthropol. 1993;2:60–73. [Google Scholar]
- Stringer C, Humphrey L, Compton T. Cladistic analysis of dental traits in recent humans using a fossil outgroup. J Hum Evol. 1997;32:389–402. doi: 10.1006/jhev.1996.0112. [DOI] [PubMed] [Google Scholar]
- Tachida H, Iizuka M. A population genetic study of the evolution of SINEs. I. Polymorphism with regard to the presence or absence of an element. Genetics. 1993;133:1023–1030. doi: 10.1093/genetics/133.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M, Paabo S, Watson E, Risch N, Jenkins T, Kidd KK. Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science. 1996;271:1380–1397. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC. African populations and the evolution of human mitochondrial DNA. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- Weiss KM, Buchanan AV, Valdez R, Moore JH, Campbell J. Amerindians and the price of modernization. In: Schell LM, Smith MT, Bilsborough A, editors. Urban ecology and health in the third world. Cambridge, UK: Cambridge University Press; 1993. pp. 221–243. [Google Scholar]