

Abstract
High-throughput fluorescent genotyping requires a considerable amount of automation for accurate and efficient processing of genetic markers. Automated DNA sequencers and corresponding software products are commercially available that contribute substantially to increased throughput rates for large-scale genotyping projects. However, some conceptually simple tasks still require time-consuming manual intervention that imposes bottlenecks on throughput capacity. One of these tasks is the conversion of imprecise DNA fragment sizes determined by commercial software programs to the underlying discrete alleles that the sizes represent. Here we describe a simple method for assigning allele sizes into their appropriate allele “bins” using least-squares minimization procedures. The method requires no special treatment of family data on plates, internal/external size standards, or electropherogram data manipulation. Tests of the method using the ABI 373A automated DNA sequencer and accompanying Genescan/Genotyper software resulted in accurate automatic classification of all alleles in >80% of 208 markers analyzed, with the remaining 20% being appropriately identified as requiring additional attention to laboratory conditions. Specific characteristics of different markers, including differences in PCR product size and inexact repeat lengths (e.g., 1.9 bp for a dinucleotide repeat), are accommodated by the method and their properties discussed.
Genome-wide screens for complex diseases such as schizophrenia, asthma, or diabetes often require genotyping of 200–400 markers on hundreds or thousands of individuals. To meet such requirements, considerable effort has been devoted to the development of high-throughput genotyping methods. Advances in robotic devices for DNA extraction, PCR multiplexing methods, fluorescent detection systems, software for fragment size analysis and data tracking, and the availability of >5000 microsatellite markers have contributed significantly to the ability to genotype large numbers of samples rapidly and accurately (e.g., Hall et al. 1996; Ghosh et al. 1997). Many of these advances have been predicated on the need for automation, as well-constructed hardware and software can increase the rate of processing genotypes as well as provide a constant environment for identifying and correcting problems. Despite the large number of significant achievements in this area, the conceptually simple task of converting alleles from real-valued DNA fragment sizes derived from traversal through a gel into the discrete segregating units they represent remains largely manual and time-consuming. Here we describe an approach for automation of this task of allele binning and calling in the context of commercially available DNA sequencing instruments and related software; specifically, the ABI 373A/377 and Genescan/Genotyper software (Applied Biosystems Division, Perkin Elmer, Foster City, CA).
Genotyping using the ABI hardware and software is semi-automated in the sense that lane tracking and allele sizing is performed using the Genescan/Genotyper software with some manual intervention (Davies et al. 1994; Reed et al. 1994). However, the results of applications of the software are often ambiguous, as sized alleles for large samples of individuals typically do not fall into discrete groups as expected genetically. Instead, data points tend to cluster in allele groupings, with variability around each allele. This variability is shown in Figure 1, with the left panel illustrating low variability, and the right panel showing a range of greater variability in allele sizes. There are many possible causes of the allelic dispersion, including plus-A amplification, gel-to-gel variability, incorrect allele sizing, nonspecific primer sequences, and others (Callen et al. 1993; Hauge and Litt 1993; Hall et al. 1996). Careful attention to primer design and PCR conditions, arrangement of individual samples on plates according to family relationships, and strategic usage of reference genotypes can be helpful for allele binning and calling in the presence of such variation. Ghosh et al. (1997), for example, have demonstrated very high accuracy of allele sizing and binning by controlling for many possible sources of variation in PCR conditions and plate organization (see also Mansfield et al. 1994; Perlin et al. 1994, 1995). However, it is occasionally not possible to arrange samples by family, as in large prospective studies, and reference genotypes take up precious lanes that can otherwise be used for genotyping the samples of interest. In addition, specific markers are sometimes essential, by virtue of their chromosomal location, for which no apparent amount of PCR optimization or primer redesign reduces their size variability. There are also situations in which investigators have no access to laboratory personnel or template, as in the case of contract genotyping services. Suboptimal data generated in these situations should not be processed in a manner solely designed for optimal data, which is comprised of nearly discrete allele sizes. In a high-throughput environment it would be useful to be able to accurately classify the allele sizes into discrete bins in the presence of such variability associated with suboptimal conditions.
Figure 1.

Frequency histograms of two markers having different quality of allele sizes. Both markers were typed on 642 individuals using the ABI 373A and accompanying Genescan/Genotyper software. Marker D10S535 has a very clear separation of alleles, while marker D13S1325 has poor separation between most alleles.
In this paper we are concerned with a specific aspect of this allele binning problem. In particular, we wish to make use of the commercially available software for defining the size of each allele and then automatically bin alleles once the initial sizing is complete. We also wish to notify investigators of potential problems in the raw data when the variability is too large for automated processing. Here we describe a simple approach for classifying data that may not meet optimal criteria for simple classification. Input required for this algorithm encompasses only real-valued allele sizes derived from existing software; output from the method is simply the categorical allele designation for each data point and a designation of accuracy for the marker/bin classification.
METHODS
Definition of Bins
We employ a least-squares minimization procedure to define the allelic bins. For any marker having repeat length ρ, let Aj represent each allele size in a data set to be binned (j = 1, … , 2 N for N samples), let T reflect the maximum number of alleles possible [i.e., T = 1 + ⌊(maxj Aj−minj Aj)/ρ⌋], and let Li represent the lower boundary of each bin, Bi. Our aim is to select the optimal Li given the allele sizes obtained from the Genescan/Genotyper software. To estimate these parameters, we begin by sorting the allele size data in ascending order (Aj ⩽ Aj − 1) and setting L1 = A1 − ρ and Li = Li−1 + ρ, for i = 2, … , T. We consider each Aj as a member of bin i if Li ⩽ Aj < Li + 1. We then calculate the average variation within bins:
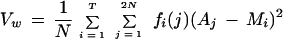 |
1 |
where fi(j) is an indicator variable containing the value 1 if Aj ∈ Bi and 0 otherwise, and Mi = Li + ρ/2.
To determine the optimal set of bins, we minimize the within-bin variance Vw. The minimization is achieved by defining a constant step parameter, s, which is set to a small value (e.g., s = 0.01). We calculate Vw over k = ρ/s trials such that Li(k) = Li(k − 1) + s and Li(k) = Li−1 (k) + ρ for the kth trial. The optimal bin set is taken as that in which Vw is smallest. This method of minimization will not necessarily find the exact global minimum for the set of bins, but as Vw is quadratic within the boundaries of the search space this procedure will locate the true minimum within the limits defined by s. Upon optimization, some internal bins may be empty, such as is observed with rare alleles. The number of individual allele sizes within each bin may be counted as
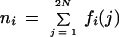 |
The number of observed alleles in the data set, omitting the empty bins, is given by
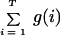 |
where g(i) is an indicator variable containing the value 1 if ni > 0 and 0 otherwise.
This approach ensures that each sized allele is contained within exactly one bin and that no bins overlap. It is important to note, however, that this method allows inter-bin distances to vary across alleles, such that the difference between allele sizes in adjacent bins may be as small as ε or as large as 2ρ − ε, where ε is an arbitrarily small number greater than 0. This type of inter-bin variability is observable as large differences in variances and/or mean and median sizes between adjacent bins. By allowing inter-bin variability the approach often yields accurate binning even under difficult conditions such as bimodal distributions associated with plus-A amplification.
Measure of Marker Quality
The measure of variation shown in equation 1 is not strictly a variance in the traditional statistical sense because it reflects dispersion from the bin median (Mi) rather than the bin mean. We use the median rather than the mean to use this measure as an indicator of allele calling accuracy or genotyping/allele sizing quality, as well as for defining the bin boundaries. As most allele sizes tend to form a normal distribution within each bin, and thus the mean and median are very close, our measure of variability is often similar to that which would be obtained using a traditional variance calculation. Situations in which the allele sizes do not tend to a normal distribution, or those in which allele boundaries are poorly defined, will result in a particularly large Vw that can be used to alert an investigator to visually examine the allele calls and/or individual genotype sizes. Also, normalizing a standard deviation, Sw, to a common scale across any repeat length (di-, tri-, tetranucleotide, etc.) as
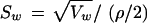 |
permits comparison of markers having variable repeat lengths. We use Sw to determine the accuracy of binning and to indicate when visual genotype inspection of the raw size data and allele bins is required.
Allelic Drift
The least-squares procedure described above appears to work well for most microsatellite markers (see Results). Occasionally, however, the optimal bin set estimated does not accurately reflect the observed data. In such cases, we have observed that the bin boundaries for some alleles are slightly shifted, causing inappropriate allele binning for adjacent alleles. The source of this inaccuracy may be attributable in part to a pattern we refer to as “allelic drift,” which is the tendency for true allele bins to differ by a value slightly different from the known repeat length. In our basic model, because we have imposed a fixed repeat length ρ (e.g., ρ = 2 for dinucleotide repeats, ρ = 3 for trinucleotide repeats, etc.), we do not allow for allele bins that actually differ by, say, 2.1 bp.
To allow for allelic drift in our model and to explore its behavior in a large empirical data set, we define an additional parameter to reflect the drift, δ. The algorithm works similarly to that described above, with the exception that at each of the k iterations for variance minimization, we evaluate δ at small increments, t, between a set of allowable drift values. Thus, at the kth iteration we conduct l = (max δ − min δ)/t trials, setting Li(k,l) = Li(k)(1 + δ(l)), where δ(1) = min δ and δ(l) = δ(l − 1) + t for l > 1. In practice, we set t = 0.01 and impose the boundary conditions of min δ = −0.10 and max δ = 0.10 so that l = 1, … , 21. Thus, the spacing between adjacent alleles is kept constant at a value ρ(1 + δ̂) rather than ρ. Further accuracy could be obtained using smaller values of t and a wider range of δ but we have not seen the need for this increased stringency.
Test Samples
To test the utility of this simple allele binning procedure, the method was applied to data for 208 markers genotyped on 642 individuals. Genotyping was performed using the ABI 373A system for DNA electrophoresis with the Genescan/Genotyper software for allele sizing as described by Hall et al. (1996). Although the DNA samples represent small nuclear families, the 96-well plates were not possible to arrange by family, thereby requiring consistent allele-calling in the absence of Mendelian transmission information. The marker set comprised a collection of highly polymorphic di-, tri-, and tetranucleotide repeats drawn from publicly available marker maps (Cooperative Human Linkage Center 1994; Reed et al. 1994; Dib et al. 1996).
Conditions of some of the test markers were varied for robust genotyping in our laboratory (by varying annealing temperature and Mg+2 concentrations only); others were simply genotyped using published conditions. For the automated allele binning tests, no special processing of the data was performed; that is, the data were simply evaluated using Genescan/Genotyper and then subjected to the allele binning method. Some of the markers performed very poorly with respect to allele-sizing prior to evaluation by our method, presumably because of over-/underpooling, variability between gels, nonspecific primer sequences, PCR contamination, or other unknown problems. Although such errors often may be detected during initial allele sizing, these markers were deliberately included in the test set to investigate the extent to which the binning procedures could identify and isolate markers of poor quality.
RESULTS AND DISCUSSION
Application of the method above to the test set of 208 markers required ∼2 min of computer time on a Sun SparcUltra1 personal workstation. For discussions of the accuracy of the method, we first describe the characteristics without the allelic drift parameter, which we describe as the “basic method,” and then discuss properties of the method accounting for allelic drift.
Basic Method
Distributions of our measure of dispersion, Sw, are presented in Figure 2. Corresponding sample statistics are given in Table 1. Several features of the basic method are apparent from these descriptive statistics. First, the mean value of Sw for all markers is very close to 0.25, indicating that the average variability around each allele bin is ±1/4 bp (assuming a dinucleotide repeat). Visual inspection of the individual markers suggests that any value of Sw ⩽ 0.30 is very likely to represent accurate allele sizing and automated binning. In fact, all markers with Sw ⩽ 0.30 have allele binning accuracy as good or better than that obtained by visual assignment. As an example, the marker profile shown in the left panel of Figure 1 has a corresponding Sw value of 0.13, which reflects the distinct nature of the specific allele sizes. In contrast, the marker in the right panel of Figure 1 has an Sw value of 0.44, reflecting the poor allele sizing and consequent binning of the individual alleles. This pattern of effects, combined with visual inspection of the raw data, suggests that some crude guidelines may be established for applications of the automated binning methods. These guidelines are given in Table 2, where it may be seen that ∼77% of markers in our test set may be binned automatically without visual inspection, irrespective of repeat type. In addition, most of the markers in the second category of Table 2 have accurate bins to the extent permitted by distinct allele sizing, and, thus, as much as 85% and 96% of di- and tetranucleotide repeats may be automatically binned. Nonetheless, because a few markers in this category do have some problems with specific bins, we have found it useful to conduct cursory evaluations of the bin boundaries prior to assignment of individual allele calls.
Figure 2.

Histogram representing standard deviations, Sw for all di- and tetranucleotide markers tested. Light shaded bars represent dinucleotide repeats and dark shaded bars represent tetranucleotide repeat markers.
Table 1.
Descriptive Statistics from Automated Allele Calling of Test Sample
| Marker type | No. | No drift parameter | With drift parameter | ||||
|---|---|---|---|---|---|---|---|
| mean | median | s.d. | mean | median | s.d. | ||
| All | 208 | 0.251 | 0.237 | 0.105 | 0.221 | 0.202 | 0.098 |
| Dinucleotide | 121 | 0.292 | 0.271 | 0.098 | 0.254 | 0.232 | 0.098 |
| Trinucleotide | 8 | 0.230 | 0.232 | 0.076 | 0.201 | 0.218 | 0.063 |
| Tetranucleotide | 79 | 0.191 | 0.176 | 0.086 | 0.174 | 0.163 | 0.078 |
Table 2.
Guidelines for Interpretation of Binning Accuracy and Marker Quality
| Percent of test sample | ||||
|---|---|---|---|---|
| di- | tetra- | |||
| Sw | Action | (nucleotide) | all | |
| 0.0–0.30 | no inspection required | 65.3 | 92.4 | 76.6 |
| 0.31–0.40 | binning likely good; check specific bins | 19.8 | 3.8 | 13.4 |
| 0.41–0.45 | binning or sizing poor; check all genotypes | 5.0 | 2.5 | 3.8 |
| >0.45 | binning and sizing unacceptable; re-genotype marker | 9.9 | 1.3 | 6.2 |
We note that all markers in the test set with Sw > 0.45 reflect severe genotyping problems such that allele sizes form a uniform distribution across all bins rather than a series of normal distributions. As noted previously, markers such as these were included in the test set as a means to evaluate the properties of the automated binning procedure under poor conditions. The method clearly identifies such markers as outliers; in no cases were these poor markers scored Sw < 0.40. However, in some of the borderline cases (0.31 ⩽ Sw ⩽ 0.40), the binning procedure does give erroneous allele assignments. Most of these are resolved by incorporation of the allelic drift parameter, described below.
Allelic Drift
In general, the basic allele binning procedure appears to capture sufficient information to automatically call specific alleles or to indicate that the marker is of such poor quality as to require further laboratory attention. Consequently, average estimates of the drift parameter are close to 0.00 (δ = −0.02, 0.01 for di- and tetranucleotide repeat markers, respectively). In certain instances, however, allowing for allelic drift results in a substantial improvement in automated binning such that a marker that otherwise would have been flagged for regenotyping can be accurately binned. Figure 3 shows an example of this improvement. The dinucleotide marker shown has a distinct series of alleles, yet the basic method performs poorly with binning (Sw = 0.40). For this marker, δ̂ = −0.05 with a corresponding Sw = 0.19. The poor performance of the basic method is attributable to differences in spacing of adjacent alleles from the expected value of 2 bp. In this case, adjacent alleles have an average spacing of 1.90 bp, and 13/15 of the alleles are separated by <2 bp (range = 1.84–2.09 bp). Consequently, poor binning results from the expectation of 2 bp separation. The δ̂ estimate of −0.05 captures the average spacing well [ρ(1 + δ̂) = 2(1 − 0.05) = 1.90].
Figure 3.

Frequency histogram of illustrative marker which has nonstandard repeat lengths between adjacent alleles. In this dinucleotide marker, the expected difference between alleles is 2 bp, yet the average spacing is 1.90 bp. The automated binning procedure accurately assigns all bins only when allowing for this discrepancy of repeat length, referred to as allelic drift.
Interestingly, this marker has a fairly large PCR product size of 288 bp at the largest allele, indicating that larger product sizes tend to require more flexibility in automated binning than do markers with shorter product sizes. Indeed, for all dinucleotide markers collectively, the drift parameter is negatively correlated with product size (r = −0.31, p = 0.0006). This suggests that spacing of adjacent alleles decreases with increases in PCR product size. This effect may reflect variability associated with the longer traversal time of large DNA fragments through gels before detection. Accounting for allelic drift is apparently unnecessary for tetranucleotide repeat markers, as no significant correlation is evident (r = 0.13, p = 0.27). This may be because the larger separation of alleles compensates for the reduced drift.
In summary, we have described a simple method for automated allele binning of markers in which optimal conditions of PCR chemistry, primer design, and internal/external standards are either unavailable or, when available, still produce considerable variability in allele sizes. In this approach, situations in which the method yields poor binning outcomes are as important as those in which it accurately classifies alleles, as one of the aims is to identify markers that are not amenable to automated processing and to alert investigators of possible problems in PCR, primers, sizing, or other conditions which require further attention in the laboratory. Indeed, Mendelian inheritance errors attributable to incorrect initial sizing were present in 1.9% of alleles in these data, which lead to subsequent classification of poor quality bins. The simple statistics and guidelines provided here are designed to assist investigators in evaluating when to visually inspect the data in the presence of such confounding factors. These guidelines should help to increase throughput capacity in large-scale genotyping projects.
Acknowledgments
We thank Dr. Susan Daniels for genotyping the samples used in these analyses and Dr. Sarah Shaw and Carrie LeDuc for helpful comments on the manuscript. A computer program written in C++ that conducts all binning assignments described here is available from the authors on request.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lon@sequana.com; FAX (619) 452-6653.
REFERENCES
- Callen DF, Thompson AD, Shen Y, Phillips HA, Richards RI, Mulley JC, Sutherland GR. Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am J Hum Genet. 1993;52:922–927. [PMC free article] [PubMed] [Google Scholar]
- Cooperative Human Linkage Center. A comprehensive human linkage map with centimorgan density. Science. 1994;265:2049–2054. doi: 10.1126/science.8091227. [DOI] [PubMed] [Google Scholar]
- Davies JL, Kawaguchi Y, Bennett ST, Copeman JB, Cordell HJ, Pritchard LE, Reed PW, Gough SCL, Jenkins SC, Palmer SM, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371:130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karanjawala ZE, Hauser ER, Ally D, Knapp JI, Rayman JB, Musick A, Tannenbaum J, Te C, Shapiro S, et al. Methods for precise sizing, automated binning of alleles, and reduction of error rates in large-scale genotyping using fluorescently labeled dinucleotide markers. Genome Res. 1997;7:165–178. doi: 10.1101/gr.7.2.165. [DOI] [PubMed] [Google Scholar]
- Hall JM, LeDuc AA, Watson AR, Roter AH. An approach to high-throughput genotyping. Genome Res. 1996;6:781–790. doi: 10.1101/gr.6.9.781. [DOI] [PubMed] [Google Scholar]
- Hauge XY, Litt M. A study of the origin of “shadow bands” seen when typing dinucleotide repeat polymorphisms by PCR. Hum Mol Genet. 1993;2:411–415. doi: 10.1093/hmg/2.4.411. [DOI] [PubMed] [Google Scholar]
- Mansfield DC, Brown AF, Green DK, Carothers AD, Morris SW, Evans HJ, Wright AF. Automation of genetic linkage analysis using fluorescent microsatellite markers. Genomics. 1994;24:225–233. doi: 10.1006/geno.1994.1610. [DOI] [PubMed] [Google Scholar]
- Perlin MW, Burks MB, Hooip RC, Hoffman EP. Toward fully automated genotyping: Allele assignment, pedigree construction, phase determination, and recombination detection in Duchenne muscular dystrophy. Am J Hum Genet. 1994;55:777–787. [PMC free article] [PubMed] [Google Scholar]
- Perlin M, Lancia G, Ng S. Toward fully automated genotyping: Genotyping microsatellite markers by deconvolution. Am J Hum Genet. 1995;57:1199–1210. [PMC free article] [PubMed] [Google Scholar]
- Reed PW, Davies JL, Copeman JB, Bennett ST, Palmer SM, Pritchard LE, Gough SCL, Kawaguchi Y, Cordell HJ, Balfour KM, et al. Chromosome-specific microsatellite sets for fluorescence-based, semi-automated genome mapping. Nat Genet. 1994;7:390–395. doi: 10.1038/ng0794-390. [DOI] [PubMed] [Google Scholar]