

Abstract
The discovery that in invertebrates, disruption of the insulin/insulin-like growth factor (IGF)-1 pathway extends life span and increases resistance to oxidative injury led to the hypothesis that IGF-1 signaling may play a role in regulating cellular reactive oxygen species production, oxidative stress resistance, and consequentially, organismal life span in mammals. However, previous studies testing this hypothesis in rodent models of IGF-1 deficiency yielded controversial results. The Lewis dwarf rat is a useful model of human growth hormone (GH)/IGF-1 deficiency as it mimics many of the pathophysiological alterations present in human GH/IGF-1–deficient patients as well as elderly individuals. Peripubertal treatment of Lewis dwarf rats with GH results in a significant extension of life span. The present study was designed to test the role of the GH/IGF-1 axis in regulating cellular oxidative stress and oxidative stress resistance, utilizing primary fibroblasts derived from control rats, Lewis dwarf rats and GH-replete dwarf rats. Measurements of cellular dihydroethidium and C-H2DCFDA fluorescence showed that cellular O2·− and peroxide production were similar in each group. Fibroblasts from control and Lewis dwarf rats exhibited similar antioxidant capacities and comparable sensitivity to H2O2, rotenone, high glucose, tunicamycin, thapsigargin, paraquat, and mitomycin, which cause apoptosis through increasing oxidative stress, mitochondrial damage, ATP depletion, and/or by damaging DNA, lipids and proteins. Fibroblasts from GH-replete rats exhibited significantly increased antioxidant capacities and superior resistance to H2O2, rotenone and bacterial lipopolysaccharide–induced cell death compared with cells from Lewis dwarf rats, whereas their sensitivity to the other stressors investigated was not statistically different. Thus, low circulating IGF-1 levels present in vivo in Lewis dwarf rats do not elicit long-lasting alterations in cellular reactive oxygen species generation and oxidative stress resistance, whereas life span–extending peripubertal GH treatment resulted in increased antioxidant capacity and increased resistance to cellular injury caused by some, but not all, oxidative stressors.
Keywords: Oxidative stress resistance, Growth hormone, Fibroblast, Free radicals
COMPARATIVE studies of short-lived and long-lived mammalian species as well as studies on long-lived mutant mice suggest that mammalian longevity is often associated with attenuated cellular production of reactive oxygen species (ROS) and/or increased cellular resistance to oxidative injury(1–12). These findings are in accord with the predictions of the oxidative stress hypothesis of aging, originally proposed by Harman (13), which posits that endogenously generated ROS cause aging through oxidative damage to macromolecules (DNA, proteins, and lipids). Yet, the mechanisms that regulate cellular ROS production and oxidative stress resistance in cells of longer living organisms are not well understood.
The discovery that disruption of the insulin/insulin growth factor (IGF)-I pathway increases life span in Caenorhabditis elegans and Drosophila melanogaster and significantly increases the resistance of these invertebrates to oxidative injury led to the hypothesis that IGF-1 signaling may have an evolutionarily conserved role in regulating cellular ROS production, oxidative stress resistance, and consequentially, organismal life span. Previous studies testing this hypothesis in rodent models of IGF-1 deficiency, however, yielded controversial results. There are mouse models in which disruption of growth hormone (GH)/IGF-1 signaling is associated with significant life span extension (eg, Ames (14) and Snell dwarf mice (15,16), Ghrhr-deficient lit/lit mice (15), growth hormone receptor knockout mice (17), and female mice heterozygous for the deletion of the IGF-1 receptor(18)). Yet, other rodent models with compromised GH/IGF-1 signaling do not exhibit a longevity phenotype (eg, male mice heterozygous for the deletion of the IGF-1 receptor (18) and a genetically GH/IGF-1–deficient strain of Lewis rats (19)). The available data on redox homeostasis and oxidative stress resistance at the cellular level in the aforementioned rodent models of IGF-1 deficiency are also controversial. In Ames dwarf mice, ROS production was reported to be attenuated in liver mitochondria (20), whereas mitochondrial ROS generation in the heart and vasculature is increased in the same model (21). Ames dwarf mice were reported to be resistant to mortality induced by treatment with the herbicide paraquat (22), which elicits cellular oxidative stress and pulmonary edema. Yet, even though the Ames dwarf mice show increased resistance to diquat-induced mortality, their livers sustained significantly greater diquat-induced damage than those of normal littermates (22). Similarly, GHR KO males are also more susceptible to paraquat toxicity as compared with control mice (23). Previous studies also suggest that the liver of the Ames dwarf also may be more sensitive to acetaminophen-induced oxidative damage (5).
Expression of major cellular antioxidant enzyme systems was reported to be increased in the liver, whereas it tends to decrease in the skeletal muscle and the vasculature of Ames dwarf mice (20,21,24–30). Cross-sectional human studies add to the controversies regarding the role of IGF-1 in regulating redox homeostasis and cellular oxidative stress resistance. Importantly, it is well documented that in human patients, GH deficiency and low circulating levels of IGF-1 tend to decrease cancer morbidity and mortality, whereas they significantly increase the risk for cardiovascular and cerebrovascular diseases, the pathogenesis of which involve increased oxidative stress (31–36).
Recently, studies utilizing primary cell cultures isolated from mouse models of IGF-1 deficiency were designed to test predictions of the oxidative stress hypothesis of aging and elucidate the role of low IGF-1 levels in oxidative stress resistance. Interestingly, these studies also yielded controversial results. In culture, H2O2-treated primary hepatocytes from Ames dwarf mice showed lower viability and a higher rate of apoptosis when compared with peroxide-treated wild-type cells (37). In contrast, fibroblasts derived from longer lived Ames and Snell dwarf mice were reported to exhibit increased cellular resistance to oxidative stressors (38,39). On the basis of the reports on fibroblasts, it was hypothesized that low IGF-1 levels in vivo may elicit long-lasting changes in cellular pathways regulating cellular oxidative stress resistance (1,38,39) that remain evident in vitro.
The present study was designed to further test the role of the GH/IGF-1 axis in regulating cellular oxidative stress and oxidative stress resistance utilizing primary fibroblast derived from Lewis dwarf rats. The Lewis dwarf rat is a useful model of human GH/IGF-1 deficiency as these animals have normal pituitary function except for a selective genetic GH deficiency, and they mimic many of the pathophysiological alterations present in human GH/IGF-1–deficient patients as well as elderly individuals (including mild cognitive impairment (40)). Importantly, GH/IGF-1 deficiency in Lewis rats significantly increases the incidence and severity of late-life strokes (19) similar to the effects of GH/IGF-1 deficiency in elderly humans (41–43). It is significant that short-term peripubertal treatment of GH deficient Lewis dwarf rats with GH results in a significant (∼14%) extension of life span (19). Thus, we determined whether life span-extending peripubertal GH replacement increases resistance of primary fibroblasts from Lewis dwarf rats to various forms of oxidative stress–induced cellular injury in vitro.
METHODS
Animals
In the present study, we used male Lewis rats that are heterozygous or homozygous for the spontaneous autosomal recessive dw-4 mutation, which causes a decrease in GH secretion from the pituitary gland (44). Lewis dwarf (dw-4/dw-4) rats have chronically low levels of GH and IGF-1 and make an excellent animal model of isolated GH/IGF-1 deficiency (44). These rats have a spontaneous mutation that results in decreased GH secretion from the pituitary beginning around postnatal Day 26 (16,44,45). Female heterozygous (dw-4/–) Lewis rats were bred with male homozygous Lewis dwarf rats (dw-4/dw-4) to generate heterozygous (dw-4/–) offspring with a normal phenotype (“control”) or homozygous rats (dw-4/dw-4) with a dwarf phenotype (“dwarf”). Classification as control or dwarf was based on their body weight as well as the serum IGF-1 levels at 33 days of age. To assess IGF-1 levels, rats were anesthetized with isofluorane, and serum was obtained via tail bleed. Total IGF-1 levels in serum were determined as previously described (40,45–47). Beginning on Day 35, dwarf rats were divided into two experimental groups: (a) dwarf rats given saline (n = 6) and (b) GH-replete dwarf rats with GH administered beginning at 5 weeks of age and continued throughout the experimental period of 30 days (termed “GH-replete,” n = 6). Saline or GH (300 μg recombinant porcine GH; Alpharma, Victoria, Australia) was injected subcutaneously twice daily. The heterozygous rats were used as controls and given saline injections twice daily from 5 weeks of age to the end of the experimental period. At the end of the experimental period, control and dwarf GH-replete rats had significantly higher serum IGF-1 levels (control: 970 ± 96 ng/mL, dwarf: 464 ± 51 ng/mL, and GH-replete: 849 ± 110 ng/mL; n = 6 in each group) compared with the untreated dwarf rats (p < .05, each), indicating that twice daily administration of GH to the dwarf rats normalized serum IGF-1. Both the control and GH-replete rats expressed the typical phenotype of adequate GH levels, indicated by comparable increases in body weight during the experimental period, whereas untreated dwarf rats gained significantly less weight than the control group (gain in body mass; control: ∼161 g, dwarf: ∼86 g, and GH-replete: ∼152 g). Rats had access to food and water ad libitum and were housed in pairs in the specific pathogen–free barrier facility of the University of Oklahoma Health Sciences Center. The rat colony was evaluated periodically by serologic tests of sentinel mice for viral antibodies and by examination for endoparasites and fur mites; all such tests were negative during the course of the studies reported. Animals were fed standard rodent chow (PicoLab Rodent Diet 20 from Purina Mills, Richmond, IN, containing 20% protein by mass and 24% protein by caloric content) and were euthanized by decapitation. All studies were approved by the Institutional Animal Care and Use Committee.
Isolation of Fibroblasts and Cell Culture Techniques
Primary fibroblast cell lines were established from rats after the 30 day treatment period using the methods of Villegas and colleagues with modifications (12,48). In brief, skin samples were digested with collagenase (at 37°C and 5% CO2 for 30 minutes), then washed twice with Minimum Essential Medium (MEM) medium, and supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, HyClone Laboratories Inc., South Logan, Utah). Cells were plated into 100-mm dishes with MEM media supplemented with 10% heat-inactivated fetal bovine serum plus penicillin/streptomycin/fungizone (at 5% CO2 and 3% O2, at 37°C). After 18 hours, the media was changed to discard unattached cells. The fibroblasts were subsequently cultured as described previously (12). Multiple cell lines were established from each experimental group (n = 6 for each group). One cell line per animal was used at the end of the third passage for measurement of cellular ROS production and antioxidant capacity and assessment of stress resistance. For each experiment, three to six replicates per cell line were used, the results of which were averaged.
Measurement of Cellular O2·− and Peroxide Production
Steady-state cellular O2·− production in cultured fibroblasts was assessed by flow cytometry using the redox-sensitive fluorescent dye dihydroethidium (DHE; 3 × 10−6 mol/L, for 30 minutes) as previously reported (49,50). The data are presented as geometric mean intensity of DHE fluorescence, normalized to the respective mean fluorescence intensities obtained in control fibroblasts.
To assess cellular peroxide levels, the cells were incubated with the cell-permeant oxidative fluorescent indicator dye 5 (and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate-acetyl ester (C-H2DCFDA; Invitrogen, Carlsbad CA, 10 μmol/L, for 30 minutes, at 37°C) as we previously reported (21). C-H2DCFDA fluorescence was assessed by flow cytometry (49,50).
Assessment of Cellular Resistance to Stressors
Cellular stress resistance was assessed in the following manner: Cells were grown in 96-well plates and treated with the following stressors—H2O2 (10, 30, and 100 μmol/L), rotenone (1–10 μmol/L), high glucose (30 mmol/L), tunicamycin (1 μg/mL), thapsigargin (1 μmol/L), paraquat (50 μmol/L), bacterial lipopolysacharide (LPS; 10 μg/mL), and mitomycin (1 μg/mL) for 24 hours. The aforementioned stressors are known to cause apoptosis through increasing oxidative stress, mitochondrial damage, ATP depletion, and/or by damaging DNA, lipids, and proteins and are useful tools to assess cellular oxidative stress resistance. High glucose induces mitochondrial oxidative stress (12,51) and H2O2, the major form of ROS released from the mitochondria in aged cells, increases the formation of highly reactive hydroxyl radicals. Rotenone induces oxidative stress by inhibiting complex I. The herbicide paraquat increases redox cycling and formation of intracellular O2·−. Thapsigargin and tunicamycin cause endoplasmic reticulum stress (tunicamycin abolishes N-linked glycosylation of proteins and thus interferes with the assembly and transport of glycoproteins from the endoplasmic reticulum to the Golgi complex). LPS is an inflammatory stressor and causes oxidative stress and apoptosis by activating toll-like receptors. Mitomycin damages DNA and the mitochondria and triggers apoptosis by activating both the intrinsic and extrinsic caspase cascades. In separate experiments, the cells were grown in 96-well plates, then the medium was removed and replaced by serum-free Dulbecco's Modified Eagle Medium supplemented with 2% bovine serum albumin, antibiotics, and fungizone for 24 hours according to the protocol of Panici and colleagues (52), followed by exposure to cytotoxic stressors (for 6–24 hours).
All incubations were at 37°C in a humidified incubator with 5% CO2 in air. After the treatment period, the ratio of nonviable cells was determined by flow cytometry (Guava HT89) using the Guava ViaCount Assay (Millipore, Billerica, MA) according to the manufacturer's protocol. The ViaCount Assay distinguishes viable and nonviable cells based on differential permeabilities of two proprietary DNA-binding dyes, including a nuclear dye, which stains only nucleated cells, and a viability dye, which stains dying cells. In experiments using the short-term treatment period (6 hours), the ratio of annexin V–positive cells, a marker of early apoptosis, was determined by flow cytometry using the Guava Nexin Assay (Millipore) according to the manufacturer's guidelines.
Cellular Antioxidant Capacity
To compare the capacity of cellular antioxidant systems to counterbalance the deleterious effects of oxidative stress in cultured fibroblasts, we assessed the Hydroxyl Radical Antioxidant Capacity (HORAC) and Oxygen Radical Absorbance Capacity (ORAC) using the OxiSelect HORAC Activity Assay (Cell Biolabs Inc., San Diego, CA) and the OxiSelect ORAC Activity Assay (Cell Biolabs Inc.) according to the manufacturer's guidelines. The HORAC Activity Assay is based on the oxidation-mediated quenching of a fluorescent probe by hydroxyl radicals produced by a hydroxyl radical initiator and Fenton reagent. The ORAC Activity Assay is based on the oxidation of a fluorescent probe by peroxyl radicals produced by a free radical initiator. Antioxidants present in cells delay the quenching of the fluorescent probe until the antioxidant activity in the sample is depleted. The antioxidant capacity of the cells was calculated on the basis of the area under the fluorescence decay curve compared with an antioxidant standard curve obtained with gallic acid (for HORAC) or the water-soluble vitamin E analog Trolox (for ORAC), respectively. Sample protein concentration was used for normalization purposes.
Data Analysis
Data were normalized to the respective control mean values and are expressed as means ± SD. Statistical analyses of data were performed by analysis of variance followed by the Tukey’s post hoc test as appropriate.
RESULTS
Similar ROS Production in Fibroblasts From Control, Dwarf, and GH-Replete Rats
In each fibroblast, cell line steady-state O2·− and peroxide production were assessed by measuring cellular DHE and C-H2DCFDA fluorescence intensity, respectively, by flow cytometry. We found that O2·− (Figure 1A) and peroxide (Figure 1B) production did not differ significantly between fibroblasts from Lewis dwarf rats and cells isolated from heterozygous controls. Peripubertal GH treatment did not significantly influence either cellular production of O2·− or peroxide levels (Figure 1A and B).
Figure 1.
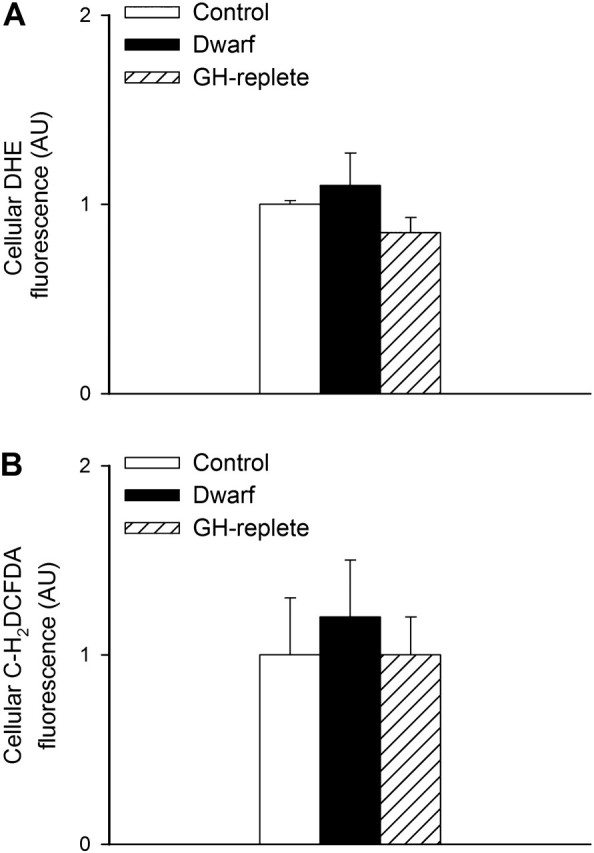
Relative cellular O2·− (panel A) and peroxide (panel B) production in primary fibroblasts derived from control rats, GH/IGF-1–deficient Lewis dwarf rats, and GH-replete dwarf rats. Cellular O2·− and peroxide levels were assessed by flow cytometry using the redox-sensitive fluorescent dyes dihydroethidium and C-H2DCFDA, respectively. Data are mean ± SD (AU: arbitrary units; n = 6 for each data point). The differences among the groups are not significant.
Fibroblasts From GH-Replete Lewis Dwarf Rats Are Resistant to Multiple Forms of Oxidative Stress-Induced Cellular Injury
To assess cellular stress resistance, cultured fibroblasts were treated with a range of stressors—H2O2, rotenone, high glucose, thapsigargin, tunicamycin, paraquat, mitomycin, and LPS. In untreated samples, the ratio of nonviable cells was low (<5%). In fibroblast cultures from control rats, treatment with H2O2 or rotenone significantly decreased cell viability (Figure 2A and B, respectively). The ratio of nonviable cells did not differ significantly between fibroblast cultures from control and Lewis dwarf rats (Figure 2A and B). In contrast, higher doses of H2O2 and rotenone elicited significantly less decline in cellular viability in fibroblast cultures from GH-replete dwarf rats as compared with Lewis dwarf cells (Figure 2A and B).
Figure 2.
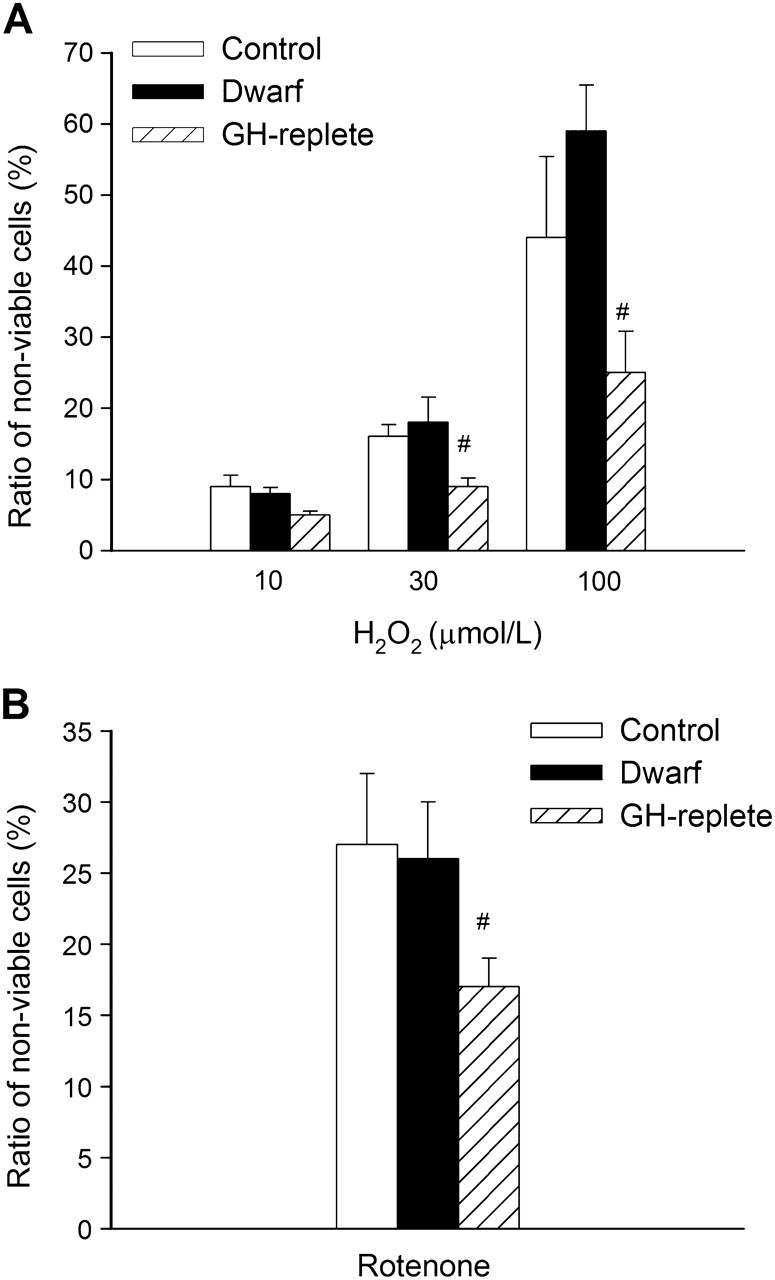
H2O2 (10, 30, and 100 μmol/L, panel A) and rotenone (10 μmol/L, panel B) -induced cell death in primary fibroblasts derived from control rats, GH/IGF-1–deficient Lewis dwarf rats, and GH-replete dwarf rats. The ratio of nonviable cells was assessed by flow cytometry using the Guava ViaCount Assay (see “METHODS”). Data are mean ± SD (n = 6 for each data point). The differences among the control and Lewis dwarf groups are not significant. #p < .05 (GH-replete vs Lewis dwarf).
Treatment of fibroblasts from control rats with high glucose significantly decreased cell viability (Figure 3A). The effect of high glucose on cell viability was comparable in fibroblasts cultures from Lewis dwarf rats and GH-replete rats (Figure 3A). Thapsigargin also induced significant cell death, the magnitude of which did not differ among the three groups of cells (Figure 3B). LPS treatment elicited significant increases in the ratio of nonviable cells in each group of fibroblast cultures. Among the three groups, dwarf cells were the most sensitive to LPS (Figure 3C), whereas LPS-induced decline in cellular viability in fibroblast cultures from GH-replete dwarf rats was significantly attenuated, reaching the levels observed in control cells (Figure 3C). Treatment with mitomycin (Figure 3D) and tunicamycin (data not shown) also resulted in a significant decline in cellular viability in each group of fibroblast cell lines. However, the magnitude of cell death induced by these stimuli did not differ significantly among the three groups. In separate experiments, cells were incubated with serum-free DMEM for 24 hours, followed by exposure to the cytotoxic stressors. We found that H2O2 or rotenone elicited comparable decreases in cell viability in serum-deprived fibroblast cultures from control and Lewis dwarf rats (Figure 4A and B, respectively), whereas H2O2 and rotenone elicited significantly less decline in cellular viability in serum-deprived fibroblast cultures from GH-replete dwarf rats as compared to Lewis dwarf cells (Figure 4A and B). Short-term (6 hours) incubation of fibroblasts with rotenone (1 μmol/L) also increased the ratio of annexin V–positive cells (control: 17% ± 6%, dwarf: 23% ± 6%), whereas rotenone elicited significantly less apoptosis in fibroblast cultures from GH-replete dwarf rats as compared with Lewis dwarf cells (GH-replete: 12% ± 2%; p < .05 vs dwarf).
Figure 3.
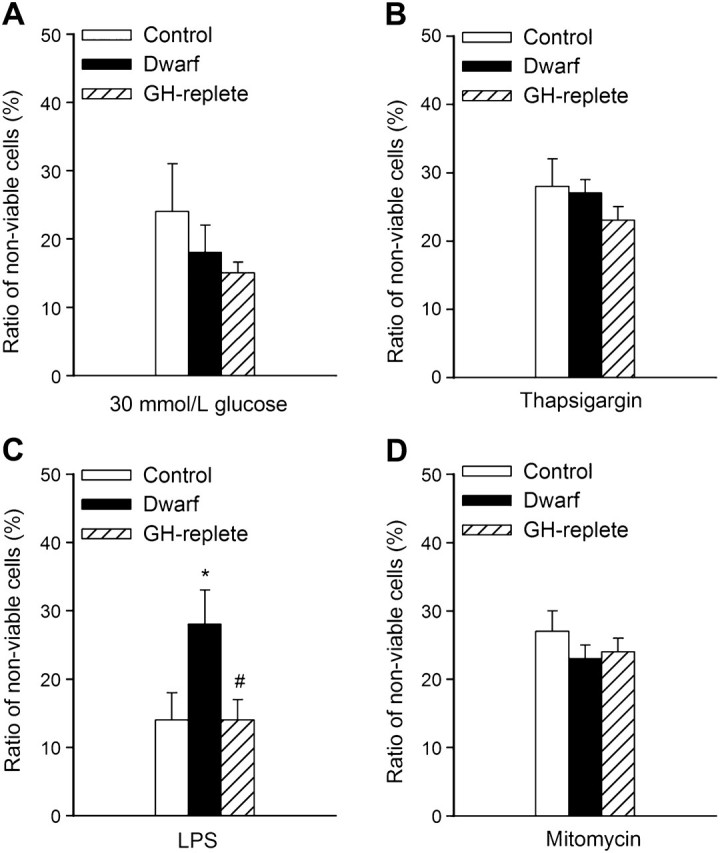
High glucose (30 mmol/L, panel A), thapsigargin (1 μmol/L, panel B), bacterial LPS (10 μg/mL, panel C), and mitomycin (1 μg/mL, Panel D) -induced cell death in primary fibroblasts derived from control rats, GH/IGF-1–deficient Lewis dwarf rats, and GH-replete dwarf rats. The ratio of nonviable cells was assessed by flow cytometry using the Guava ViaCount Assay (see “METHODS”). Data are mean ± SD (n = 6 for each data point) *p < .05 (Lewis dwarf vs control), #p < .05 (GH replete vs Lewis dwarf).
Figure 4.
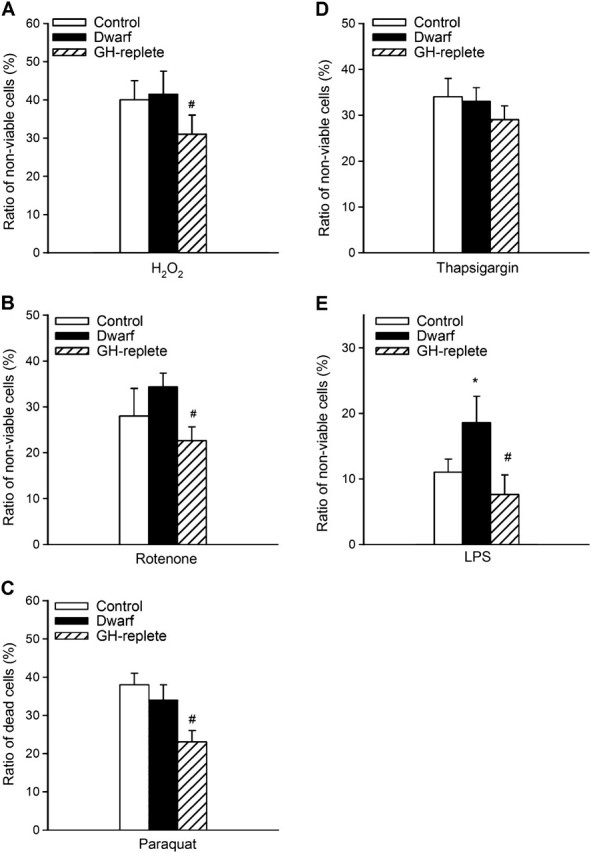
H2O2 (100 μmol/L, panel A), rotenone (1 μmol/L, panel B), paraquat (50 μmol/L, panel C), thapsigargin (1 μmol/L, panel D), and bacterial LPS (10 μg/mL, panel E) -induced cell death in serum-deprived primary fibroblasts derived from control rats, GH/IGF-1–deficient Lewis dwarf rats, and GH-replete dwarf rats. Cells were maintained in serum-free DMEM medium supplemented with 2% bovine serum albumin for 24 hour prior to exposure to oxidative stressors (see “METHODS”). The ratio of nonviable cells was assessed by flow cytometry using the Guava ViaCount Assay (see “METHODS”). Data are mean ± SD (n = 6 for each data point) *p < .05 (Lewis dwarf vs control), #p < .05 (GH replete vs Lewis dwarf).
Cellular Antioxidant Capacity
There were no differences between HORAC (Figure 5A) and ORAC (Figure 5B) in fibroblasts derived from control rats and Lewis dwarf rats. Peribupertal GH treatment of Lewis dwarf rats resulted in a significant increase in cellular ORAC (Figure 5B). Compared with control rats, HORAC was also increased in fibroblasts derived from GH-replete rats; however, the difference between HORAC in Lewis dwarf cells and cells from GH-replete dwarf rats did not reach statistical significance as (Figure 5A).
Figure 5.
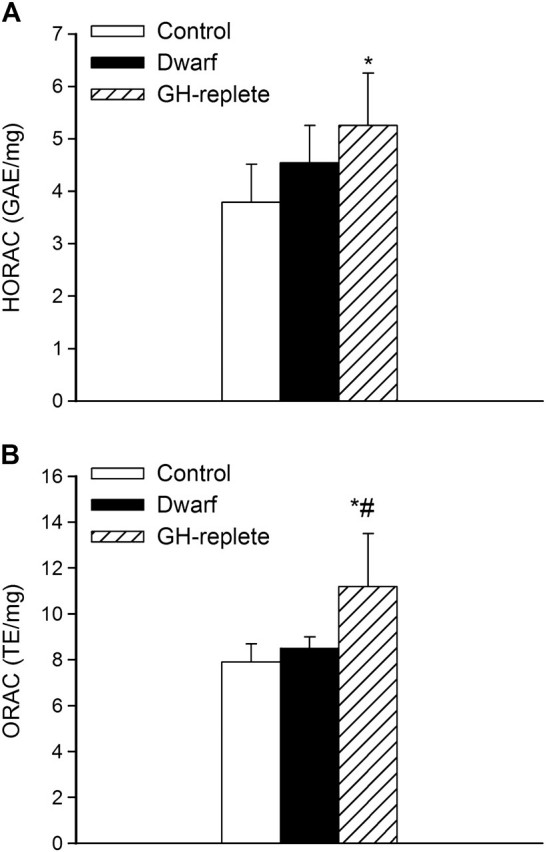
Hydroxyl Radical Antioxidant Capacity (HORAC, panel A) and Oxygen Radical Absorbance Capacity (ORAC, panel B) in primary fibroblasts derived from control rats, GH/IGF-1–deficient Lewis dwarf rats, and GH-replete dwarf rats. Data are mean ± SD (n = 6 for each data point). GAE and TE: gallic acid equivalent and trolox equivalent, respectively, normalized to milligrams protein in sample. *p < .05 (Lewis dwarf vs control), #p < .05 (GH replete vs Lewis dwarf).
DISCUSSION
Postnatal GH and IGF-1 levels are low but increase to higher concentrations immediately before puberty and then progressively decline with increasing age (53–56). Late-life changes in the endocrine milieu, including the age-related decline of the GH/IGF-1 axis, have led to the formulation of a number of neuroendocrine theories of aging, which attempt to explain peripheral aging on the basis of endocrine dysregulation (57). These theories implicate low IGF-1 levels during aging in development of multiple age-related pathological alterations (neurocognitive decline, heart disease, atherosclerosis, and sarcopenia). These theories are in contrast to the hypothesis that low IGF-1 levels induce evolutionarily conserved mechanisms to extend life span, attenuate oxidative stress, increase oxidative stress resistance, and prevent diseases of aging. However, if absence of IGF-1 resulted in a reduced cellular ROS production and an increase in cellular oxidative stress resistance, one would expect that these effects would be manifest throughout the life span. Clearly, this is not the case. There is solid evidence that cellular ROS production significantly increases, whereas oxidative stress resistance declines with advancing age (58), despite the substantial decreases in IGF-1 levels (11). In recent years, significant advances have been made toward the reconciliation of the aforementioned two contradictory theories on the role of IGF-1 in the aging process. Recent studies by the Sonntag, Bartke, and Miller laboratories (19,52) have provided strong evidence that in rodents, levels of GH/IGF-1 during a critical developmental time window play a key role in determining life span and/or development of age-related diseases later in life. Yet, the role of GH/IGF-1 levels early in life on cellular redox homeostasis and stress resistance remains less understood.
This is the first study to characterize the role of GH/IGF-1 during a critical developmental time window on cellular ROS production, antioxidant capacity, and oxidative stress response signatures using cultured fibroblasts derived from Lewis dwarf rats as a model. Interestingly, in the present study, low circulating IGF-1 levels present in vivo in Lewis dwarf rats do not elicit long-lasting alterations in basal cellular ROS generation in cultured fibroblasts (Figure 1). Our findings are in accordance with the conclusions of recent studies showing that cellular and mitochondrial ROS production as well as basal levels of lipid peroxidation do not differ between primary skin-derived fibroblasts from the long-lived Snell dwarf mutant mouse and cells derived from wild-type mice (1) (of note, Snell-derived fibroblasts exhibit lower levels of lipid peroxidation after exposure to organic peroxides). Rotenone treatment also results in comparable increases in mitochondrial ROS production in fibroblasts derived from IGF-1 deficient and control animals (1), suggesting that low in vivo IGF-1 levels per se do not render cells resistant to oxidative stress associated with complex I inhibition. It is important to note that our recent studies demonstrate that vascular and/or cardiac tissues freshly isolated from GH/IGF-1 deficient dwarf rats (59) and Ames dwarf mice (21) exhibit an increased, rather than a decreased, O2·− and H2O2 production. Moreover, treatment of cultured endothelial cells and cardiomyocytes with IGF-1 results in a significant attenuation of mitochondrial ROS production in both cell types (21). Collectively, these findings argue against the concept, based on the oxidative stress hypothesis of aging, that in mammalian cells, low IGF-1 levels alone exert prosurvival effects by attenuating cellular ROS production.
Interestingly, we find that primary fibroblasts derived from GH/IGF-1–deficient Lewis rats are not generally resistant to multiple forms of oxidative stress-induced cellular injury (Figures 2–4). These findings are in contrast with the conclusions of earlier studies demonstrating that fibroblasts derived from long-lived Ames and Snell dwarf mice exhibit increased cellular resistance to H2O2 and some other oxidative stressors (ultraviolet light, paraquat) (38,39,52). Interestingly, previous studies from the Brown-Borg laboratory demonstrated that primary hepatocytes from Ames dwarf mice exhibit increased sensitivity to H2O2-induced apoptosis compared with cells of wild-type mice (37). At present, the factors responsible for the discrepancy between the aforementioned findings are unknown. One possibility is that technical differences in the stress methodology used in these studies may partially explain the observed discrepancies. Importantly, the methods of the detection of loss in cellular viability differ between our study and important previous reports (2,3,7,38). Specifically, we used flow cytometry to assay cell viability, whereas in earlier studies, survival was measured by a plate reader using a test based on reductive cleavage of a tetrazolium dye (2,3,7,38). Another technical difference of potential importance is that we exposed the fibroblasts to stress for a 24-hour period, which is longer than the short-term time period (6 hours) used in many previous studies (4). We chose to study cell viability at the 24 hour timepoint because oxidative stress–induced apoptosis is fully manifest as reflected by the time course of H2O2-induced caspase 3 activity in various fibroblast strains (60). Although using a 6-hour time period of stress exposure in Lewis dwarf rat fibroblasts we observed stress response signatures similar to that obtained using a 24-hour stress exposure, further studies are warranted to contrast the time course of oxidative stress–induced apoptosis in various cell lines derived from different animal models of IGF-1 deficiency. Because many of the stress assays used previously in the Miller laboratory (3–5,7,8,38) included 18–24 hours of culture in medium without serum prior to exposure to stress, we also tested this protocol in our experiments as well. Because we found that low-serum preincubation did not alter the differences between Lewis dwarf and control fibroblasts in stress resistance (Figure 4), we believe that the discrepancy in fibroblast stress resistance between our work and previous studies represents a variation between the animal models of GH/IGF-1 deficiency used rather than differences in the assay methodology.
It is significant that short-term replacement of GH has major effects on cellular oxidative stress resistance that persists in cell culture. Specifically, we found that peripubertal GH treatment in Lewis dwarf rats increases cellular resistance to diverse oxidative stressors (including H2O2, rotenone, paraquat, and LPS; Figures 2A and B and 3C). Because the same treatment was shown to extend life span in Lewis dwarf rats (19), it is tempting to speculate that peripubertal GH treatment of Lewis dwarf rats during a critical developmental time window also results in long-lasting increases in cellular oxidative stress resistance, which contributes to the extension of life span in these animals. Recent important data from the Miller laboratory (52) are also consistent with the concept that patterns of cellular stress resistance can be influenced by GH and/or IGF-1 signals in the first two months of life in Ames dwarf mice. However, in contrast to our findings in the Lewis dwarf rat, GH treatment of Ames dwarf mice during early postnatal development decreased, rather than increased, resistance of skin-derived fibroblasts to cadmium and paraquat (but not to rotenone) (52), producing a set of cellular traits normally seen in fibroblasts derived from wild-type control mice. Our current data and previous results from the Miller laboratory (3,4,38) showing that rodent fibroblasts retain their unique oxidative stress resistance signatures in culture through many rounds of mitosis are consistent with the presence of epigenetic control mechanisms induced in vivo by neurohormonal factors and maintained in extended culture. This concept is further supported by the finding that the stress resistance signatures fundamentally differ between cells taken from newborn Snell dwarf mice and cells derived from ∼3-month-old Snell dwarf mice in which postnatal changes in circulating hormone levels are manifest (52).
The factors responsible for the differential oxidative stress response signatures in Lewis dwarf rats and mouse models of GH/IGF-1 deficiency are unknown. It should be noted that the endocrine defects in the aforementioned animal models are not identical. Ames and Snell dwarf mice exhibit chronic GH/IGF-1 deficiency and other complex endocrine defects (including low thyroid-stimulating hormone and prolactin levels) throughout early development, which may modulate cellular stress resistance. In contrast, Lewis dwarf rats have a specific GH/IGF-1 deficiency that appears around 4 weeks of age. Although when compared with Ames dwarf mice, GH receptor knockout mice (which lack a functional GH receptor) are similar in many respects including undetectable circulating IGF-1 levels and their cellular stress resistance signatures (3), GHR KO mice differ from Ames mice in their decreased antioxidative enzyme expression and functionality (23,61,62). It is possible that the cellular changes induced by alterations in GH/IGF-1 levels depend on the onset/duration of GH/IGF-1 deficiency (eg, the effects of perinatal, peripubertal, or adult-onset hormone deficiencies are likely different). For example, short-term GH treatment of GH/IGF-1–deficient dwarf animals with GH delays the appearance of cognitive decline and development of intracerebral hemorrhage (19). Thus, our data are consistent with the hypothesis that the effects of blood levels of GH/IGF-1 are unique to specific cell types, species, and the age at which the hormonal changes occur.
The GH/IGF-1–sensitive molecular mechanisms involved in regulation of cellular stress resistance may include antioxidant enzymes, low molecular weight antioxidants, the plasma membrane antioxidant redox system, heat shock proteins, cellular repair factors (including DNA repair pathways and factors involved in protein homeostasis), Phase II detoxification proteins, and regulators of apoptosis. In the present study, we determined whether in the Lewis dwarf rats peripubertal alterations in GH/IGF-1 levels elicit changes in cellular antioxidant capacities that persist in culture. Interestingly, we found that antioxidant capacity was not decreased in Lewis dwarf fibroblasts, whereas peripubertal GH treatment increased cellular antioxidant capacity in fibroblasts of Lewis dwarf rats (Figure 5A and B). Further studies are warranted to elucidate the role of higher basal levels of free radical detoxification systems in oxidative stress response signatures in GH-treated Lewis dwarf rats. Because expression of antioxidant genes regulated by the transcription factor NF-E2–related factor 2 (Nrf2), a key regulator of the adaptive antioxidant defense response, is decreased in the vasculature of adult Lewis dwarf rats (59), future studies should also investigate the regulation of this pathway by peripubertal changes in the somatotropic axis at the cellular level. In that regard, it is interesting that expression of Nrf2 and Nrf2 target genes was reported to increase in parenchymal tissues of Snell dwarf mice (1), which may explain the differential stress response signatures observed in cells of Snell dwarf mice and Lewis dwarf rats. In addition to the role of antioxidant enzymes and low molecular weight antioxidants, other cellular mechanisms that may be involved in regulation of cellular stress resistance by the somatotropic axis include heat shock proteins (63), cellular repair factors (including DNA repair pathways and factors involved in protein homeostasis), and regulators of apoptosis (eg, Bcl2). Further studies are warranted to determine how these cellular mechanisms are altered in the Lewis dwarf rat and whether peripubertal changes in GH/IGF-1 levels in this model elicit epigenetic changes in the regulation of the aforementioned pathways that would affect life span and healthspan in these animals.
FUNDING
This work was supported by grants from the American Diabetes Association (to ZU), American Federation for Aging Research (to AC), the University of Oklahoma College of Medicine Alumni Association (to AC) and the NIH (AG031085 to AC; AT006526 and HL077256 to ZU; P01 AG11370 to WES).
References
- 1.Leiser SF, Miller RA. Nrf2 signaling, a mechanism for cellular stress resistance in long-lived mice. Mol Cell Biol. 2010;30(3):871–884. doi: 10.1128/MCB.01145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maynard SP, Miller RA. Fibroblasts from long-lived Snell dwarf mice are resistant to oxygen-induced in vitro growth arrest. Aging Cell. 2006;5(1):89–96. doi: 10.1111/j.1474-9726.2006.00187.x. [DOI] [PubMed] [Google Scholar]
- 3.Salmon AB, Murakami S, Bartke A, Kopchick J, Yasumura K, Miller RA. Fibroblast cell lines from young adult mice of long-lived mutant strains are resistant to multiple forms of stress. Am J Physiol Endocrinol Metab. 2005;289(1):E23–E29. doi: 10.1152/ajpendo.00575.2004. [DOI] [PubMed] [Google Scholar]
- 4.Murakami S, Salmon A, Miller RA. Multiplex stress resistance in cells from long-lived dwarf mice. FASEB J. 2003;17(11):1565–1566. doi: 10.1096/fj.02-1092fje. [DOI] [PubMed] [Google Scholar]
- 5.Harper JM, Salmon AB, Chang Y, Bonkowski M, Bartke A, Miller RA. Stress resistance and aging: influence of genes and nutrition. Mech Ageing Dev. 2006;127(8):687–694. doi: 10.1016/j.mad.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salmon AB, Sadighi Akha AA, Buffenstein R, Miller RA. Fibroblasts from naked mole-rats are resistant to multiple forms of cell injury, but sensitive to peroxide, ultraviolet light, and endoplasmic reticulum stress. J Gerontol A Biol Sci Med Sci. 2008;63(3):232–241. doi: 10.1093/gerona/63.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Page MM, Salmon AB, Leiser SF, et al. Mechanisms of stress resistance in Snell dwarf mouse fibroblasts: enhanced antioxidant and DNA base excision repair capacity, but no differences in mitochondrial metabolism. Free Radic Biol Med. 2009;46(8):1109–1118. doi: 10.1016/j.freeradbiomed.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leiser SF, Salmon AB, Miller RA. Correlated resistance to glucose deprivation and cytotoxic agents in fibroblast cell lines from long-lived pituitary dwarf mice. Mech Ageing Dev. 2006;127(11):821–829. doi: 10.1016/j.mad.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Csiszar A, Labinskyy N, Zhao X, et al. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6(6):783–797. doi: 10.1111/j.1474-9726.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 10.Ungvari Z, Krasnikov BF, Csiszar A, et al. Testing hypotheses of aging in long-lived mice of the genus Peromyscus: association between longevity and mitochondrial stress resistance, ROS detoxification pathways and DNA repair efficiency. 30(2-3):121–133. doi: 10.1007/s11357-008-9059-y. (Dordr). 2008 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ungvari Z, Buffenstein R, Austad SN, Podlutsky A, Kaley G, Csiszar A. Oxidative stress in vascular senescence: lessons from successfully aging species. Front Biosci. 2008;13:5056–5070. doi: 10.2741/3064. [DOI] [PubMed] [Google Scholar]
- 12.Labinskyy N, Mukhopadhyay P, Toth J, et al. Longevity is associated with increased vascular resistance to high glucose-induced oxidative stress and inflammatory gene expression in Peromyscus leucopus. Am J Physiol Heart Circ Physiol. 2009;296(4):H946–H956. doi: 10.1152/ajpheart.00693.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 14.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384(6604):33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 15.Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98(12):6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter CS, Ramsey MM, Sonntag WE. A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends Genet. 2002;18(6):295–301. doi: 10.1016/S0168-9525(02)02696-3. [DOI] [PubMed] [Google Scholar]
- 17.Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm IGF Res. 2008;18(6):455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 19.Sonntag WE, Carter CS, Ikeno Y, et al. Adult-onset growth hormone and insulin-like growth factor I deficiency reduces neoplastic disease, modifies age-related pathology, and increases life span. Endocrinology. 2005;146(7):2920–2932. doi: 10.1210/en.2005-0058. [DOI] [PubMed] [Google Scholar]
- 20.Brown-Borg H, Johnson W, Rakoczy S, Romanick M. Mitochondrial oxidant generation and oxidative damage in Ames dwarf and GH transgenic mice. J Am Aging Assoc. 2001;24:85–96. doi: 10.1007/s11357-001-0012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Csiszar A, Labinskyy N, Perez V, et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008;295(5):H1882–H1894. doi: 10.1152/ajpheart.412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bokov AF, Lindsey ML, Khodr C, Sabia MR, Richardson A. Long-lived ames dwarf are resistant to chemical stressors. J Gerontol A Biol Sci Med Sci. 2009;64(8):819–827. doi: 10.1093/gerona/glp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hauck SJ, Aaron JM, Wright C, Kopchick JJ, Bartke A. Antioxidant enzymes, free-radical damage, and response to paraquat in liver and kidney of long-living growth hormone receptor/binding protein gene-disrupted mice. Horm Metab Res. 2002;34(9):481–486. doi: 10.1055/s-2002-34787. [DOI] [PubMed] [Google Scholar]
- 24.Brown-Borg HM, Rakoczy SG, Romanick MA, Kennedy MA. Effects of growth hormone and insulin-like growth factor-1 on hepatocyte antioxidative enzymes. Exp Biol Med (Maywood, NJ) 2002;227(2):94–104. doi: 10.1177/153537020222700203. [DOI] [PubMed] [Google Scholar]
- 25.Brown-Borg HM, Rakoczy SG. Glutathione metabolism in long-living Ames dwarf mice. Exp Gerontol. 2005;40(1–2):115–120. doi: 10.1016/j.exger.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Brown-Borg HM, Rakoczy SG, Uthus EO. Growth hormone alters components of the glutathione metabolic pathway in Ames dwarf mice. Ann N Y Acad Sci. 2004;1019:317–320. doi: 10.1196/annals.1297.053. [DOI] [PubMed] [Google Scholar]
- 27.Romanick MA, Rakoczy SG, Brown-Borg HM. Long-lived Ames dwarf mouse exhibits increased antioxidant defense in skeletal muscle. Mech Ageing Dev. 2004;125(4):269–281. doi: 10.1016/j.mad.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Brown-Borg HM, Rakoczy SG. Catalase expression in delayed and premature aging mouse models. Exp Gerontol. 2000;35(2):199–212. doi: 10.1016/s0531-5565(00)00079-6. [DOI] [PubMed] [Google Scholar]
- 29.Brown-Borg HM, Rakoczy SG, Uthus EO. Growth hormone alters methionine and glutathione metabolism in Ames dwarf mice. Mech Ageing Dev. 2005;126(3):389–398. doi: 10.1016/j.mad.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Brown-Borg HM, Rakoczy SG. Growth hormone administration to long-living dwarf mice alters multiple components of the antioxidative defense system. Mech Ageing Dev. 2003;124(10–12):1013–1024. doi: 10.1016/j.mad.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Rosen T, Bengtsson BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990;336(8710):285–288. doi: 10.1016/0140-6736(90)91812-o. [DOI] [PubMed] [Google Scholar]
- 32.Bates AS, Van’t Hoff W, Jones PJ, Clayton RN. The effect of hypopituitarism on life expectancy. J Clin Endocrinol Metab. 1996;81(3):1169–1172. doi: 10.1210/jcem.81.3.8772595. [DOI] [PubMed] [Google Scholar]
- 33.Bulow B, Hagmar L, Mikoczy Z, Nordstrom CH, Erfurth EM. Increased cerebrovascular mortality in patients with hypopituitarism. Clin Endocrinol (Oxf) 1997;46(1):75–81. doi: 10.1046/j.1365-2265.1997.d01-1749.x. [DOI] [PubMed] [Google Scholar]
- 34.Tomlinson JW, Holden N, Hills RK, et al. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet. 2001;357(9254):425–431. doi: 10.1016/s0140-6736(00)04006-x. [DOI] [PubMed] [Google Scholar]
- 35.Vasan RS, Sullivan LM, D’Agostino RB, et al. Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the Framingham Heart Study. Ann Intern Med. 2003;139(8):642–648. doi: 10.7326/0003-4819-139-8-200310210-00007. [DOI] [PubMed] [Google Scholar]
- 36.Laughlin GA, Barrett-Connor E, Criqui MH, Kritz-Silverstein D. The prospective association of serum insulin-like growth factor I (IGF-I) and IGF-binding protein-1 levels with all cause and cardiovascular disease mortality in older adults: the Rancho Bernardo Study. J Clin Endocrinol Metab. 2004;89(1):114–120. doi: 10.1210/jc.2003-030967. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy MA, Rakoczy SG, Brown-Borg HM. Long-living Ames dwarf mouse hepatocytes readily undergo apoptosis. Exp Gerontol. 2003;38(9):997–1008. doi: 10.1016/s0531-5565(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 38.Harper JM, Salmon AB, Leiser SF, Galecki AT, Miller RA. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6(1):1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh CC, Papaconstantinou J. Dermal fibroblasts from long-lived Ames dwarf mice maintain their in vivo resistance to mitochondrial generated reactive oxygen species (ROS) Aging (Albany NY) 2009;1(9):784–802. doi: 10.18632/aging.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nieves-Martinez E, Sonntag WE, Wilson A, et al. Early-onset GH deficiency results in spatial memory impairment in mid-life and is prevented by GH supplementation. J Endocrinol. 2010;204(1):31–36. doi: 10.1677/JOE-09-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denti L, Annoni V, Cattadori E, et al. Insulin-like growth factor 1 as a predictor of ischemic stroke outcome in the elderly. Am J Med. 2004;117(5):312–317. doi: 10.1016/j.amjmed.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 42.Johnsen SP, Hundborg HH, Sorensen HT, et al. Insulin-like growth factor (IGF) I, -II, and IGF binding protein-3 and risk of ischemic stroke. J Clin Endocrinol Metab. 2005;90(11):5937–5941. doi: 10.1210/jc.2004-2088. [DOI] [PubMed] [Google Scholar]
- 43.Bondanelli M, Ambrosio MR, Onofri A, et al. Predictive value of circulating insulin-like growth factor I levels in ischemic stroke outcome. J Clin Endocrinol Metab. 2006;91(10):3928–3934. doi: 10.1210/jc.2006-1040. [DOI] [PubMed] [Google Scholar]
- 44.Charlton HM, Clark RG, Robinson IC, et al. Growth hormone-deficient dwarfism in the rat: a new mutation. J Endocrinol. 1988;119(1):51–58. doi: 10.1677/joe.0.1190051. [DOI] [PubMed] [Google Scholar]
- 45.Carter CS, Ramsey MM, Ingram RL, et al. Models of growth hormone and IGF-1 deficiency: applications to studies of aging processes and life-span determination. J Gerontol A Biol Sci. 2002;57(5):B177–B188. doi: 10.1093/gerona/57.5.b177. [DOI] [PubMed] [Google Scholar]
- 46.Hua K, Forbes ME, Lichtenwalner RJ, Sonntag WE, Riddle DR. Adult-onset deficiency in growth hormone and insulin-like growth factor-I alters oligodendrocyte turnover in the corpus callosum. Glia. 2009;57(10):1062–1071. doi: 10.1002/glia.20829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsey MM, Ingram RL, Cashion AB, et al. Growth hormone-deficient dwarf animals are resistant to dimethylbenzanthracine (DMBA)-induced mammary carcinogenesis. Endocrinology. 2002;143(10):4139–4142. doi: 10.1210/en.2002-220717. [DOI] [PubMed] [Google Scholar]
- 48.Villegas J, McPhaul M. Establishment and culture of human skin fibroblasts. Curr Protoc Mol Biol. 2005 doi: 10.1002/0471142727.mb2803s71. Chapter 28:Unit 28.3. DOI: 10.1002/0471142727.mb2803s71. [DOI] [PubMed] [Google Scholar]
- 49.Csiszar A, Labinskyy N, Jimenez R, et al. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009;130(8):518–527. doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ungvari Z, Bagi Z, Feher A, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010;299(1):H18–H24. doi: 10.1152/ajpheart.00260.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2(9):2295–2301. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panici JA, Harper JM, Miller RA, Bartke A, Spong A, Masternak MM. Early life growth hormone treatment shortens longevity and decreases cellular stress resistance in long-lived mutant mice. FASEB J. 2010;24:1–7. doi: 10.1096/fj.10-163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Argente J, Barrios V, Pozo J, et al. Normative data for insulin-like growth factors (IGFs), IGF-binding proteins, and growth hormone-binding protein in a healthy Spanish pediatric population: age- and sex-related changes. J Clin Endocrinol Metab. 1993;77(6):1522–1528. doi: 10.1210/jcem.77.6.7505288. [DOI] [PubMed] [Google Scholar]
- 54.Groban L, Pailes NA, Bennett CD, et al. Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin II expression in senescent rats. J Gerontol A Biol Sci Med Sci. 2006;61(1):28–35. doi: 10.1093/gerona/61.1.28. [DOI] [PubMed] [Google Scholar]
- 55.Sonntag WE, Lynch CD, Cooney PT, Hutchins PM. Decreases in cerebral microvasculature with age are associated with the decline in growth hormone and insulin-like growth factor 1. Endocrinology. 1997;138(8):3515–3520. doi: 10.1210/endo.138.8.5330. [DOI] [PubMed] [Google Scholar]
- 56.Breese CR, Ingram RL, Sonntag WE. Influence of age and long-term dietary restriction on plasma insulin-like growth factor-1 (IGF-1), IGF-1 gene expression, and IGF-1 binding proteins. J Gerontol. 1991;46(5):B180–B187. doi: 10.1093/geronj/46.5.b180. [DOI] [PubMed] [Google Scholar]
- 57.Everitt AV. The neuroendocrine system and aging. Gerontology. 1980;26(2):108–119. doi: 10.1159/000212403. [DOI] [PubMed] [Google Scholar]
- 58.Miyoshi N, Oubrahim H, Chock PB, Stadtman ER. Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proc Natl Acad Sci U S A. 2006;103(6):1727–1731. doi: 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ungvari Z, Gautam T, Koncz P, et al. Vasoprotective Effects of Life Span-Extending Peripubertal GH Replacement in Lewis Dwarf Rats. J Gerontol A Biol Sci Med Sci. 2010;65(11):1145–1156. doi: 10.1093/gerona/glq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugano N, Ito K, Murai S. Cyclosporin A inhibits H2O2-induced apoptosis of human fibroblasts. FEBS Lett. 1999;447(2–3):274–276. doi: 10.1016/s0014-5793(99)00312-9. [DOI] [PubMed] [Google Scholar]
- 61.Brown-Borg HM, Rakoczy SG, Sharma S, Bartke A. Long-living growth hormone receptor knockout mice: potential mechanisms of altered stress resistance. Exp Gerontol. 2009;44(1–2):10–19. doi: 10.1016/j.exger.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chandrashekar V, Bartke A, Coschigano KT, Kopchick JJ. Pituitary and testicular function in growth hormone receptor gene knockout mice. Endocrinology. 1999;140(3):1082–1088. doi: 10.1210/endo.140.3.6557. [DOI] [PubMed] [Google Scholar]
- 63.Swindell WR, Masternak MM, Kopchick JJ, Conover CA, Bartke A, Miller RA. Endocrine regulation of heat shock protein mRNA levels in long-lived dwarf mice. Mech Ageing Dev. 2009;130(6):393–400. doi: 10.1016/j.mad.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]